

Abstract
3-Aryltropanes have been widely explored for potential medications for remediation of cocaine abuse. Research has focused predominantly on 8-azatropanes and it is now well recognized that these compounds can be designed to manifest varied selectivity and potency for inhibition of the dopamine, serotonin and norepinephrine uptake systems. We had reported that the 8-nitrogen atom present in the 3-aryltropanes is not essential for tropanes to bind to monoamine uptake systems. We demonstrated that compounds in which the amine had been exchanged for an ether or a thioether retained binding potency and selectivity. We have now designed bivalent compounds in which two tropane moieties are linked by an intervening chain. These 8-homo- and 8-heterotropane bivalent compounds allowed a search for adjacent tropane binding sites on the DAT as well as a further exploration of whether the binding sites for 8-azatropanes are the same as those for other 8-heterotropanes. A comparison of these compounds with their progenitor tropanes cast into doubt the existence of proximal binding sites on the DAT, and offered support for the existence of different binding sites for the 8-azatropanes compared with 8-oxa- and 8-thiatropanes. Indeed, 8-aza bivalent tropanes inhibited DAT with potency about 10-fold lower (DAT: IC50 = 31 nM) than their monovalent counterparts. Furthermore, bivalent ligands in which one or both of the tropanes was devoid of an amine suffered a further loss of inhibitory potency. We conclude that it is unlikely that there exist two tropane binding sites in close proximity to one another on either the DAT or SERT.
Keywords: Monoamine transporter ligands, Cocaine medications, Bivalent ligands
1. Introduction
The search for a pharmacotherapy for the treatment of cocaine addiction has not yet yielded a compound of proven clinical utility. Cocaine is a potent stimulant of the mammalian central nervous system and its stimulatory activity is a consequence of its interactions with monoamine uptake systems located in the striatum.1, 2 The dopamine transporter (DAT) and serotonin transporter (SERT), located presynaptically on neurons in the striatum, have been specifically implicated in the reinforcing and stimulatory properties of cocaine.3-9 Cocaine binds to both the DAT and SERT and inhibits reuptake of synaptic dopamine and serotonin. The excess dopamine in the synapse then stimulates postsynaptic receptors, which in turn leads to the pharmacological sequellae of cocaine addiction. The search for potential medications for cocaine abuse has consequently focused largely on the design of compounds that target the DAT and SERT, and a considerable effort has been placed on the search for DAT and SERT selective molecules.10-16 The DAT, the predominant mechanism for reuptake of dopamine in the synapse, has been the focus of particular attention. Many moderately selective DAT inhibitors have been reported.12, 16-20 Far fewer compounds that manifest exclusive DAT inhibitory potency have been discovered.12, 21 The class of bicyclo[3.2.1]octanes has been a focus of much attention in the design of prospective medications for cocaine abuse.13, 14, 22, 23 8-Azabicyclo[3.2.1]octanes (8-azatropanes),24 8-oxabicyclo[3.2.1])octanes (8-oxatropanes),11 8-thiabicyclo[3.2.1])octanes, (8-thiatropanes),13 and 8-carbabicyclo[3.2.1])octanes, (8-carbatropanes)25 have all been evaluated. These 8-heterotropanes (8-aza, 8-oxa, 8-thia, 8-carba) have provided a broad array of promising DAT inhibitors.
The reinforcing and addictive properties of cocaine are thought to be related to its pharmacokinetic profile of extremely rapid onset and short duration of action.26 Therefore, a biological rationale that has guided much of the search for pharmacotherapies has been to seek “replacement agonist therapies” that inhibit cocaine binding but that manifest a slow onset and a long duration of action. Another approach to such “cocaine replacement therapies” might utilize the possible existence of proximal cocaine binding sites on the DAT. If such proximal binding sites exist, it may be possible to design highly selective inhibitors of cocaine binding. Furthermore, if these proximal sites are located at a different distance from one another with respect to dopamine uptake and cocaine binding, it may conceivably be possible to design a DAT selective, dopamine sparing partial agonist with prolonged duration of action as well as slow onset of activity. Therefore the existence of proximal binding sites could offer a unique window on the design of cocaine medications.
This bivalent ligand concept has been presented and validated by Portoghese27, 28 in his evaluation of molecules that target opioid receptors. In that work he demonstrated that potent but non-selective monovalent ligands that inhibit more than one opioid receptor could be linked to one another, through a molecular spacer, to provide bivalent ligands that then proved highly selective. In this fashion, the linker itself provided a new “dimension for the recognition process”27 because each component of the linked ligands bound to proximal binding sites on the target receptors, and further, these sites were localized at different distances from one another on different receptors.
Tamiz et al.29 later explored the introduction of a spacer in a class of bivalent 4-arylmethypiperidines in an effort to obtain selective SERT versus DAT inhibitors. They reported the striking finding that while the parent monovalent methyl 4β-(4-chlorophenyl)-1-methylpiperidine-3α-carboxylate manifested poor inhibitory potency at SERT (Ki = 3,600 nM), a bivalent analog with a C5 spacer showed considerable SERT potency (Ki = 1.2 nM). They concluded that proximal binding sites probably exist on the SERT.
Christopoulus et al.30 reported the design and evaluation of bivalent muscarinic acetylcholine receptor antagonists. They found that the binding potency of the bivalent compounds was increased by as much as 300-fold (M1) and 700-fold (M2) over the monovalent parent compound. Furthermore, M1 and M2 selectivity versus M3 and M5 receptors could be attained. The implication of these results was that two proximal binding sites existed for inhibition of M1 and M2 receptors.
The bivalent approach has also been explored by Fandrick et al.31 They reported that while the parent monovalent 8-azatropane RTI-3132 inhibited the DAT with a potency of about Ki = 1 nM, the bivalent RTI-31 ligands manifested considerably poorer DAT inhibitory potency. However, they reported that bivalent compounds with longer intervening linkers showed greater inhibitory potency than those with shorter interlinking chains. Furthermore, they observed that the length of the interlinking spacers influenced the effect on the discrimination ratios (Ki/IC50) between inhibition of radioligand binding to the DAT inhibition and inhibition of DA reuptake. The longest spacer provided as much as a 130-fold preference for DAT inhibition over DA reuptake inhibition. Cashman consequently suggested that “certain bivalent aryltropanes analogues bind to different domains on the hDAT”.31
The DAT has been proposed to exist as a dimer or tetramer.33-37 However, the premise that we have explored, and on which we now report, is the possible existence of two proximal tropane binding regions on a single dopamine transporter.31 In addition, selected bivalent ligands have provided an opportunity to further explore whether 8-azatropanes, 8-oxatropanes and 8-thiatropanes bind at identical domains11 on the DAT.
2. Chemistry
2.1. Compound Design
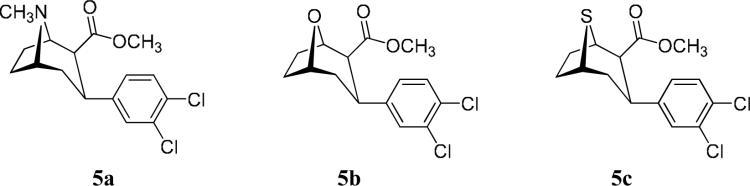
The parent compounds selected for this study were based upon the 2β-carbomethoxy-3-aryl-8-N,O, or S-bicyclo[3.2.1]octane skeleton. This skeleton was specifically selected from among the available stereoisomers because it generates compounds of comparable nanomolar potency at DAT and SERT.13 In contrast, the 2β-carbomethoxy-3α-aryl-8-N,O, or S-bicyclo[3.2.1]octane and the 2-carbomethoxy-3-aryl-8-N,O, or S-bicyclo[3.2.1]oct-2-enes are considerably more potent inhibitors of the DAT than the SERT. The 3β-(3,4-dichlorophenyl)-substitution was selected because this 3,4-dichloro motif has offered among the most potent DAT and SERT inhibitors available.13, 38, 39 The linking-chain lengths were restricted to a maximum of 10 intervening methylene groups. Consequently interaction of each of the tropane moieties of the bivalent ligands with tropane binding sites on adjacent DATs of a DAT dimer would be highly unlikely. The parent C2-ester compounds and their inhibitory potencies at DAT and SERT are presented in Table 1.
Table 1.
Monovalent ligands: Inhibition of [3H]WIN 35,428 binding to the hDAT and [3H]citalopram binding to the hSERT for 5a, 5b and 5ca,b
 | ||||
|---|---|---|---|---|
|
8-Position |
Compound |
Number |
DAT |
SERT |
| IC50 (nM) | ||||
| 8-NCH3 | 5a | O-401 | 1.1 | 2.5 |
| 8-O | 5b | O-1072 | 3.3 | 4.7 |
| 8-S | 5c | O-3516 | 2.0 | 3.0 |
Each value is the mean of 2 or more independent experiments each conducted in different brains and in triplicate. Errors generally do not exceed 10% between replicate experiments.
Data from Pham-Huu et al. Bioorg. & Med. Chem. 15, 1067, 2007.
2.2 Synthesis
The starting 2β-carbomethoxy-3β-(3,4-dichlorophenyl)-8-aza-, 8-oxa-, and 8-thia-bicyclo[3.2.1]octanes (8-azatropane, 8-oxatropane and 8-thiatropane) were synthesized (Scheme 1) as described previously. In brief, a critical ketoester 19 served as the starting point for the synthesis. The 8-aza-, 8-oxa- and 8-thia-keto esters were resolved10, 11, 13, 40 to provide enantiomerically pure keto esters 1R-2. The enoltriflates 3, obtained upon reaction with N-phenyltriflimide and bis(trimethylsilyl)sodium imide, were then coupled under Suzuki conditions41 with 3,4-dichlorophenylboronic acid and palladium tetrakis(triphenylphosphine) to provide the unsaturated 3-aryl compounds 4 in good yields. Reduction to provide the saturated compounds 5 was achieved with samarium iodide in methanol at low temperature. The desired 3β-aryl (chair) configured compounds 5 were accompanied by formation of the 3α-aryl configured analogs (generally, the major products of this reduction) and were separated from them by column chromatography to provide the 2β-carbomethoxy-3β-(3,4-dichlorophenyl) analogs in low yields (ca. 20%). The C2-esters 5 were reduced with LiAlH4 in THF at 0 °C to provide the 2β-hydroxymethyl precursors 6a, b, and c in about 85−95% yields.
Scheme 1.

Synthesis of 8-heterobicyclo[3.2.1]octanes. Reagents and conditions: (i) (a) NaN(TMS)2; (b) (1S)-Camphanyl chloride, (c) Recrystallization from CH2Cl2 or THF/hexanes, 64%; (d) NaOCH3,MeOH, 90%; (ii) NaN(TMS)2, PhN(Tf)2, 75%; (iii) 3,4-Cl2-PhB(OH)2, Pd(PPh3)4,LiCl, Na2CO3, 74−96%; (iv) Sml2, THF, MeOH, −60°C, 20%; (v) LiAlH4, THF, −78°C, 85−95%. See: Pjam-Huu et al. Bioorg. & Med. Chem. 15, 1067, 2007; Meltzer et al. J. Med. Chem. 40, 2661, 1997.
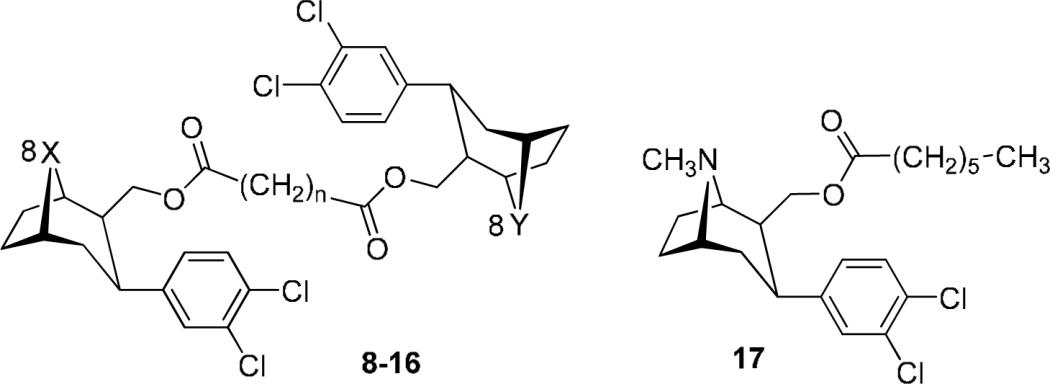
In an attempt to obtain all three N, N; N, S; and S, S (n=2) bivalent ligands simultaneously, 8-aza 6a and 8-thia 6c were reacted with succinyl chloride in the presence of DMAP in one pot. However, only the S, S-16 (n=2) was obtained, and in excellent yield (93%). Consequently two routes were adopted for synthesis of the bivalent ligands. The homobivalent ligands were prepared via Route 1 in which tandem coupling of the hydroxymethyl precursor 6a (or 6c) was achieved with the appropriate linker. The heterobivalent ligands were prepared via Route 2 in which the intermediacy of 7b or 7c avoided homobivalent side products.
Thus, the homobivalent ligands 8 (32%) and 16 (93%) were obtained (Route 1) by reaction of the 6a and 6c respectively with succinnic anhydride in the presence of DMAP, EDCI and HOBT, or succinyl chloride. The bivalent ligands were obtained (Route 2) upon reaction of the intermediate acids 7b and 7c (prepared from the primary alcohols, 6b and 6c upon reaction with succinnic anhydride in the presence of ethyldimethylamine) with 6a or 6b in the presence of DMAP and EDCI to provide the target compounds in yields of 79% (13), 40% (14) and 55% (15). The monovalent 17 (57%) was prepared by coupling of 6a with heptanoic acid in the presence of DMAP and EDCI.
3. Biology
The affinities (IC50 values) for the DAT and SERT were determined in competition studies with tritium labeled ligands. The DAT was labeled with [3H]3β-(4-fluorophenyl)tropane-2β-carboxylic acid methyl ester ([3H]WIN 35,428 or [3H]CFT (4 nM)) and non-specific binding was defined as the difference in binding in the presence and absence of (-)-cocaine (100 μM). [3H]Citalopram was used to label the SERT and non-specific binding was measured in the presence of fluoxetine (10 μM). Competition studies were conducted with a fixed concentration of radioligand and a range of concentrations of the test drug. All drugs inhibited [3H]WIN 35,428 and [3H]citalopram binding in a concentration-dependent manner.
4. Discussion
Table 1 presents the parent compounds 5a, 5b and 5c on which the bivalent ligands of this study are based. The data show that these compounds are potent inhibitors of both DAT (IC50 = 1 − 4 nM) and SERT (IC50 = 2 − 5 nM). Furthermore, these compounds are non-selective inhibitors, with SERT/DAT ratios of between 1 and 2. It should be anticipated that bivalent analogs of these parent compounds would display a considerable enhancement of inhibitory potency. In fact, at least a 10 to 100-fold increase in inhibitory potency might be expected to signify bivalent binding.28, 30
Since the distance between putative binding sites on the DAT was unknown, a study of the effect of increasing linking chain length was important. In order to optimize the chances of both tropane moieties encountering, and therefore binding to proximal binding sites, the linker was designed to be “flexible”. Therefore a linker composed of methylene units was selected. Linking chain lengths were less than 20 Å. Consequently binding to two sites on different DATs within a DAT dimer34 would not be possible. For ease of synthesis, ester linkages were chosen for attachment of the linking chain to the two tropane moieties.
The 8-azabicyclo[3.2.1]octane series was utilized to explore the impact of the length of the spacer with respect to inhibitory potency of the DAT and SERT. Five compounds (8 − 12) with linking chains consecutively differing by two-carbon atoms were evaluated. This provided a linker of between 8 bond lengths (ca. 8.4 Å) and 16 bond lengths (ca. 18.4 Å). It is evident from the data in Table 2 that the length of the linking chain had little, if any, significant impact upon inhibitory potency at the DAT. Thus, the two-methylene linked 8 had an IC50 = 12 nM, the four-methylene linked 9 manifested IC50 = 28 nM, the six-methylene compound 10 had IC50 = 11 nM and the 8-methylene compound 11 manifested IC50 = 31 nM. The longest linker investigated 12 (10-methylene units: ca. 18.4 Å) had an IC50 = 115 nM. It is therefore evident that linker length had little bearing upon inhibitory potency at the DAT. Inhibition of the SERT was somewhat more consistent in that inhibitory potency increased as the chain linker increased in length from n = 2 (8) to n = 8 (11). However, when n = 10 (12) the potency of inhibition of radioligand binding to the SERT decreased to a value similar to that obtained when n = 2.
Table 2.
Bivalent ligands: Inhibition of [3H]WIN 35,428 binding to the hDAT and [3H]citalopram binding to the hSERT for 8 − 17a
 | ||||||
|---|---|---|---|---|---|---|
|
8X-Position |
8Y-Position |
n |
Compound |
Number |
DAT |
SERT |
| Homo-Bivalent | IC50 (nM) | |||||
| 8-NCH3 | 8-NCH3 | 2 | 8 | O-4222 | 12 ± 3 | 106 ± 27 |
| 8-NCH3 | 8-NCH3 | 4 | 9 | O-4245 | 28 ± 5 | 56 ± 16 |
| 8-NCH3 | 8-NCH3 | 6 | 10 | O-4623 | 11 ± 3 | 31 ± 11 |
| 8-NCH3 | 8-NCH3 | 8 | 11 | O-5178 | 31 ± 8 | 17 ± 1 |
| 8-NCH3 | 8-NCH3 | 10 | 12 | O-4380 | 115 | 140 |
| Hetero-Bivalent | ||||||
| 8-NCH3 | 8-O | 2 | 13 | O-4023 | 67 ± 11 | 405 ± 171 |
| 8-NCH3 | 8-S | 2 | 14 | O-3996 | 55 ± 19 | 593 ± 188 |
| 8-O | 8-S | 2 | 15 | O-4022 | 2,960 | 992 |
| 8-S | 8-S | 2 | 16 | O-3962 | 1,540 | 2,200 |
| Monovalent Ligand | ||||||
| 8-NCH3 | 6 | 17 | O-5784 | 8 ± 2 | 53 ± 27 | |
Each value is the mean +/− SEM of at least 3 independent experiments, each conducted in triplicate.
The most important observation among these compounds was that none of the bivalent 8-azatropanes manifested DAT potency better than, or even equal to, that of the parent compound 5a (DAT IC50 = 1.1 nM; SERT IC50 = 2.5 nM). Therefore it can be concluded that the second tropane in the bivalent ligand did not contribute to binding. Therefore a second tropane binding site on the DAT, within the range of the chain lengths explored in these compounds, does not exist. The reduction (10 to 30-fold) of binding potency at DAT for 8 − 12 was most likely a consequence of steric hindrance offered by the unbound second tropane. To confirm the contention that the second tropane offered only a steric impediment, we prepared the monovalent hexanoyl analogue 17. The binding potencies observed for 17 (DAT IC50 = 8 nM and SERT IC50 = 53 nM) were very similar to those manifested by the homobivalent ligand with six methylene groups, 10, (DAT IC50 = 11 nM and SERT IC50 = 31 nM). Therefore, the steric effect presented by the hexanoyl group in 17 was similar to that offered by the second tropane of homobivalent 10.
Second, the inhibitory potency of the bivalent ligands at SERT was also considerably weaker (17 − 140 nM) than that evidenced by the parent compound 5a (SERT IC50 = 2.5 nM). Therefore a similar conclusion can be reached that no second tropane binding site within the range of the test compounds exists on the SERT. However in this case, it is possible that the consistent improvement in binding as chain length is extended (n = 2 to 8) was a consequence of reduced steric encumbrance at the SERT tropane binding site as the second tropane extends further away from that actual binding site.
In summary, the bivalent 8-azatropanes manifested potent DAT and SERT inhibition thus providing conclusive evidence that these compounds interacted and bound at the cocaine binding site on both transporters. However, the fact that these homobivalent compounds manifested much poorer inhibitory potencies than did their monovalent progenitors negates the existence of dual proximal tropane (cocaine) DAT or SERT binding sites.
We have previously established that 8-oxatropanes and 8-thiatropanes inhibit DAT and SERT with potencies similar to those of their 8-azatropane counterparts.10, 11, 13 However, the question remained: do the bivalent-8-heterotropanes interact with the DAT and SERT similarly? In order to explore this question, the two-methylene linked hetero-N, O (13), hetero-N,S (14), hetero-S,O (15), and homo-S,S (16) were compared with the homobivalent two-methylene linked 8-azatropane, 8. As presented in Table 2, the compounds that contained an 8-azatropane and an 8-oxatropane (13) or 8-thiatropane (14) retained DAT inhibitory potency (IC50 = 55 − 67 nM) but lost significant SERT potency (IC50 = 593 − 405 nM). Those compounds that did not possess an 8-azatropane (15, 16) lost all potency at DAT (IC50 = 1.5 − 3 μM) and (SERT: IC50 = 1 − 2.2 μM). It is therefore likely that within this class of bivalent ligands, an 8-azatropane is required to maintain binding potency at DAT and SERT, and it can be presumed that the 8-azatropane binding sites (“acceptor sites”)11 on both DAT and SERT are not available to either the 8-oxatropane or 8-thiatropane. Further, it can be concluded that there is no 8-oxatropane or 8-thiatropane binding site in the proximity of the 8-azatropane binding site because both 13 and 14 manifested much weaker potency (DAT: IC50 = ∼60 nM; SERT IC50 = ∼500 nM) than either of their parent compounds (8-oxa: 5b DAT: IC50 = 3.3 nM; SERT IC50 = 4.7 nM) and 8-thia: 5c DAT: IC50 = 2.0 nM; SERT: IC50 = 3.0 nM). It is also evident that the acceptor site for 8-oxatropanes and 8-thiatropanes is considerably more sensitive to steric factors than is the 8-azatropane site because the bivalent 8-heterotropanes 15 and 16 were completely inactive at DAT and SERT.
5. Conclusions
8-Homobivalent and 8-heterobivalent tropanes, comprised of two tropane moieties linked by intervening chains of different lengths, were evaluated in a search for adjacent tropane binding sites on the DAT and SERT. These bivalent ligands were also used to explore whether the binding sites on the DAT and the SERT for 8-azatropanes were the same as those for 8-oxatropanes or 8-thiatropanes. A comparison of these bivalent ligands with their progenitor tropanes has cast into doubt the existence of proximal binding sites on the DAT or SERT. Indeed, 8-aza-bivalent tropanes inhibited DAT and SERT with potencies about 10-fold lower at DAT (IC50 = 11 − 31 nM) and about 17-fold lower (IC50 = 17 − 106 nM) at SERT than their monovalent counterparts (DAT: IC50 = 1.1 − 3.3 nM; SERT IC50 = 2.5 − 4.7 nM). Furthermore, bivalent ligands in which one or both of the tropanes was devoid of an amine suffered a further loss of DAT and SERT inhibitory potency. It is therefore unlikely that there exist two tropane binding sites in close proximity to one another on either the DAT or SERT. It is also likely that different binding sites exist on both the DAT and SERT for 8-azatropanes compared with 8-oxatropanes or 8-thiatropanes.
6. Experimental
NMR spectra were recorded on a Jeol 300 NMR spectrometer with tetramethylsilane (TMS) as internal standard and CDCl3 as solvent unless otherwise mentioned. Melting points are uncorrected and were measured on a Mel-Temp melting point apparatus. Room temperature is 22 °C ± 1 °C. Thin layer chromatography (TLC) was carried out on Baker Si 250F plates. Visualization was accomplished with iodine vapor, UV exposure or treatment with phosphomolybdic acid (PMA). Flash chromatography was carried out on Baker Silica Gel 40 μM (silica gel). CombiFlash Chromatography was conducted on a Teledyne ISCO Sq 16 system. All reactions were conducted under an inert atmosphere of dry nitrogen. Reagents and solvents were obtained from commercial suppliers and used as received. Reaction solvents were anhydrous. Yields have not been optimized. Elemental analyses were performed by Atlantic Microlab, Norcross, GA. HPLC and MS data were obtained on an Agilent 1100 LC/MSD system. Abbreviations: HOBT (N-hydroxybenzotriazole), DMAP (dimethylaminopyridine), EDCI (1-ethyl-3-(3'-dimethylaminopropyl)carbodiimide), DCC (dicyclohexylcarbodiimide), brine (saturated aqueous NaCl). A Beckman 1801 Scintillation Counter and Wallac beta-plate and microbeta-plate readers were used for scintillation spectrometry. 0.1% Bovine serum albumin and (-)-cocaine were purchased from Sigma Chemicals. ([3H]WIN35,428, 2β-carbomethoxy-3β-(4-fluorophenyl)-N-[3H]methyltropane, 79.4−87.0 Ci/mmol) and [3H]citalopram (86.8 Ci/mmol) were purchased from Perkin Elmer (Boston, MA). (-)-Cocaine hydrochloride for the pharmacological studies was donated by the National Institute on Drug Abuse. Fluoxetine was purchased from Sigma Chemicals (St. Loius, MO).
(1R)-3β-(3,4-Dichlorophenyl)-2β-hydroxymethyl-8-methyl-8-azabicyclo[3.2.1]octane (6a)
Compound 5a24 (1.15 g, 5.51 mmol) in dry THF (39 mL) under a nitrogen atmosphere was cooled to −78 °C and LiAlH4 in THF (2M, 2.1 mL) was added via syringe. The reaction was then warmed to 0 °C and stirred for 1h with complete consumption of starting material (TLC). The reaction was quenched by slow addition of a few drops of brine to the cold solution. When gas evolution ceased, the reaction mixture was diluted with aq. 10% NaOH (50 mL) and EtOAc (75 mL) and transferred to a separatory funnel. The aqueous layer was extracted with EtOAc (3 × 75 mL) and the organic layers combined, dried over MgSO4, filtered, and evaporated in vacuo to provide a white solid. The solid was purified by flash chromatography (ratio 32:1; eluent: 48:48:4 EtOAc:hexanes:NEt3) to provide 6a as a white solid (0.89 g, 84%): mp 99−100 °C; Rf0.21 (48:48:4 EtOAc:hexanes:NEt3); 1H NMR: δ 7.42 (d, 1H), 7.37 (d, 1H), 7.25 (dd, 1H), 7.20−7.10 (bs, OH, 1H), 3.77 (dd, J = 11.3, 1.9 Hz, 1H), 3.45 (d, J = 2.6 Hz, 1H), 3.35 − 3.31 (m, 2H), 3.03 (dt, J = 13.2, 6.0 Hz, 1H), 2.47 (td, J = 14.4, 3.1 Hz, 1H), 2.27 (s, 3H), 2.24 − 2.04 (m, 2H), 1.76 − 1.57 (m, 3H), 1.50−1.44 (m, 1H); MS (CI, m/z), 300.0 [(M+H)+].
(1R)-3β-(3,4-Dichlorophenyl)-2β-(hydroxymethyl)-8-oxabicyclo[3.2.1]octane (6b)
Compound 6b was prepared by LiAlH4 reduction of methyl ester 5b11 as described above for 6a. Compound 6b was obtained as a thick yellow oil (0.73 g, 96%): Rf 0.12 (40:60 EtOAc:hexanes); 1H NMR: δ7.39 (d, 1H), 7.36 (d, 1H), 7.15 (dd, 1H), 4.68 (d, J = 7.2 Hz, 1H), 4.60 (m, 1H), 3.54 (m, 2H), 3.25 (dt, J = 13.1, 5.6 Hz, 1H), 2.75 (dd, J = 7.6, 3.2 Hz, 1H), 2.40 (td, J = 13.2, 3.6 Hz, 1H), 2.24 − 1.51 (m, 5 H); MS (CI, m/z), 287.1 [M+H]+).
3β-(3,4-Dichlorophenyl)-2β-(hydroxymethyl)-8-thiabicyclo[3.2.1]octane (6c)
Compound 6c was prepared by LiAlH4 reduction of 5c13 as described for 6a above. Compound 6c was obtained as a white solid (0.59 g, 95%): Rf0.62 (50:50 EtOAc:hexanes); 1H NMR: δ7.37 (d, 1H), 7.30 (d, 1H), 7.08 (dd, 1H), 3.83 (t, J = 5.0 Hz, 1H), 3.74−3.65 (m, 2H), 3.38 (dt, J = 11.0, 3.7 Hz, 1H), 3.15 (dt, J = 13.1, 5.6 Hz, 1H), 2.41−1.97 (m, 6H), 1.88 (dt, J = 13.5, 4.9 Hz, 1H).
(1R)-3β-(3,4-Dichlorophenyl)-2β-(3-carboxypropionyloxymethyl)-8-oxabicyclo[3.2.1]octane (7b)
Compound 6b (108 mg, 0.376 mmol) was dissolved in CH2Cl2 (10 mL). Ethyldimethylamine (241 mg, 3.29 mmol) was added via syringe and the reaction was allowed to stir for 5 min at room temperature. Solid succinic anhydride (46.3 mg, 0.463 mmol) was then added and the reaction stirred at room temperature for 19 h. The reaction was quenched with 1.5N HCl (25 mL) and allowed to stir for 5 min. The crude reaction mixture was then diluted with CHCl3 (50 mL) and aqueous layer was separated and extracted with CHCl3 (3 × 50 mL). The combined CHCl3 extracts were washed with water followed by brine, dried (MgSO4), filtered, and evaporated in vacuo to obtain 7b as a yellow oil (190 mg, quant.). This material was used in subsequent steps without further purification. Rf 0.09 (20:80 MeOH:EtOAc); 1H NMR: δ 7.39 (d, 1H), 7.25 (d, 1H), 7.02 (dd, 1H), 4.57 − 4.55 (m, 1H), 4.50 (d, J = 6.6 Hz, 1H), 4.18 (t, J = 10.3 Hz, 1H), 3.73 (dd, J = 10.7, 5.0 Hz, 1H), 3.28 (dt, J = 12.8, 5.1 Hz, 1H), 2.62−2.55 (m, 2H), 2.51−2.43 (m, 2H), 2.23−1.75 (m, 6H), 1.57−1.48 (m, 1H); MS (CI, m/z), 385.1 [(M-H)−].
(1R)-3β-(3,4-Dichlorophenyl)-2β-(3-carboxypropionyloxymethyl)-8-thiabicyclo[3.2.1]octane (7c)
Compound 7c was obtained from 6c as described for 7b above and used in subsequent steps without purification: Rf0.23 (50:50 EtOAc:hexanes);. 1H NMR: δ 7.37 (d, 1H), 7.23 (d, 1H), 7.00 (dd, 1H), 4.31 (dd, J = 10.9, 9.2 Hz, 1H), 3.75−3.69 (m, 2H), 3.64−3.57 (m, 1H), 3.18 (dt, J = 13.3, 5.5 Hz, 1H), 2.61−2.53 (m, 2H), 2.47−2.39 (m, 2H), 2.36−2.01 (m, 6H), 1.86 (dt, J = 13.4, 4.8 Hz, 1H).
General procedure for the synthesis of homobivalent ligands 8−12 (n=2, 4, 5, 8, 10): Bis[(1R)-(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl]methyl succinate (8: n=2), Bis[(1R)-(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)]methyl adipate (9: n=4), Bis[(1R)-(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)]methyl octanedioate (10: n=6), Bis[(1R)-(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)]methyl decanedioate (11: n=8), Bis[(1R)-(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)]methyl dodecanedioate (12: n=10)
To a solution of (3-(3,4-dichlorophenyl)-8-methyl-8-aza-bicyclo[3.2.1]octan-2-yl)methanol 6a (0.33 mmol) in anhydrous CH2Cl2 (4 − 5 mL) at room temperature was added Me2NEt (1.7 mmol), followed by addition of succinic anhydride (0.17 mmol) (compound 8) or the corresponding alkyldioyldichlorides (0.17 mmol) at 0 °C (compounds 9−12). The reaction mixture was stirred for 1.5 − 2 h at room temperature. DMAP (0.17 mmol) was added, followed by addition of EDCI (0.23 mmol) (16) and HOBT (0.23 mmol) (compounds 9−12). The reaction mixture was stirred for 1−3 days at room temperature and monitored by TLC and mass spectrometry. The reaction mixture was diluted with CH2Cl2, filtered, washed with aq. Na2CO3, brine, dried (Na2SO4), filtered, and concentrated. Gravity column chromatography (eluent: acetone) provided compounds 8−12 (19 − 52 %) (Route 1).
Bis[(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2 β-yl)]methyl succinate (8: n=2)
White solid: (32%), mp 148.5− 149.5 °C, Rf 0.31 (acetone); 1H NMR: δ 7.35 (d, 2H), 7.25 (d, 2H), 7.08 (dd, 2H), 4.25 (dd, J = 10.7, 8.8 Hz, 2H), 3.67 (dd, J1 = 10.7, 5.2 Hz, 2H), 3.15 − 3.30 (m, 4H), 3.00 − 3.13 (m, 2H), 1.95 − 2.38 (m, 18H), 1.50 −1.70 (m, 6H); 13C NMR: δ 24.8, 26.1, 28.9, 33.7, 34.3, 41.9, 45.2, 61.8, 63.6, 63.8, 126.9, 129.6, 130.0, 130.2, 132.3, 142.9, 171.9; MS (CI, m/z), 681.2 [(M + H)+]. Anal. C34H40Cl4N2O4
Bis[(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2 β-yl)]methyl adipate (9: n=4)
White solid: (53%), mp 130.7− 131.6 °C; Rf 0.33 (acetone); 1H NMR: δ 7.35 (d, 2H), 7.25 (d, 2H), 7.08 (dd, 2H), 4.17 (dd, J = 11, 8 Hz, 2H), 3.70 (dd, J = 11.0, 5.4 Hz, 2H), 3.20 − 3.30 (m, 4H), 3.05 (dt, J = 13.4, 5.4 Hz, 2H), 1.95 − 2.23 (m, 18H), 1.50−1.60 (m, 6H), 1.25 − 1.48 (m, 4H); 13C NMR: δ 24.3, 24.8, 26.1, 33.7, 33.8, 34.3, 42.0, 45.3, 61.8, 63.6, 63.9, 126.9, 129.6, 130.0, 130.2, 132.0, 143.0, 173.0; MS (CI, m/z), 709.2 [(M + H)+]. Anal. C36H44Cl4N2O4.
Bis[(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2 β-yl)]methyl octanedioate (10: n=6)
White solid: (19%), mp 136.0− 137.0 °C; Rf0.32 (acetone); 1H NMR: δ 7.33 − 7.40 (m, 2H), 7.20 − 7.30 (m, 2H), 6.97 − 7.10 (m, 2H), 4.15 − 4.26 (m, 2H), 3.65 − 3.80 (m, 2H), 3.17 − 3.35 (m, 4H), 3.05 − 3.15 (m, 2H), 1.95 − 2.25 (m, 18H), 1.35 − 1.72 (m, 10H), 1.15 − 1.28 (m, 4H); 13C NMR: δ 24.8, 24.9, 26.2, 28.9, 33.8, 34.2, 34.4, 42.1, 45.4, 61.9, 63.6, 64.0, 127.0, 129.7, 130.1, 130.3, 143.1, 173.6; MS (CI, m/z), 737.3 [M + H]+). Anal. C38H48Cl4N2O4
Bis[(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2 β-yl)]methyl decanedioate (11: n=8)
Colorless oil: (37%); Rf0.27 (acetone); 1H NMR: δ 7.35 (d, 2H), 7.23 (d, 2H), 7.01 (dd, 2H), 4.18 (dd, J = 11.0, 8.2 Hz, 2H), 3.71 (dd, J = 11.0, 5.4 Hz, 2H), 3.20 − 3.30 (m, 4H), 3.05 (dt, J = 13.2, 5.4 Hz, 2H), 1.97 − 2.25 (m, 18H), 1.40 − 1.68 (m, 10H), 1.15 − 1.28 (m, 8H); 13C NMR: δ 24.9, 26.2, 29.2, 33.8, 34.3, 34.4, 42.1, 45.4, 61.9, 63.5, 64.0, 127.0, 129.7, 139.3, 143.1, 173.6; MS (CI, m/z), 765.3 [M + H]+ Anal. C40H52N2O4Cl4
Bis[(3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)]methyl dodecanedioate (12: n=10)
White solid: (44%); mp 115− 116 °C; Rf0.32 (acetone); 1H NMR: δ 7.35 (d, 2H), 7.26 (m, 2H), 7.02 (dd, 2H), 4.20 (dd, J = 10.7, 8.3 Hz, 2H), 3.71 (dd, J = 11.0, 5.5 Hz, 2H), 3.34−3.22 (m, 4H), 3.11−3.01 (m, 2H), 2.31−1.96 (m, 18H), 1.75 − 1.14 (m, 22H); 13C NMR: δ 24.8, 24.9, 26.2, 29.1, 29.2, 29.4, 33.7, 34.2, 34.3, 42.0, 45.5, 61.8, 63.4, 63.9, 126.9, 129.6, 130.0, 130.2, 143.0, 173.6; MS (CI, m/z), 793.3 [M + H]+). Anal. C42H56N2O4Cl4
((1R)-3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)methyl, (3β-(3,4-dichlorophenyl)-8-thiabicyclo[3.2.1]octan-2β-yl)methyl succinate (14)
(Route 2) 3β-(3,4-Dichlorophenyl)-2β-(3-carboxy-propionyloxymethyl)-8-thiabicyclo[3.2.1]octane 7c (109 mg, 0.269 mmol) was dissolved in CH2Cl2 (6 mL). To the resulting solution was added DMAP (107 mg, 0.874 mmol), followed by 6a (82.4 mg, 0.274 mmol), and finally EDCI (106 mg, 0.555 mmol). The reaction was stirred at room temperature for 25 h under nitrogen atmosphere and then diluted with water (25 mL) and CH2Cl2 (25 mL). The organic layer was separated and washed with water and brine, dried (MgSO4), filtered, and evaporated in vacuo to obtain a yellow solid that contained significant amounts of DMAP and desired product. The crude 14 was then purified by CombiFlash chromatography (40 g silica; eluent CHCl3/MeOH) to obtain a yellow oil, which was crystallized from EtOAc/heptane to provide 14 as a white solid (73.5 mg, 40%): mp 165 − 166 °C; Rf 0.51 (10:90 MeOH:CH2Cl2); 1H NMR: δ 7.36 (d, 1H), 7.34 (d, 1H), 7.25−7.20 (m, 2H), 7.02 − 6.95 (m, 2H), 4.32−4.18 (m, 2H), 3.70 − 3.58 (m, 4H), 3.27−3.00 (m, 3H), 2.36 − 1.94 (m, 17H), 1.85 (dt, J = 13.5, 5.0 Hz, 1H), 1.65 − 1.49 (m, 4H). Anal. (C33H37Cl4NO4S).
((1R)-3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)methyl, ((1R)-3β-(3,4-dichlorophenyl)-8-oxabicyclo[3.2.1]octan-2β-yl)methyl succinate (13)
Route 2) (1R)-3β-(3,4-Dichlorophenyl)-2β-(3-carboxy-propionyloxymethyl)-8-oxabicyclo[3.2.1]octane 7b (145 mg, 0.376 mmol) was dissolved in CH2Cl2 (9 mL). DMAP (146 mg, 1.19 mmol) was added to the solution, followed by 6a (113 mg, 0.378 mmol), and EDCI (144 mg, 0.753 mmol). The reaction was stired at room temperature for 18.5 h, then quenched with water (10 mL), and extracted with CH2Cl2 (50 mL). The CH2Cl2 layer was washed with saturated aqueous NaHCO3 (50 mL), water (50 mL), and brine (50 mL), dried (MgSO4), filtered, and evaporated in vacuo to yield a yellow oil which was purified by CombiFlash chromatography (40 g silica; eluent: CH2Cl2/MeOH). Like fractions were combined and evaporated to provide an oil that crystallized from Et2O/pentane to provide the product 13 as a white solid (200 mg, 79%): mp 117 − 119 °C; Rf0.49 (10:90 MeOH:CH2Cl2); 1H NMR: δ 7.38 (d, 1H), 7.34 (d, 1H), 7.24 (s, 2H), 7.03 − 6.98 (m, 2H), 4.58−4.52 (m, 1H), 4.46 (d, J = 7.2 Hz, 1H), 4.26 − 4.11 (m, 2H), 3.71−3.64 (m, 2H), 3.31 − 3.16 (m, 3H), 3.04 (dt, J = 13.5, 5.1 Hz, 1H), 2.42 − 2.25 (m, 4H), 2.22 − 1.48 (m, 17H); MS (CI, m/z), 668.2 [(M+H)+]. Anal. (C33H37Cl4NO5)
3β-(3,4-dichlorophenyl)-8-oxabicyclo[3.2.1]octan-2β-yl)methyl, 3β-(3,4-dichlorophenyl)-8-thiabicyclo[3.2.1]octan-2β-yl)methyl succinate (15)
Compound 15 was prepared from 7c and 6b as described above for 13, except that DCC was used in place of EDCI. White solid: (55%); mp 195− 196 °C; Rf0.39 (40%EtOAc/Hex); 1H NMR: δ 7.35 − 7.4 (m, 2H), 7.23 −7.27 (m, 2H), 6.96 − 7.05 (m, 2H), 4.52 − 4.60 (m, 1H), 4.45 (d, J = 6.8 Hz, 1H), 4.28 (t, J = 10.1 Hz, 1H), 4.10 − 4.20 (m, 1H), 3.60 − 3.78 (m, 4H), 3.22 − 3.35 (m, 1H), 3.10 − 3.20 (m, 1H), 1.80 − 2.40 (m, 17H), 1.47 − 1.60 (m, 1H); MS (CI, m/z), 671.3 [(M − H−)]; 704.1 [(M+MeOH)−]. Anal. C32H34SO5Cl
(3β-(3,4-dichlorophenyl)-8-thiabicyclo[3.2.1]octan-2β-yl)methyl, (3β-(3,4-dichlorophenyl)-8-thiabicyclo[3.2.1]octan-2β-yl)methyl succinate (16)
Succinyl chloride (144 mg, 0.931 mmol) was added dropwise to a solution of DMAP (284 mg, 2.33 mmol) in CH2Cl2 (3 mL) at 0 °C. The mixture was stirred at 0 °C for 15 min. A solution of 6a (233 mg, 0.776 mmol) in CH2Cl2 (3 mL) was added to the DMAP solution and the reaction was allowed to warm to room temperature over 2 h. The mixture was then cooled to 0 °C and a solution of 6c (235 mg, 0.776 mmol) in CH2Cl2 (3 mL) was added to the reaction mixture which was stirred overnight at room temperature. A second portion of succinyl chloride (36 mg, 0.23 mmol) was added and the reaction mixture was stirred for an additional 2 h. The reaction was quenched with water (30 mL) followed by dropwise addition of aqueous NH4OH until a pH of 8 was achieved. The crude mixture was extracted with CHCl3, (3 × 30 mL) and the organic layers combined, dried (Na2SO4) and evaporated under reduced pressure to give a product which was purified by flash chromatography (eluent: 20% ethyl acetate/80% hexanes). Evaporation of appropriate fractions provided compound 16 as a white solid (249 mg, 93%): mp: 188−190 °C; Rf0.26 (hexanes:ethyl acetate; 20:1); 1H NMR: δ 7.36 (d, 2H), 7.22 (d, 2H), 6.98 (dd, 2H), 4.28 (m, 2H), 3.70 − 3.58 (m, 6H), 3.16 (dt, J = 13.5, 5.4 Hz, 2H), 2.36 − 2.01 (m, 16H), 1.85 (dt, J = 13.2, 4.7 Hz, 2H). Anal. (C32H34Cl4O4S2)
((1R)-3β-(3,4-dichlorophenyl)-8-methyl-8-azabicyclo[3.2.1]octan-2β-yl)methyl heptanoate (17)
Compound 6a (105 mg, 0.351 mmol) was dissolved in CH2Cl2 (3 mL). DMAP (134 mg, 1.09 mmol) was added to the solution, followed by a solution of heptanoic acid (64.1 mg, 0.492 mmol) in CH2Cl2 (4 mL). EDCI (134.4 mg, 0.7011 mmol) was then added and the reaction allowed stirred at room temperature for 19 h. The reaction was quenched with water (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The organic extracts were combined and washed with saturated aqueous NaHCO3 water, and brine. The organic layer was dried (MgSO4), filtered, and evaporated in vacuo to obtain a solid (211 mg) which was purified by flash chromatography (ratio: 30:1; eluent: 2:98 MeOH:CH2Cl2 to 8:92 MeOH:CH2Cl2). Compound 17 was obtained as an amber oil (82.3 mg, 57%): Rf0.45 (49:49:2 EtOAc:Hexanes:NEt3); 1H NMR: δ 7.35 (d, 1H), 7.25 (d, 1H), 7.02 (dd, 1H), 4.20 (dd, J = 11.0, 8.3 Hz, 1H), 3.71 (dd, J = 11.0, 5.5 Hz, 1H), 3.31 − 3.20 (m, 2H), 3.05 (dt, J = 13.2, 5.3 Hz, 1H), 2.30 − 1.97 (m, 9H), 1.75 − 1.19 (m, 12H), 0.93 − 0.85 (m, 2H). Anal. (C22H31Cl2NO2).
Dopamine transporter assay
Assays were conducted by minor modifications of the methods of Eshleman, et al.42 and Pham-Huu et al.13 In assays involving the human transporters, membranes from transfected cells stably expressing the DAT were labeled with [3H]WIN35,428 ([3H]CFT, 2β-carbomethoxy-3β-(4-fluorophenyl)-N-[3H]methyltropane, 81 − 84 Ci/mmol, Perkin Elmer, Boston , MA). The affinities of drugs at the [3H]WIN35,428 binding site on the DAT were determined by incubating tissue with a fixed concentration of [3H]WIN35,428 and a range of drug concentrations. The assay tubes received, in Tris.HCl buffer (50 mM, pH 7.4 at 0 − 4 °C; NaCl 100 mM), the following constituents at a final assay concentration: [3H]WIN35,428 (4 nM) and cell membrane preparation 0.2 mL. The 1 h incubation at 22 °C was initiated by addition of membranes and terminated by rapid filtration over Whatman GF/B glass fiber filters pre-soaked in 0.1% polyethyleneimine (Sigma Chem. Co., St. Louis MO). The filters were washed twice with 5 mL Tris.HCl buffer (50 mM), dried, scintillation fluid added to each spot, and radioactivity was measured on a Wallac beta plate or microbeta plate reader. Non-specific binding was defined as [3H]WIN35,428 bound in the presence of an excess (100 μM) of (-)-cocaine. Stock solutions of drugs were dissolved in DMSO. The stock solutions were diluted serially in the assay buffer and added (0.2 mL) to the assay medium as described above. In assays involving brain tissue, 1 nM radioligand was used, and filters were presoaked in 0.1% bovine serum albumin (Sigma Chem. Co.). In addition, non-specific binding was defined as [3H]WIN35,428 bound in the presence of an excess (30 μM) of (-)-cocaine.
Serotonin transporter assay
Assays were conducted essentially as described for the DAT above. The affinity of drugs at the [3H]citalopram (spec. act.: 82 Ci/mmol, Perkin Elmer, Boston) binding site on the SERT was determined in experiments by incubating HEK-293 cell membranes stably expressing the cDNA for the SERT with a fixed concentration of [3H]citalopram and a range of concentrations of unlabeled drugs. The assay tubes received, in Tris.HCl buffer (50 mM, pH 7.4 at 0 − 4 °C; NaCl 100 mM), the following constituents at a final assay concentration: [3H]citalopram (1 nM) and membrane preparation . The 2 h incubation (0−4 °C) was initiated by addition of membranes and terminated by rapid filtration over Whatman GF/B glass fiber filters pre-soaked in 0.1% polyethyleneimine. The filters were washed twice with 5 mL Tris.HCl buffer (50 mM), and radioactivity was measured as described above. Non-specific binding was defined as [3H]citalopram bound in the presence of an excess (10 μM) of fluoxetine.
Data Analysis
Data were analyzed by the EBDA computer software programs (Elsevier-Biosoft, U. K.) or the PRIZM software program. (San Diego, CA) Baseline values for the individual drugs were established from the competition curves and these generally were similar to baseline values established by 30 μM (-)-cocaine or 1 μM fluoxetine. For assays involving brain tissue, the SERT was assayed in caudate-putamen membranes using conditions similar to those for the dopamine transporter, above.
Scheme 2.

Stnthesis of Bivalent 8-Aza-, 8-Oxa, and 8-Thiatropanes 8 − 16. Reagents and Conditions: (i) Heptanoic acid, DMAP, EDCI; (ii) DCC, DMAP, EtNMe2 or EDCI, DMAP, EtNMe2; (iii) DMAP, EDCI, HOBT, EtNMe2.
Acknowledgements
This work was supported by the National Institute on Drug Abuse: DA18825 (PM), DA11542 (PM), DAO18165 (AJ) and by the VA Merit Review and Research Career Scientist Programs (AJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
References
- 1.Ritz MC, Cone EJ, Kuhar MJ. Life Sciences. 1990;46:635. doi: 10.1016/0024-3205(90)90132-b. [DOI] [PubMed] [Google Scholar]
- 2.Kuhar MJ, Ritz MC, Boja JW. Trends Neurosci. 1991;14:299. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy LT, Hanbauer IJ. Neurochem. 1983;34:1137. doi: 10.1111/j.1471-4159.1983.tb13666.x. [DOI] [PubMed] [Google Scholar]
- 4.Schoemaker H, Pimoule C, Arbilla S, Scatton B, Javoy-Agid F, Langer SZ. Naunyn-Schmiedeberg's Arch. Pharmacol. 1985;329:227. doi: 10.1007/BF00501873. [DOI] [PubMed] [Google Scholar]
- 5.Reith MEA, Meisler BE, Sershen H, Lajtha A. Biochem. Pharmacol. 1986;35:1123. doi: 10.1016/0006-2952(86)90148-6. [DOI] [PubMed] [Google Scholar]
- 6.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Science. 1987;237:1219. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 7.Madras BK, Fahey MA, Bergman J, Canfield DR, Spealman RD. J. Pharmacol. Exp. Ther. 1989;251:131. [PubMed] [Google Scholar]
- 8.Spealman RD. Psychopharmacol. 1993;112:93. doi: 10.1007/BF02247368. [DOI] [PubMed] [Google Scholar]
- 9.Meltzer PC, Blundell P, Huang H, Liu S, Yong YF, Madras BK. Bioorg. & Med. Chem. 2000;8:581. doi: 10.1016/s0968-0896(99)00322-3. [DOI] [PubMed] [Google Scholar]
- 10.Meltzer PC, Blundell P, Madras BK. Med. Chem. Res. 1998;8:12. [Google Scholar]
- 11.Meltzer PC, Liang AY, Blundell P, Gonzalez MD, Chen Z, George C, Madras BK. J. Med. Chem. 1997;40:2661. doi: 10.1021/jm9703045. [DOI] [PubMed] [Google Scholar]
- 12.Meltzer PC, Wang B, Chen Z, Blundell P, Jayaraman M, Gonzalez MD, George C, Madras BK. J. Med. Chem. 2001;44:2619. doi: 10.1021/jm0101242. [DOI] [PubMed] [Google Scholar]
- 13.Pham-Huu DP, Deschamps JR, Liu S, Madras BK, Meltzer PC. Bioorg. & Med. Chem. 2006;15:1067. doi: 10.1016/j.bmc.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh S. Chem. Rev. 2000;100:925. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]
- 15.Newman AH. Med. Chem. Res. 1998;8:1. [Google Scholar]
- 16.Runyon SP, Carroll FI. Curr. Topics in Med. Chem. 2006;6:1825. doi: 10.2174/156802606778249775. [DOI] [PubMed] [Google Scholar]
- 17.Zou M-F, Kopajtic T, Katz JL, Newman AH. J. Med. Chem. 2003;46:2908. doi: 10.1021/jm0300375. [DOI] [PubMed] [Google Scholar]
- 18.Petukhov PA, Zhang J, Kozikowski AP, Wang CZ, Ye YP, Johnson KM, Tella SR. J. Med. Chem. 2002;45:3161. doi: 10.1021/jm0200153. [DOI] [PubMed] [Google Scholar]
- 19.Xu L, Izenwasser S, Katz JL, Kopajtic T, Klein-Stevens C, Zhu N, Lomenzo SA, Winfield L, Trudell ML. J. Med. Chem. 2002;45:1203. doi: 10.1021/jm010453u. [DOI] [PubMed] [Google Scholar]
- 20.Deutsch HM. Med. Chem. Res. 1998;8:91. [Google Scholar]
- 21.Meltzer PC, Butler D, Deschamps J, Madras BK. J. Med. Chem. 2006;49:1420. doi: 10.1021/jm050797a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman AH. Med. Chem. Res. 1998;8:1–113. [Google Scholar]
- 23.Carroll FI, Pawlush N, Kuhar MJ, Pollard GT, Howard JL. J. Med. Chem. 2004;47:296. doi: 10.1021/jm030453p. [DOI] [PubMed] [Google Scholar]
- 24.Meltzer PC, Liang AY, Brownell A-L, Elmaleh DR, Madras BK. J. Med. Chem. 1993;36:855. doi: 10.1021/jm00059a010. [DOI] [PubMed] [Google Scholar]
- 25.Meltzer PC, Blundell P, Yong YF, Chen Z, George C, Gonzalez MD, Madras BK. J. Med. Chem. 2000;43:2982. doi: 10.1021/jm000191g. [DOI] [PubMed] [Google Scholar]
- 26.Rothman RB, Mele A, Reid AA, Akunne HC, Greig N, Thurkauf A, deCosta BR, Rice KC, Pert A. Pharmacol. Biochem. Behav. 1991;40:387. doi: 10.1016/0091-3057(91)90570-r. [DOI] [PubMed] [Google Scholar]
- 27.Portoghese PS. Trends. Pharm. Sci. 1989;10:230. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- 28.Xie Z, Bhusan RG, Daniels DJ, Portoghese PS. J. Pharm. Exp. Ther. 2005;68:1079. doi: 10.1124/mol.105.012070. [DOI] [PubMed] [Google Scholar]
- 29.Tamiz AP, Bandyopadhyay BC, Zhang J, Flippen-Anderson JL, Zhang M, Wang CZ, Johnson KM, Tella S, Kozikowski AP. J. Med. Chem. 2001;44:1615. doi: 10.1021/jm000552s. [DOI] [PubMed] [Google Scholar]
- 30.Christopoulos A, Grant MKO, Ayoubzadeh N, Kim ON, Sauerberg P, Jeppesen L, El-Fakahany EEJ. Pharm. & Exp. Ther. 2001;298:1260. [PubMed] [Google Scholar]
- 31.Fandrick K, Feng X, Janowsky A, Johnson R, Cashman JR. Bioor. & Med. Chem. Lett. 2003;13:2151. doi: 10.1016/s0960-894x(03)00386-x. [DOI] [PubMed] [Google Scholar]
- 32.Carroll FI, Kotian P, Dehghani A, Gray JL, Kuzemko MA, Parham KA, Abraham P, Lewin AH, Boja JW, Kuhar MJ. J. Med. Chem. 1995;38:379. doi: 10.1021/jm00002a020. [DOI] [PubMed] [Google Scholar]
- 33.Kilic F, Rudnick G. Proc. Natl. Acad. Sci. USA. 2000:97. doi: 10.1073/pnas.060408997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hastrup H, Karlin A, Javitch JA. Proc. Natl. Acad. Sci. USA. 2001;98:10055. doi: 10.1073/pnas.181344298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin AJ. Biol. Chem. 2003;278:28274. doi: 10.1074/jbc.M210652200. [DOI] [PubMed] [Google Scholar]
- 36.Javitch JA. Mol Pharmacol. 2004;66:1077. doi: 10.1124/mol.104.006320. [DOI] [PubMed] [Google Scholar]
- 37.Sitte HH, Farhan H, Javitch JA. Mol Interv. 2004;4:38. doi: 10.1124/mi.4.1.38. [DOI] [PubMed] [Google Scholar]
- 38.Meltzer PC, Wang P, Blundell P, Madras BK. J. Med. Chem. 2003;46:1538. doi: 10.1021/jm0205292. [DOI] [PubMed] [Google Scholar]
- 39.Kim DK, Deutsch HM, Ye X, Schweri MM. J. Med. Chem. 2007:50. doi: 10.1021/jm061354p. [DOI] [PubMed] [Google Scholar]
- 40.Meltzer PC, Liang AY, Madras BK. J. Med. Chem. 1996;39:371. doi: 10.1021/jm950463t. [DOI] [PubMed] [Google Scholar]
- 41.Oh-e T, Miyaura N, Suzuki AJ. Org. Chem. 1993;58:2201. [Google Scholar]
- 42.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. J Pharmacol Exp Ther. 1999;289:877. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.