

Abstract
Changes in genome structure and gene expression have been documented in both resynthesized and natural allopolyploids that contain two or more divergent genomes. The underlying mechanisms for rapid and stochastic changes in gene expression are unknown. Arabidopsis suecica is a natural allotetraploid derived from the extant A. thaliana and A. arenosa genomes that are homeologous in the allotetraploid. Here we report that RNAi of met1 reduced DNA methylation and altered the expression of ∼200 genes, many of which encode transposons, predicted proteins, and centromeric and heterochromatic RNAs. Reduced DNA methylation occurred frequently in promoter regions of the upregulated genes, and an En/Spm-like transposon was reactivated in met1-RNAi A. suecica lines. Derepression of transposons, heterochromatic repeats, and centromeric small RNAs was primarily derived from the A. thaliana genome, and A. arenosa homeologous loci were less affected by methylation defects. A high level of A. thaliana centromeric small RNA accumulation was correlated with hypermethylation of A. thaliana centromeres. The greater effects of reduced DNA methylation on transposons and centromeric repeats in A. thaliana than in A. arenosa are consistent with the repression of many genes that are expressed at higher levels in A. thaliana than in A. arenosa in the resynthesized allotetraploids. Moreover, non-CG (CC) methylation in the promoter region of A. thaliana At2g23810 remained in the resynthesized allotetraploids, and the methylation spread within the promoter region in natural A. suecica, leading to silencing of At2g23810. At2g23810 was demethylated and reactivated in met1-RNAi A. suecica lines. We suggest that many A. thaliana genes are transcriptionally repressed in resynthesized allotetraploids, and a subset of A. thaliana loci including transposons and centromeric repeats are heavily methylated and subjected to homeologous genome-specific RNA-mediated DNA methylation in natural allopolyploids.
POLYPLOIDY is a prominent genomic feature for many plants and some animals (Becak and Becak 1998; Wolfe 2001). Over 95% of angiosperms and ∼70% of ferns went at least one event of polyploidization (Masterson 1994; Leitch and Bennett 1997), indicating an evolutionary advantage for being polyploids (Wendel 2000; Soltis et al. 2003; Comai 2005). Increase in genome dosage may increase gene expression levels and cell sizes as well as generate “buffering” effects on deleterious mutations. Moreover, stable meiotic transmission of homeologous genomes in allopolyploids provides a means for permanent fixation of heterozygosity or hybrid vigor, leading to broad adaptation to environmental niches (Soltis and Soltis 2000; Wendel 2000; Comai 2005).
Arabidopsis allotetraploids can be resynthesized by pollinating Arabidopsis thaliana with A. arenosa pollen (Comai et al. 2000; Bushell et al. 2003; Wang et al. 2004). A. suecica is a natural and selfing allotetraploid native to northern Europe (Hylander 1957; Love 1961; O'Kane et al. 1995; Lind-Hallden et al. 2002). The 26 chromosomes of A. suecica (2n = 4x = 26) are derived from A. thaliana (2n = 2x = 10) and tetraploid outcrossing species A. arenosa (2n = 4x = 32) (Price et al. 1994; Kamm et al. 1995; O'Kane et al. 1995). Although they are very closely related species, A. thaliana and A. arenosa exhibit 5–10% DNA sequence variation in protein-coding sequences (Hanfstingl et al. 1994; Lee and Chen 2001) and 30–40% nucleotide sequence changes in 180-bp centromeric repeats (Kamm et al. 1995; Comai et al. 2003).
DNA methylation changes are observed in newly formed allopolyploids of Arabidopsis (Madlung et al. 2002) and wheat (Shaked et al. 2001) and are responsible for the maintenance of silenced rRNA genes and protein-coding genes in Arabidopsis allopolyploids (Chen and Pikaard 1997; Lee and Chen 2001). Blocking DNA methylation using either chemical inhibitors or RNA interference derepresses the silenced genes in the allopolyploids (Lee and Chen 2001; Lawrence et al. 2004; Wang et al. 2004), suggesting causal effects of DNA methylation on silencing homeologous genes in Arabidopsis allopolyploids.
To test the general and specific effects of DNA methylation loss and species hybridization on gene expression, we compared gene expression changes in met1-RNAi A. suecica lines (Wang et al. 2004) and in resynthesized allotetraploids (Wang et al. 2006). Our data suggest that there is a little overlap between the genes that are affected by DNA methylation in the met1-RNAi lines and the genes that are expressed nonadditively in the resynthesized allotetraploids. Many expression changes in the met1-RNAi lines are derived from transposable elements and repetitive DNA, whereas the genes that are nonadditively expressed in the resynthesized allotetraploids encode proteins that are involved in various biological pathways important to plant growth and development (Wang et al. 2006). The reactivated transposons and repetitive DNA in the met1-RNAi lines are preliminarily derived from the A. thaliana parent, suggesting repression of these elements in natural allotetraploids, which is reminiscent of genomewide repression of A. thaliana genes in the resynthesized allotetraploids (Wang et al. 2006). Therefore, both interspecific hybridization (or allopolyploidization) and DNA methylation affect the A. thaliana genes that are generally repressed in the allotetraploids (Chen 2007), but the interspecific hybridization affects primarily the protein-coding genes, whereas RNAi of met1 affects transposons and centromeric repeats. Moreover, disruption of MET1 expression induces higher levels of short interfering RNA (siRNA) accumulation and DNA methylation in A. thaliana centromeres than in A. arenosa centromeres. Compared to A. arenosa, the A. thaliana homeologous genome (as a maternal parent) is hypersensitive to interspecific hybridization and DNA methylation perturbation in allopolyploids.
MATERIALS AND METHODS
Plant materials:
All plants were grown in vermiculite mixed with 30% soil in a growth chamber with growth conditions of 22°/18° (day/night) and 16 hr of illumination per day. The accessions included A. thaliana diploid ecotype Landsberg erecta (Ler), tetraploid A. arenosa (Arabidopsis Biological Resource Center, accession no. 3901, 2n = 4x = 32), and natural A. suecica (9502) (2n = 4x = 26). Autotetraploid A. thaliana (2n = 4x = 20) was obtained through colchicine treatment of Ler (accession no. CS3900). Transgenic met1-RNAi lines were generated in A. suecica (9502) as previously described (Wang et al. 2004). A. suecica was also transformed using a GUS reporter as a control (AsGUS) (Wang et al. 2004). Resynthesized allotetraploids were derived from previous studies (Wang et al. 2004, 2006). Rosette leaves were collected prior to bolting for all plants. For DNA and RNA extraction, leaves were harvested from individual plants for RNAi transgenic lines and were pooled from 10 plants for nontransgenic plants.
DNA and RNA analysis:
Genomic DNA was isolated using a published protocol (Chen et al. 1994). DNA probes were obtained from PCR using the primers shown in supplemental Table 1 followed by purification [QIAGEN (Valencia, CA) PCR purification kit] and were prepared by a random-priming method (Amersham, Arlington Heights, IL) with radioactive labeling ([α-32P]ATP). Hybridization and washing were performed using a published method (Church and Gilbert 1984).
RNA was extracted using TRIzol reagent (Invitrogen Life Technologies, San Diego), following the manufacturer's instructions. Total RNA (25 μg) was subjected to electrophoresis in a polyacrylamide gel (15%, polyacrylamide:bisacrylamide = 38:1) and transferred onto Hybond-N+ membrane (Amersham Pharmacia, Piscataway, NJ) for small RNA blot analysis. The DNA oligonucleotide probes (supplemental Table 2) were end labeled using T4 polynucleotide kinase as previously described (Chen and Pikaard 1997) and hybridized with the small RNA blot using a published protocol (Church and Gilbert 1984). Signals were detected by exposure to Kodak X-ray films or analyzed by the PhosphorImager BAS1800II system (Fuji, Tokyo).
DNA microarrays and data analysis:
Spotted oligo-gene microarrays were used for genomewide gene expression assays following a published protocol (Wang et al. 2005). The 70-mer oligos hybridized equally well with both A. thaliana and A. arenosa genes (Lee et al. 2004; Wang et al. 2006) because they share ∼95% nucleotide sequence identity in coding regions where the oligos are designed. A total of eight slides were used for two biological and four technical replications. Each biological replication consists of two technical replications (four slides). Within a technical replication, one slide was hybridized with an equal amount of Cy3-labled A. suecica control cDNA and Cy5-labeled met1-RNAi cDNA as probes and another with the same two cDNA samples labeled in a reverse Cy3 and Cy5 combination. We used 500 ng of mRNA in each labeling reaction using Cy3- or Cy5-dCTP (Amersham Biosciences). The dye-swap experiment was replicated using an independently isolated RNA sample. Raw data were collected using Genepix Pro4.1 after the slides were scanned using Genepix 4000B (Axon Instruments/Molecular Devices, Sunnyvale, CA). The data were processed using a lowess function to remove nonlinear components and analyzed using a linear model (Wang et al. 2006). This linear model was employed to partition variation in the observed data relative to technical and biological variation. Given that each feature is represented once on an array, the linear model is
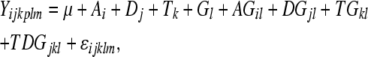 |
where μ represents the overall mean effect; A, D, T, F, and G represent main fixed effects from the array, the dye, the treatment (e.g., RNA from two species), and the gene, respectively; and i = 1, …, 8, k = 1, 2, j = 1, 2, and l = 1, …, 27,648 (including 26,090 70-mer Arabidopsis oligos plus controls). The interaction terms AG, DG, TG, and TDG represent array-by-gene, dye-by-gene, treatment-by-gene, and treatment-by-dye-by-gene interactions, and ɛijklm denotes the random error and is used to test for significance of main and interaction effects in the model. The hypotheses that reflect whether a gene, g, has undergone differential expression between treatments, t and t′ (e.g., control and met1-RNAi A. suecica), are
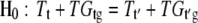 |
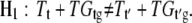 |
A standard t-test statistic is used for this comparison, based on the normality assumption for the residuals. The false discovery rate (FDR) (α = 0.05) of Benjamini and Hochberg (1995) was employed to control multiple testing errors. All analyses of variance models were fit using standard statistical packages (SAS, R, and Matlab) (Moser et al. 1988; Ihaka and Gentleman 1996).
We selected the genes that were differently expressed at a statistically significant level (FDR, α = 0.05) under both common variance and per-gene variance. The microarray data were deposited in GEO (http://www.ncbi.nlm.nih.gov/geo/) (accession no. GSE9512).
Functional categories of up- and downregulated genes were classified using ftp://ftp.arabidopsis.org/home/tair/Ontologies/Gene_Ontology/OLD/ATH_GO_GOSLIM.20070317.txt (TAIR release on March 17, 2007) and compared using Venn diagrams. The differentially expressed genes (identities) in met1-RNAi lines compared to A. suecica control were mapped to oligonucleotide sequences using Perl scripts. The oligos were mapped to genomic coordinates using high (red) and low (blue) gradients corresponding to gene densities. Vertical lines above and below the chromosomes showed up- and downregulation, respectively, and the length was proportional to the logarithm-fold changes in differential gene expression.
RT–PCR and SSCP analyses:
Approximately 5 μg of total RNA was treated with DNase I, and first-strand cDNA was synthesized using reverse transcriptase (RT) Superscript II (Invitrogen) according to the manufacturer's recommendations. An aliquot (1/100) of cDNA was used as a template in quantitative (or real-time) RT–PCR (qRT–PCR), single-strand conformation polymorphism (SSCP) (Adams et al. 2003), and cleaved amplified polymorphism sequence (CAPS) analyses (Wang et al. 2004). qRT–PCR was performed in an ABI7500 machine (ABI Biosystems, Forest City, CA), using the primers (supplemental Table 3) and the SYBER green dye method as previously described (Lee et al. 2004), except that ACT2 was used as a control to estimate the relative expression levels of the genes tested. For SSCP analysis, the primers were from A. thaliana loci and used to amplify both A. thaliana and A. arenosa loci. The PCR reactions were performed using one cycle of 94° for 2 min followed by 25–30 cycles of amplification at 94° for 30 sec, 53° for 30 sec, and 72° for 90 sec. The amplified products were denatured in a loading buffer and resolved in a 0.5× mutation detection enhancement (MDE) gel (SSCP analysis). The images were captured, and band intensities were quantified using a Fujifilm Phosphorimager. In brief, a 12% polyacrylamide gel is made containing 0.5× TBE, 0.5% of 10% APS, 0.1% of TEMED, and ddH2O. The gel is allowed to solidify and is run in 0.5× TBE for the appropriate time for good separation. We use 200 V for 10 hr running for a 20 × 20-cm gel.
Bisulfite sequencing:
Bisulfite sequencing was performed according to published protocols (Frommer et al. 1992; Cao et al. 2003) and a protocol that is published online at http://www.methods.info/Methods/DNA_methylation/Bisulphite_sequencing.html. Primer sequences are shown in supplemental Table 4. The percentage of cytosine methylation was estimated using average ratios of signal peak of C's to T's at the same site in three independent experiments.
RESULTS
Reactivation of transposable elements and heterochromatic genes in met1-RNAi A. suecica lines:
To study the effects of DNA methylation on gene regulation in allopolyploids, we generated transgenic A. suecica lines that overexpressed double-stranded met1 (Wang et al. 2004). The expression level of endogenous MET1 was ∼5% of the wild type in the met1-RNAi A. suecica plants. Reduced DNA methylation caused severe growth and developmental abnormalities, and the severity increased in selfing progeny. This is probably because hypomethylation induces other changes in the genome as observed in the selfing progeny of ddm1 mutants (Stokes and Richards 2002; Stokes et al. 2002). The data suggest that disruption of met1 expression affects gene regulation and development in allopolyploids.
We used spotted oligo-gene microarrays (Wang et al. 2005) to detect transcriptome changes in one of the met1-RNAi A. suecica lines generated (met1-1-RNAi) (Wang et al. 2004). A total of 197 genes (199 oligo probes) were expressed statistically significantly different (P ≤ 0.05) in the met1-RNAi A. suecica plants compared to a control A. suecica line that was transformed with a GUS reporter (AsG) (Wang et al. 2004) (supplemental Table 5). Two loci (At5g32511 and At2g05510) each had duplicate oligo probes in microarrays. Approximately equal numbers of genes were up- and downregulated, indicating that DNA methylation affects silencing and activation of a subset of genes through direct effects on specific targets and indirect effects on downstream genes, respectively.
To determine chromosomal location and distribution of up- or downregulated genes in the met1-RNAi line, we mapped 196 genes onto five chromosomes in Arabidopsis (Figure 1A). Upregulated genes were clustered toward centromeric and heterochromatic regions, suggesting that these regions are generally repressed by DNA methylation. In contrast, the downregulated genes were evenly distributed along the chromosomes, indicating that these genes are probably located in enchromatic regions, and their expression changes are associated indirectly with DNA methylation or inversely with changes in DNA methylation.
Figure 1.—

(A) Chromosomal distribution of the 221 genes that were up- or downregulated in met1-RNAi A. suecica. Colors represent oligonucleotide/gene densities along five chromosomes of A. thaliana from high (red) to low (blue). Blue-colored regions with gaps are locations of low gene densities and coincide with centromeres. Vertical bars represent genes that are up- or downregulated, and the length is proportional to the logarithm-fold changes. The length of chromosomes is shown above chromosome 1. (B) RT–PCR validation of gene expression changes detected by microarrays. The colors of hybridization signals in two biological replicates are shown on the left, and the RT–PCR results are shown on the right. ACT2, action control; control, A. suecica transgenic plants transformed using a GUS reporter; met1-RNAi, A. suecica transgenic plants that overexpress double-stranded MET1.
We used RT–PCR to validate a subset of genes that displayed up- and downregulation in met1-RNAi A. suecica lines. Similar to a previous study (Wang et al. 2006), 18 of 20 genes (90%) tested could be validated by RT–PCR. For example, a gene encoding transposon related (TPR) was upregulated, and three genes encoding VSP1, pathogen related (PAR), and expressed protein (EXP) were repressed (Figure 1B). Two genes encoding transposon En/spm related (TES) and serine carboxypeptidase (SCP) that were detected by statistical tests using common variance but not per-gene variance were also validated by RT–PCR, suggesting that the number of differentially expressed genes may be underestimated using both per-gene and common variances (Wang et al. 2006).
The differentially expressed genes detected in the met1-RNAi A. suecica lines were classified into 14 categories according to the GOSlim biological process annotation (TAIR release on March 17, 2007) (Figure 2A). The observed percentage of the genes that are differentially expressed in the met1-RNA1 lines in all 14 functional classifications was low, suggesting that DNA methylation affects a small fraction (∼1%) of the genes in A. suecica. Compared to the expected percentage of an entire Arabidopsis genome (Figure 2A, dashed line), transposons and pseudogenes are significantly enriched in differentially expressed genes in met1-RNAi A. suecica (Pearson's chi-square test with Yates' continuity correction, χ2 = 23.78, d.f. = 1, P = 1.07 × 10−6). Of 197 differentially expressed genes, 33 (17%) genes are transposons and pseudogenes, whereas only ∼7% (1801 of 24,377) of oligo probes in microarrays represent pseudogenes and transposable elements. The data indicate that reduced DNA methylation disrupts the regulation of a subset of transposons and heterochromatic genes. The proportions of genes in response to abiotic and biotic stresses were also overrepresented in the met1-RNAi lines (supplemental Table 5) as in the resynthesized allotetraploids (Wang et al. 2006), suggesting downregulation of MET1 and that polyploidization affects stress-responsive pathways. This supports the role of DNA methylation in defense mechanisms (Yoder and Bestor 1996).
Figure 2.—

(A) The percentages of the genes in each functional category that are differentially expressed in met1-RNAi A. suecica and two resynthesized allotetraploids (Allo733 and -738) (Wang et al. 2006). The ratios in the y-axis were calculated using the observed percentage of the genes that are differentially expressed in each functional category in a microarray experiment divided by the expected percentage of all annotated genes in the Arabidopsis genome. The dashed line shows the observed percentage of the genes that are differentially expressed in a microarray experiment (e.g., the met1-RNAi line) equal to that of the expected genes in the whole genome (100%). Original microarray data were deposited in GEO (accession no. GSE9512). (B) Venn diagram showing comparison among the numbers of up- or downregulated genes in met1-RNAi A. suecica (blue) and nonadditively expressed genes in two allotetraploids, Allo733 (red) and Allo738 (green). AsG, A. suecica transformed with a GUS reporter.
Effects of DNA methylation and allopolyploidization on gene expression in allotetraploids:
Treating resynthesized and natural Arabidopsis allotetraploids with 5-aza-DC, a chemical inhibitor for DNA methyltransferases, induces dramatic changes in phenotypes (Madlung et al. 2002) and reactivation of protein-coding genes (Lee and Chen 2001), suggesting that DNA methylation is responsible for reactivation of some genes in the allotetraploids. It is unknown how changes in gene expression in the newly formed allotetraploids correlate with those in reduced methylation lines in natural allotetraploids. One hypothesis is that allopolyploidization induces DNA methylation changes in the genes that are repressed in synthetic allopolyploids, and reduced DNA methylation induces activation of repressed genes in natural allopolyploids. To test this, we compared the genes that are up- or downregulated in the met1-RNAi A. suecica line with the genes that are expressed nonadditively (different from the midparent value) in two independent allotetraploids lineages (Allo733 and -738). Totals of 1362 and 1469 genes are nonadditively expressed in Allo733 and -738, respectively (Wang et al. 2006). By comparing the gene lists, we found that 63 genes (Figure 2B) were common in the met1-RNAi line and one or both allotetraploid lines, suggesting that expression of these genes is affected by DNA methylation and interspecific hybridization or allopolyploidization (supplemental Table 6). Among them, 34 genes were common in the met1-RNAi line and both allotetraploids, and 11 and 18 were detected in the met1-RNAi line and in allotetraploids, respectively. This relatively small number of overlapping genes between the two experiments indicates that DNA methylation affects a subset of heterochromatic genes and transposons, whereas interspecific hybridization or allopolyploidization affects the majority of protein-coding genes in various biological pathways (Wang et al. 2006). A few genes showed opposite changes in Allo733 and allo738. For example, At2g14580 was downregulated in Allo733 but upregulated in Allo738. The differential gene expression patterns in independent allopolyploid lines may give rise to epigenetically associated natural variation over time (Chen 2007).
If DNA methylation is responsible for gene repression in diploids (Martienssen and Colot 2001; Richards and Elgin 2002) as in allotetraploids, the genes should be downregulated in the new allotetraploids but upregulated in the met1-RNAi A. suecica line. Indeed, 24 of 34 (∼70%) genes belonged to this category (supplemental Table 6). The other 10 genes belonged to three categories: (1) 6 genes were downregulated in both allotetraploids and the met1-RNAi line, (2) 1 gene was upregulated in both allotetraploids and the met1-RNAi line, and (3) 2 genes were upregulated in the allotetraploids but downregulated in the met1-RNAi line. Moreover, 134 of 197 genes (∼68%) that showed expression changes in the met1-RNAi line did not alter expression in resynthesized allotetraploids. The data suggest that DNA methylation plays a role in gene repression in resynthesized allotetraploids but is not solely responsible for A. thaliana gene repression in the resynthesized allotetraploids.
Reactivation of A. thaliana genes in met1-RNAi A. suecica:
Gene activation or silencing can occur in one or both homeologous loci in an allotetraploid. To determine the progenitors' origin of expression variation in A. suecica, we analyzed allelic expression patterns of 11 genes that were up- or downregulated in the resynthesized allotetraploids and met1-RNAi A. suecica lines, using SSCP. Primers for RT–PCR analysis were designed exclusively in exons of A. thaliana sequences such that both A. thaliana and A. arenosa genes can be amplified.
The results obtained from RT–PCR (Figure 1B) and SSCP (Figure 3) analyses confirmed the data obtained using microarray analysis in both met1-RNAi lines and resynthesized allotetraploids. Among eight genes examined (supplemental Table 7), five of six genes that were upregulated in the met1-RNAi line were derived from A. thaliana homeologs, whereas two downregulated genes were associated with A. arenosa homeologs, suggesting that A. thaliana genes are generally repressed in the natural allotetraploids and are sensitive to changes in DNA methylation. An example is shown in Figure 3. The controls showed that genomic DNA of At1g36470 was equally amplified in A. thaliana (lane 9), A. arenosa (lane 10), and A. suecica (lane 11) and that ACT2 expression levels were equal in all lines tested (Figure 3A, right). Compared to the corresponding genomic loci, the upregulated alleles of At1g36470 in two met1-RNAi lines (met1-1 and met-1-12, bottom two arrows) were derived from A. thaliana origin (Figure 3A, left). Expression of new alleles (top two arrows) was also detected probably due to derepression of the alleles in met1-RNAi A. suecica lines. Similarly, At2g23810 was downregulated in resynthesized allotetraploids and upregulated in met1-RNAi lines. The downregulated alleles were mainly from an A. thaliana parent (Figure 3B, solid arrows), whereas upregulated alleles were of both A. thaliana and A. arenosa origins (Figure 3B, open arrows).
Figure 3.—

Parental origins of the up- or downregulated genes in met1-RNAi A. suecica. (A) SSCP analysis of At1g36470 encoding a Tnp2/En/Spm CACTA-element that is located close to the centromeric region. The RT–PCR SSCP products of At1g36470 are shown on the left, and genomic amplification and RT–PCR SSCP results of ACT2 are shown on the right. The cDNA samples are A. thaliana diploid (At2, lanes 1 and 12), A. thaliana autotetraploid (At4, lanes 2, 9, and 13), A. arenosa (Aa, lanes 3, 10, and 14), an equal mixture of At4 and Aa (lanes 4 and 15), A. suecica (lanes 5 and 16), A. suecica 9502-GUS (lanes 6, 11, and 16), met1-1-RNAi A. suecica (lanes 7 and 18), and met1-12 RNAi A. suecica (lanes 8 and 19). Arrows indicate A. thaliana origin of cDNA fragments. Molecular size markers are shown in the first lane. (B) RT–PCR and SSCP analyses of At2g23810 that encodes a putative senescence-associated family protein. Solid and open arrows indicate the origins of cDNA fragments from A. thaliana and A. arenosa, respectively.
Hypomethylation-induced reactivation of the genes and transposable elements in met1-RNAi A. suecica:
To test if loss of DNA methylation is associated with gene activation in allopolyploids, we analyzed DNA methylation changes in reactivated genes and transponsons using DNA blot analysis. HpaII and MspI are isoschizomers that have the same recognition site (5′-CCGG-3′) but different digestibility due to DNA methylation. MspI does not cut if external cytosine is methylated, whereas HpaII fails to cut if internal cytosine is methylated and cuts poorly if external cytosine is methylated (Mann and Smith 1977).
The genes encoding a serine carboxypeptidase gene (SCP, At5g36180) family member and two CACTA-like transposase (En/Spm) family members (At4g08010 and At1g44070) were upregulated 30-, 12-, and 9-fold, respectively (supplemental Table 5). In the met1-RNAi lines, all three genes were cut by HpaII, whereas they were undigested by HpaII in A. thaliana diploid (2x) and tetraploid (4x), A. arenosa, and A. suecica control (lanes 6–9) (Figure 4A, At4g08010, and data not shown). Multiple fragments detected indicate multiple copies of this locus (At4g08010) in the genome. The data suggest that reactivation of these three genes is associated with reduced CG methylation. No differences were detected among MspI-digested patterns in these genes, suggesting that MET1 affects mainly the CG methylation in allotetraploids as in diploids.
Figure 4.—

DNA methylation changes in the En/Spm-like transposon (At4g08010) and the RD22-like gene (At5g25610) and transposon mobility in met1-1 A. suecica. Left: At4g08010 encoding a transposon En/Spm-like (TES) displayed reduced DNA methylation in met1 A. suecica lines. Multiple bands detected suggest multiple copies of this element. A diagram of DNA fragments is shown at the top. Open boxes and solid lines indicate coding and noncoding regions, respectively. The upstream promoter region is located toward the centromere. The location of DNA probes used in the DNA blot analysis is shown below the diagram. DNA samples are met1-1-I RNAi (lanes 1 and 9), met1-1-II RNAi (lanes 2 and 10), met1-12-I (lanes 3 and 11), met1-12-II (lanes 4 and 12), A. suecica 9502-GUS (AsG, lanes 5 and 13), A. thaliana diploid (At2, lanes 6 and 14), A. thaliana autotetraploid (At4, lanes 7 and 15), and A. arenosa (Aa, lanes 8 and 16). N, NdeI; E, EcoRI; H/M, HpaII/MspI. Molecular size markers are shown in the leftmost lane. Right: mobility of TES in met1-RNAi transgenic lines. Arrows indicate new TES fragments detected. (B) Increased methylation occurs in the 5′ region of a downregulated gene At5g25610 (encoding dehydration-responsive protein, RD22) in met1 lines. The upstream promoter region is located toward the centromere. Allo(F1) and Allo(F2) indicate allotetraploids in the first and second generations, respectively. The labels of all other materials are the same as in A. Arrows indicate an enhanced level of DNA methylation (approximately threefold increase related to the control in lane 4 using the common bands in individual lanes at the bottom of the gel as loading controls).
To test whether activation of transposons induces their mobility, we performed genomic DNA blot analysis using EcoRI (G/AATTC) and NdeI (CA/TATG) that are insensitive to CG methylation for digestion (McClelland et al. 1994). Two new fragments were detected in both met1-1 and met1-12 lines (Figure 4A, arrows) but not in the A. suecica control, A. arenosa, and A. thaliana diploid or autotetraploid lines (Figure 4A, right), suggesting that reduced CG methylation induces activation and mobility of CACTA-like transposons in met1-RNAi A. suecica lines. On the basis of the restriction map, the small fragment corresponded to the EcoRI–NdeI fragment, whereas the large fragment may be due to the NdeI or the EcoRI site that is present in A. suecica but not in A. thaliana (Ler) or from another locus.
Approximately 50% of the genes that display expression changes in the met1-RNAi line were downregulated, which is contrary to a general correlation between reduced DNA methylation and increased gene expression (Cao et al. 2000; Cao and Jacobsen 2002). To determine how reduced DNA methylation represses gene expression, we analyzed At5g25610, a gene encoding a dehydration-responsive protein RD22 precursor, that was downregulated in met1-RNAi A. suecica lines. The intensity of a promoter fragment increased in HpaII-digested genomic DNA located in the 5′ promoter region of the gene in met1-RNAi lines (Figure 4B). A relatively low level of methylation was also detected in the control plants (AsGUS) and new allotetraploids (F1 and F2), suggesting sensitivity of this site to the changes in DNA methylation and polyploidization. MET1 primarily reduces CG methylation. Increase in DNA methylation is likely caused by an effect that is indirectly associated with a met1 defect. The increase in DNA methylation in the promoter region is probably associated with the downregulation of this gene in an overall demethylation genetic background, which is reminiscent of another observation that dense methylation is present around the transcription start site and within the coding region of the SUPERMAN (SUP) gene in A. thaliana that overexpresses antisense MET1 (Kishimoto et al. 2001). In natural and met1-RNAi A. suecica (9502) lines, the HpaII site is heavily methylated. CG methylation of this site is either undetectable in A. thaliana diploid, tetraploid, and resynthesized allotetraploid (At2, At4, and allotetraploids F1) lines or at a very low level in the early generation of allotetraploid (F2) lines. The data suggest a gain of DNA methylation at some specific sites during allopolyploid formation and evolution. Alternatively, some downregulated genes may be caused indirectly by decrease in DNA methylation. For example, derepression of a transcription repressor may induce downregulation of many genes in the hypomethylation genetic background. Low hybridization signals detected in A. arenosa (Figure 4B, lanes 3 and 11) are probably due to sequence divergence because an A. thaliana DNA probe was used (Figure 4B).
Upregulation of A. thaliana homeologous genes and decrease of DNA methylation in the A. thaliana promoters:
Both the density and the specific sites of DNA methylation can affect gene regulation. To further dissect the effects of DNA methylation on gene activation, we analyzed promoter methylation of At2g23810 and At2g38470 that were activated and repressed, respectively, in the met1-RNAi lines using the bisulfite sequencing method (Frommer et al. 1992; Jacobsen et al. 2000). This method allows a precise analysis of methylation in a specific region by converting all unmethylated cytosines into uracils, whereas methylated cytosines remain unchanged. Thus, the methylated and unmethylated cytosines can be distinguished after sequencing. After bisulfite reactions, we directly sequenced PCR amplicons to determine the degree of methylation in the amplicon population.
We amplified ∼500-bp fragments upstream of the ATG codon for promoter methylation analysis. Primers were designed from A. thaliana promoter sequences, and A. arenosa promoter fragments may not be amplified because of sequence divergence. We analyzed cytosine methylation changes in met1-RNAi lines, resynthesized and natural allotetraploids, and A. thaliana lines. At2g23810 encodes a putative senescence-associated protein (SAP1). SAP1 was expressed at high levels in A. thaliana and repressed in resynthesized allotetraploids (F5) (Wang et al. 2006), and silenced in natural allotetraploid A. suecica. Silencing of this gene correlated with hypermethylation of both CG and non-CG sites predominately from −350 to −430 bp and from −450 to −480 bp (Figure 5E). In met1-RNAi A. suecica lines, the overall methylation levels in both CG and non-CG sites were dramatically reduced (Figure 5F), and SAP1 was reactivated (supplemental Table 6). Interestingly, non-CG methylation occurred at only one site (CC, position −450) in A. thaliana diploids and autotetraploids, and ∼5% of the cytosines in this site were methylated (Figure 5, A and B). The methylation levels of this site increased to ∼80% in the fifth generation of resynthesized allotetraploids (Figure 5D), which may lead to the repression of this gene. In addition, two other sites (−420 and −350) were methylated at low levels in the second generation of resynthesized allotetraploids (Figure 3C). These sites coincided with two methylation peaks observed in A. suecica (Figure 5E, dashed lines). In met1-RNAi lines, non-CG methylation at position −450 remained, compared to other sites, suggesting an important role of this site for methylation initiation and spreading. This does not exclude a possibility for the role of A. arenosa SAP1 in DNA methylation and expression variation.
Figure 5.—

Site-specific non-CG methylation spreading within the promoter region of A. thaliana SAP (At2g23810) in allotetraploids. Bisufite sequencing analysis of DNA methylation in a promoter fragment is shown (positions −150 to −479 from the transcription start site). The percentage of DNA methylation (y-axis) is shown as average C/T ratios in three independent experiments. (A) A. thaliana diploid. (B) A. thaliana autotetraploid. (C) Allotetraploid in the second generation (F2). (D) Allotetraploid in the fifth generation (F5). (E) A. suecica GUS. (F) met1-1-RNAi A. suecica. Arrows indicate the site-specific non-CG methylation (CC). Circles in E and F indicate CG or GC methylation sites that are shown in E, and dashed lines in E show methylation peaks.
At2g38470 that encodes a putative WRKY family transcription factor was downregulated in the met1-RNAi line. The cytosine methylation levels in the promoter region of this gene were consistent in all lines examined (data not shown), suggesting that met1 defects and allopolyploidy do not alter its methylation and expression patterns.
Roles of RNA interference in maintaining differential methylation patterns of A. thaliana and A. arenosa centromeres in met1-RNAi A. suecica:
Preferential activation of A. thaliana genes in met1-RNAi lines and DNA methylation changes in resynthesized allotetraploids let us hypothesize that the homeologous genomes in natural allotetraploids have different sensitivities to the perturbation of DNA methylation. Centromeres in Arabidopsis are heavily methylated and packed as facultative heterochromatin (Fransz et al. 2000). To study demethylation effects on centromeres, we analyzed methylation changes in centromeres in met1-RNAi lines using A. thaliana and A. arenosa centromere-specific probes (Comai et al. 2003). A. thaliana and A. arenosa share ∼60% nucleotide sequence identity in centromeric satellite repeats. The DNA blot was hybridized with an A. thaliana centromeric probe (Figure 6, A and B). After stripping off the probe, the blot was rehybridized with an A. arenosa centromeric probe (Figure 6B). In the met1-RNAi A. suecica lines, centromeric repeats from both A. thaliana and A. arenosa were demethylated, and A. arenosa centromeres were less methylated than A. thaliana centromeres. Moreover, A. arenosa centromeres were moderately demethylated in natural A. suecica (HpaII-AsG, Figure 6C), suggesting that polyploidization or interspecific hybridization plays a role in differential DNA methylation of homeologous centromeres during evolution.
Figure 6.—

Preferential DNA methylation and siRNA accumulation in A. thaliana centromeres over A. arenosa centromeres in allotetraploids. (A) Diagram of probes used for DNA and RNA blot analyses. Full-length repeats of A. thaliana (At, 1) and A. arenosa (Aa, 8) were used for DNA blot analysis. Three pairs of antisense and sense DNA oligo probes (∼20 bp) are shown, and the numbers with an asterisk indicate the sequences derived from A. arenosa. (B) A. thaliana centromeric DNA is heavily methylated in met1-RNAi A. suecica lines. Low hybridization signals detected in lane “Aa” suggest the DNA probe is specific to A. thaliana centromeres. The lines are A. thaliana 2x (At2), A. thaliana 4x (At4), A. arenosa (Aa), A. suecica-GUS (AsG), and met1-1 and met1-12-RNAi A. suecica lines. (C) DNA blot analysis showing hypomethylation of A. arenosa centromeres in met1-RNAi A. suecica lines. Low hybridization signals in lanes “At2” and “At4” suggest the DNA probe is specific to the A. arenosa centromere. (D) High accumulation levels of small RNAs derived from A. thaliana centromeres in met1-RNAi A. suecica lines. The small RNA blot was hybridized with an antisense A. thaliana centromeric probe (3). The RNA materials are At2 (lane 1), At4 (lane 2), Aa (lane 3), AsG (lane 4), allotetraploid F1 (lane 5), allotetraploid (F2) (lane 6), allotetraploid F5 (lane 7), met1-1-RNAi A. suecica (lane 8), and met1-12-RNAi A. suecica (lane 9). RNA size markers are shown in lane “M,” and the sizes of small RNAs are indicated in the right. (E) Low accumulation levels of small RNAs derived from A. arenosa centromeres in met1-RNAi A. suecica. The same blot was stripped off the probe and rehybridized with an antisense A. arenosa centromeric probe (3*). (F) Accumulation of antisense small RNAs derived from the A. thaliana centromere in met1-RNAi A. suecica. The same blot was used except that a sense A. thaliana centromeric probe (6) was used for hybridization. (G) The small RNA blot was hybridized with an antisense U6 probe.
Centromeric heterochromatin is established and maintained by siRNAs (Bender 2001; Lippman and Martienssen 2004). However, it is unknown whether differential centromeric methylation in the allopolyploids is associated with progenitors' centromeric siRNAs. In the met1-1 and -12-RNAi A. suecica lines, we detected sense centromeric siRNAs (24–26 nt) in A. thaliana and A. arenosa centromeres (lanes 8 and 9 in Figure 6, D and E, respectively) but not in other lines, suggesting a role of demethylation in siRNA accumulation. Using U6 RNA as a loading control (Figure 6G), we found that A. thaliana centromeric siRNAs accumulated ∼10 times more than A. arenosa centromeric siRNAs in both met1-RNAi A. suecica lines. This may explain the high level of A. thaliana centromeric DNA methylation in the met1-RNAi lines that is generated by RNA-directed DNA methylation (RdDM) (Hamilton et al. 2002). In contrast, A. arenosa centromeres accumulated a small amount of siRNAs, which is associated with a relatively low level of DNA methylation. Similar results were obtained using sense and antisense probes derived from the left and right ends of centromeric repeats (data not shown), suggesting that the entire centromeric repeats are transcribed in both orientations. In addition to processed 24- to 26-nt siRNAs, additional transcripts of ∼80, ∼100, and ∼150 nt in size detected in both met1-1 and met1-12-RNAi A. suecica lines (Figure 6F) were likely the processing intermediates derived from centromeric RNA. Similar intermediates were observed in tomato when potato spindle tuber viroid (PSTVd) RNA was expressed (Denti et al. 2004). Transcripts of 160 nt in size matched the length of individual centromeric repeats that may serve as the original trigger for RNA interference and RNA-directed DNA methylation pathways (Lippman and Martienssen 2004; Matzke and Birchler 2005).
DISCUSSION
Effects of DNA methylation and allopolyploidy on changes in gene expression:
Although rapid and dynamic changes in genome organization and gene expression in allopolyploids have been documented, mechanisms for gene regulation in allopolyploids remain poorly understood (Osborn et al. 2003; Chen 2007). Gene expression in allopolyploids may be mediated by dosage-dependent regulation, regulatory interactions, rapid genomic changes including elimination or rearrangement of homeologous DNA fragments, and/or epigenetic alterations such as histone modification, DNA methylation, and RNA-mediated pathways (Song et al. 1995; Pikaard 2000; Shaked et al. 2001; Levy and Feldman 2002; Osborn et al. 2003; Adams and Wendel 2005; Chen 2007). One hypothesis suggests that epigenetic regulation of duplicate genes and genomes in allopolyploids may contribute to flexibility in expressing homeologous genes to improve fitness. Indeed, stochastic and rapid changes in gene expression are observed in newly synthesized Arabidopsis allotetraploids (Wang et al. 2004). Some changes in gene expression at early generations can be maintained during evolution as the same expression patterns are detected in natural allotetraploids, suggesting similar mechanisms for gene activation and silencing in early stages and during evolution of allopolyploids.
Our data suggest that allopolyploidy and DNA methylation may have distinct and some overlapping effects on gene regulation in allopolyploid genomes. In the resynthesized allopolyploids, ∼1400 genes in various biological pathways and regulatory networks displayed expression changes, whereas only ∼200 genes including many transposons and repetitive elements are affected by reduced DNA methylation in met1-RNAi A. suecica lines. Both demethylation and allopolyploidization affect a large number of stress-related genes, suggesting a common mechanism for defense responses (Ha et al. 2007; Yoder et al. 1997). A small number of transposable elements showed expression changes in resynthesized allotetraploids, suggesting that many transposons after “genomic shock” (McClintock 1984) are “settled down” during selfing. Consistent with this notion, many transposons and heterochromatic genes are repressed in natural allotetraploids. Disruption of DNA methylation in natural A. suecica lines releases chromatin constraints and consequently reactivates these heterochromatic genes. Notably, the majority of reactivated genes in met1-RNAi A. suecica lines are derived from an A. thaliana parent, which is reminiscent of genomewide transcriptome repression in which many genes that are expressed at high levels in A. thaliana are repressed in resynthesized allotetraploids (Wang et al. 2006). The data suggest that allopolyploidization induces homeologous genome-specific changes in DNA methylation and gene expression. Although changes in A. arenosa genomes are relatively unknown, the A. thaliana homeologous genome tends to be suppressed in the resynthesized allotetraploids. Repression of transposons and centromeric repeats are subjected to stable modifications including DNA methylation in natural A. suecica. Bisulfite sequencing analysis reveals that reactivation of A. thaliana-specific transcripts correlates with hypomethylation of A. thaliana-specific promoter in met1-RNAi A. suecica lines (Figure 5).
A role of DNA methylation in epigenetic silencing of homeologous genes:
Cytosine methylation is involved in epigenetic phenomena such as imprinting and X-inactivation as well as in dynamic responses to environmental cues and developmental programs (Bird 1992; Richards and Elgin 2002). In Arabidopsis and Brassica allotetraploids, DNA methylation is involved in the maintenance of silenced homeologous rRNA loci and protein-coding genes (Chen and Pikaard 1997; Lee and Chen 2001; Lawrence et al. 2004; Wang et al. 2004). We demonstrated that downregulation of met1 induces the expression of a subset of transposons, repetitive elements, and protein-coding genes. Compared to the control, ∼50% of 197 genes were either up- or downregulated in met1-RNAi A. suecica. Several upregulated genes studied are associated with the loss of methylation in the promoter regions, whereas a few downregulated genes examined are correlated with increased levels of DNA methylation or unrelated to methylation changes, suggesting an indirect effect. The data are consistent with the genomewide methylation analysis in A. thaliana diploid. DNA methylation may not directly affect gene transcription, but loss of methylation in promoters (Zhang et al. 2006) or gene body (Zilberman et al. 2007) leads to enhanced transcription.
Increase in DNA methylation in a genetic background with an overall reduced level of DNA methylation is likely associated with locus-specific modifications. Alternatively, cytosine methylation including de novo CG and non-CG may not be affected because other genes including DOMAINS REARRANGED METHYLTRANSFERASES (DRMs) (Cao et al. 2000; Cao and Jacobsen 2002), CHROMOMETHYLASE3 (CMT3) (Bartee et al. 2001; Lindroth et al. 2001; Tompa et al. 2002), and DDM1 (Vongs et al. 1993) remain functional in the met1-RNAi A. suecica lines. Although both met1- and ddm1-RNAi A. suecica plants display similar levels of siRNA accumulation in centromeres (data not shown), met1-RNAi lines had severer developmental defects than ddm1-RNAi lines (Wang et al. 2004), suggesting that other changes in these A. suecica transgenic plants are yet to be discovered.
Some silenced genes are not reactivated in the A. suecica lines overexpressing the double-stranded methyltransferase 1 (MET1) gene (Wang et al. 2004), implying that other mechanisms independent of DNA methylation are responsible for silencing homeologous genes in allopolyploids (Chen and Ni 2006; Chen 2007). Alternatively, non-CG methylation may be important to gene silencing (Martienssen and Colot 2001; Jackson et al. 2002) because MET1 is essential for the maintenance of methylation in symmetrical CG dinucleotides in A. thaliana (Finnegan and Kovac 2000). It is also likely that RNAi does not completely knock out MET1 expression such that residual methylation remains in the genome. MET1 may also contribute to de novo CG methylation in the presence of RNA signal (Aufsatz et al. 2004) and in the maintenance of CG methylation after removal of the RNA signals (Jones et al. 2001). Loss-of-function Arabidopsis mutants (met1-1∼4) display severe reduction in CG methylation and moderate loss of non-CG methylation in the genome (Kankel et al. 2003; Saze et al. 2003). In the plants that possess a single mutation of either met1 or cmt3, transcription and mobility levels of transposons were relatively low. In met1 and cmt3 double mutants, high frequencies of transposition can be detected (Kato et al. 2003), indicating MET1 and CMT3 are partially redundant in gene function.
In the allopolyploids, downregulation of MET1 leads to an overall reduction of CG methylation and increased levels of transcription in SCP repetitive gene family and En/Spm transposons and centromeric repeats, suggesting a causal link between CG methylation maintained by MET1 and transposon repression. Moreover, an increased level of CG methylation is found at the 5′-transcriptional region of a RD22 gene family member, corresponding to the suppression of this gene in the met1-RNAi plants. Both En/Spm transposons are capable of transposition in the homeologous genomes, which is in contrast to immobility of CACTA elements in spite of accumulated transcripts in the A. thaliana lines that overexpress antisense MET1 (Lippman et al. 2003). This may suggest different effects of met1 defects on transposon activities in diploid and tetraploid transgenic plants. However, we do not know whether A. thaliana En/Spm elements can move into A. arenosa chromosomes in the allotetraploids.
Differential DNA methylation and homeologous-specific regulation of centromeres and other loci in Allopolyploids:
The differential DNA methylation patterns in A. thaliana and A. arenosa centromeres detected in met1-RNAi A. suecica lines suggest that centromeric repeats are modified differently from other genes and heterochromatic regions in the allotetraploids. Modest demethylation levels detected in the A. arenosa centromeres in wild-type A. suecica strains indicate that repressive chromatin constraints were leaky during polyploid formation and evolution. This may be associated with relatively recent events in the formation of natural allopolyploids (Jakobsson et al. 2006). The levels of centromeric sensitivity to perturbation in DNA methylation may increase over generational time and hence disrupt normal patterns of meiosis and/or gametogenesis as previously observed (Comai et al. 2003), which leads to the increased severity of phenotypic effects in met1-RNAi A. suecica plants. Moreover, increased mobility of transposable elements and disruption of imprinting patterns may induce abnormal embryo development and a high rate of mortality in the sefling progeny in met1-RNAi lines (Grini et al. 2002; Xiao et al. 2003; Berger 2004).
Changes in DNA methylation have been observed in allopolyploids in Arabidopsis (Madlung et al. 2002), wheat (Shaked et al. 2001), cotton (Salmon et al. 2005), and Brassica (Gaeta et al. 2007) and in interspecific hybrids in marsupials (O'Neill et al. 1998), although global methylation changes in eutherian hybrids are debatable (Roemer et al. 1999). Reduction in DNA methylation may result in activation of transposons and other genes in interspecific hybrids or allopolyploids (Lee and Chen 2001; Madlung et al. 2002). Activation of retrotransposons may also affect expression of neighboring genes via antisense or sense transcripts as observed in wheat hybrids (Kashkush et al. 2003).
The different levels of centromere demethylation in A. thaliana and A. arenosa are associated with siRNAs preferentially accumulated in one genome (e.g., A. thaliana), which correlated with RdDM for transcriptional silencing of the heterochromatic region (Bender 2001; Lippman and Martienssen 2004). In met1 A. thaliana mutants, transcriptional gene silencing (TGS) cannot be established by siRNA signal through the RdDM pathway (Jones et al. 2001; Aufsatz et al. 2004). In the allotetraploids, RdRp may convert A. thaliana- or A. arenosa-specific transcripts from heterochromatic DNA into a double-stranded form that serves as the precursor of siRNA formation (Hamilton et al. 2002). Bidirectional transcription from heterochromatin is detectable in met1 and ddm1 mutants in Arabidopsis (Lippman et al. 2003; Lippman and Martienssen 2004) and in met1-RNAi A. suecica lines, arguing that transcription from both the sense and the antisense strand may be the source of dsRNA production. Moreover, readthrough transcription of inverted repeats may account for the occurrence of dsRNA from heterochromatin (Sijen et al. 2007). The production of dsRNAs from A. thaliana and A. arenosa centromeric regions may be differentially regulated in the allotetraploids, which leads to different levels of methylation accumulation in centromeric regions in the wild-type A. suecica and in the A. suecica transgenic lines in which MET1 is downregulated.
In Drosophila virilis, endogenous siRNAs are derived from the transposon Penelope in both males and females but only maternally loaded in embryos, which may suggest maternal transmission of Penelope siRNA in the repression of transposition (Blumenstiel and Hartl 2005). A. thaliana homeologous-specific changes in DNA methylation and small RNA accumulation in allopolyploids may be similar to the maternal repression of transposons because A. thaliana is used as a maternal parent in production of allotetraploids (Comai et al. 2000; Wang et al. 2004). We failed to produce interspecific hybrids or allotetraploids using A. arenosa as the maternal parent, which is reminiscent of hybrid dysgenesis in Drosophila (Engels and Preston 1979; Kidwell 1981; Bingham et al. 1982) and Peromyscus (Vrana et al. 2000). It is unknown how similar siRNAs in the same allotetraploid cells would lead to homeologous genome-specific methylation patterns. One possibility is that entire centromeric repeats are transcribed and processed into siRNAs (24–26 nt), which couple with the RNA-dependent DNA methylation pathways (Hamilton et al. 2002). Indeed, we found that probes derived from anywhere within the centromeric repeats detected a consistent amount of siRNA transcripts in A. thaliana or A. arenosa centromeres in the allotetraploids (data not shown). The level of sequence divergence between A. thaliana and A. arenosa centromeres (∼40%) may determine the specificity of RdDM pathways for two homeologous genomes in allotetraploids. Our data also suggest that centromeric DNA methylation levels are directly correlated with siRNA accumulation levels, supporting the model of RNA interference and heterochromatin maintenance (Lippman and Martienssen 2004).
In addition to homeologous-specific centromeric DNA methylation, methylation of some homeologous loci (e.g., At2g23810) is correlated with gene repression in Arabidopsis resynthesized and natural allotetraploids. It will be interesting to investigate whether and how siRNAs affect the initiation and maintenance of DNA methylation and transcript accumulation of homeologous loci and centromeres in resynthesized and natural allopolyploids.
Acknowledgments
We thank Dorothy Shippen, David Stelly, and Mary Byrk for critical suggestions. This work was supported by a grant from the National Science Foundation Plant Genome Research Program (DBI0077774 and DBI0733857) and a grant from the National Institutes of Health (GM067015).
References
- Adams, K. L., and J. F. Wendel, 2005. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8 135–141. [DOI] [PubMed] [Google Scholar]
- Adams, K. L., R. Cronn, R. Percifield and J. F. Wendel, 2003. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 100 4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufsatz, W., M. F. Mette, A. J. Matzke and M. Matzke, 2004. The role of MET1 in RNA-directed de novo and maintenance methylation of CG dinucleotides. Plant Mol. Biol. 54 793–804. [DOI] [PubMed] [Google Scholar]
- Bartee, L., F. Malagnac and J. Bender, 2001. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 15 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becak, M. L., and W. Becak, 1998. Evolution by polyploidy in Amphibia: new insights. Cytogenet. Cell Genet. 80 28–33. [DOI] [PubMed] [Google Scholar]
- Bender, J., 2001. A vicious cycle: RNA silencing and DNA methylation in plants. Cell 106 129–132. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57 289–300. [Google Scholar]
- Berger, F., 2004. Plant sciences. Imprinting—a green variation. Science 303 483–485. [DOI] [PubMed] [Google Scholar]
- Bingham, P. M., M. G. Kidwell and G. M. Rubin, 1982. The molecular basis of P-M hybrid dysgenesis: the role of the P element, a P-strain-specific transposon family. Cell 29 995–1004. [DOI] [PubMed] [Google Scholar]
- Bird, A., 1992. The essentials of DNA methylation. Cell 70 5–8. [DOI] [PubMed] [Google Scholar]
- Blumenstiel, J. P., and D. L. Hartl, 2005. Evidence for maternally transmitted small interfering RNA in the repression of transposition in Drosophila virilis. Proc. Natl. Acad. Sci. USA 102 15965–15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell, C., M. Spielman and R. J. Scott, 2003. The basis of natural and artifical postzygotic hybridization barriers in Arabidopsis species. Plant Cell 15 1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, X., and S. E. Jacobsen, 2002. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12 1138–1144. [DOI] [PubMed] [Google Scholar]
- Cao, X., N. M. Springer, M. G. Muszynski, R. L. Phillips, S. Kaeppler et al., 2000. Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc. Natl. Acad. Sci. USA 97 4979–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, X., W. Aufsatz, D. Zilberman, M. F. Mette, M. S. Huang et al., 2003. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr. Biol. 13 2212–2217. [DOI] [PubMed] [Google Scholar]
- Chen, Z., M. Devey, N. a. Tuleen and G. E. Hart, 1994. Use of recombinant substitution lines in the construction of Rflp-based genetic maps of chromosomes 6a and 6B of tetraploid wheat (Triticum-turgidum L). Theor. Appl. Genet. 89 703–712. [DOI] [PubMed] [Google Scholar]
- Chen, Z. J., 2007. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58 377–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. J., and Z. Ni, 2006. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. BioEssays 28 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. J., and C. S. Pikaard, 1997. Epigenetic silencing of RNA polymerase I transcription: a role for DNA methylation and histone modification in nucleolar dominance. Genes Dev. 11 2124–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church, G. M., and W. Gilbert, 1984. Genomic sequencing. Proc. Natl. Acad. Sci. USA 81 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai, L., 2005. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6 836–846. [DOI] [PubMed] [Google Scholar]
- Comai, L., A. P. Tyagi, K. Winter, R. Holmes-Davis, S. H. Reynolds et al., 2000. Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12 1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai, L., A. P. Tyagi and M. A. Lysak, 2003. FISH analysis of meiosis in Arabidopsis allopolyploids. Chromosome Res. 11 217–226. [DOI] [PubMed] [Google Scholar]
- Denti, M. A., A. Boutla, M. Tsagris and M. Tabler, 2004. Short interfering RNAs specific for potato spindle tuber viroid are found in the cytoplasm but not in the nucleus. Plant J. 37 762–769. [DOI] [PubMed] [Google Scholar]
- Engels, W. R., and C. R. Preston, 1979. Hybrid dysgenesis in Drosophila melanogaster: the biology of female and male sterility. Genetics 92 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan, E. J., and K. A. Kovac, 2000. Plant DNA methyltransferases. Plant Mol. Biol. 43 189–201. [DOI] [PubMed] [Google Scholar]
- Fransz, P. F., S. Armstrong, J. H. de Jong, L. D. Parnell, C. van Drunen et al., 2000. Integrated cytogenetic map of chromosome arm 4S of A. thaliana: structural organization of heterochromatic knob and centromere region. Cell 100 367–376. [DOI] [PubMed] [Google Scholar]
- Frommer, M., L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt et al., 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 89 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeta, R. T., J. C. Pires, F. Iniguez-Luy, E. Leon and T. C. Osborn, 2007. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19 3403–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grini, P. E., G. Jurgens and M. Hulskamp, 2002. Embryo and endosperm development is disrupted in the female gametophytic capulet mutants of Arabidopsis. Genetics 162 1911–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, M., W. H. Li and Z. J. Chen, 2007. External factors accelerate expression divergence between duplicate genes. Trends Genet. 23 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, A., O. Voinnet, L. Chappell and D. Baulcombe, 2002. Two classes of short interfering RNA in RNA silencing. EMBO J. 21 4671–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanfstingl, U., A. Berry, E. A. Kellogg, J. T. Costa, 3rd, W. Rudiger et al., 1994. Haplotypic divergence coupled with lack of diversity at the Arabidopsis thaliana alcohol dehydrogenase locus: Roles for both balancing and directional selection? Genetics 138 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylander, N., 1957. Cardaminopsis suecica (Fr.) Hiit., a northern amphidiploid species. Bull. Jard. Bot. Etat. Brux. 27 591–604. [Google Scholar]
- Ihaka, R., and R. Gentleman, 1996. A language for data analysis and graphics. J. Comput. Graph. Stat. 5 299–314. [Google Scholar]
- Jackson, J. P., A. M. Lindroth, X. Cao and S. E. Jacobsen, 2002. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416 556–560. [DOI] [PubMed] [Google Scholar]
- Jacobsen, S. E., H. Sakai, E. J. Finnegan, X. Cao and E. M. Meyerowitz, 2000. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 10 179–186. [DOI] [PubMed] [Google Scholar]
- Jakobsson, M., J. Hagenblad, S. Tavare, T. Sall, C. Hallden et al., 2006. A unique recent origin of the allotetraploid species Arabidopsis suecica: evidence from nuclear DNA markers. Mol. Biol. Evol. 23 1217–1231. [DOI] [PubMed] [Google Scholar]
- Jones, L., F. Ratcliff and D. C. Baulcombe, 2001. RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Curr. Biol. 11 747–757. [DOI] [PubMed] [Google Scholar]
- Kamm, A., I. Galasso, T. Schmidt and J. S. Heslop-Harrison, 1995. Analysis of a repetitive DNA family from Arabidopsis arenosa and relationships between Arabidopsis species. Plant Mol. Biol. 27 853–862. [DOI] [PubMed] [Google Scholar]
- Kankel, M. W., D. E. Ramsey, T. L. Stokes, S. K. Flowers, J. R. Haag et al., 2003. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush, K., M. Feldman and A. A. Levy, 2003. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 33 102–106. [DOI] [PubMed] [Google Scholar]
- Kato, M., A. Miura, J. Bender, S. E. Jacobsen and T. Kakutani, 2003. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr. Biol. 13 421–426. [DOI] [PubMed] [Google Scholar]
- Kidwell, M. G., 1981. Hybrid dysgenesis in Drosophila melanogaster: the genetics of cytotype determination in a neutral strain. Genetics 98 275–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto, N., H. Sakai, J. Jackson, S. E. Jacobsen, E. M. Meyerowitz et al., 2001. Site specificity of the Arabidopsis METI DNA methyltransferase demonstrated through hypermethylation of the superman locus. Plant Mol. Biol. 46 171–183. [DOI] [PubMed] [Google Scholar]
- Lawrence, R. J., K. Earley, O. Pontes, M. Silva, Z. J. Chen et al., 2004. A concerted DNA methylation/histone methylation switch regulates rRNA gene dosage control and nucleolar dominance. Mol. Cell 13 599–609. [DOI] [PubMed] [Google Scholar]
- Lee, H. S., and Z. J. Chen, 2001. Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc. Natl. Acad. Sci. USA 98 6753–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. S., J. L. Wang, L. Tian, H. M. Jiang, M. A. Black et al., 2004. Sensitivity of 70-mer oligonucleotides and cDNAs for microarray analysis of gene expression in Arabidopsis and its related species. Plant Biotechnol. J. 2 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch, I. L., and M. D. Bennett, 1997. Polyploidy in angiosperms. Trends Plant Sci. 2 470–476. [Google Scholar]
- Levy, A. A., and M. Feldman, 2002. The impact of polyploidy on grass genome evolution. Plant Physiol. 130 1587–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind-Hallden, C., C. Hallden and T. Sall, 2002. Genetic variation in Arabidopsis suecica and its parental species A. arenosa and A. thaliana. Hereditas 136 45–50. [DOI] [PubMed] [Google Scholar]
- Lindroth, A. M., X. Cao, J. P. Jackson, D. Zilberman, C. M. McCallum et al., 2001. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292 2077–2080. [DOI] [PubMed] [Google Scholar]
- Lippman, Z., and R. Martienssen, 2004. The role of RNA interference in heterochromatic silencing. Nature 431 364–370. [DOI] [PubMed] [Google Scholar]
- Lippman, Z., B. May, C. Yordan, T. Singer and R. Martienssen, 2003. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 1 E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, A., 1961. Hylandra—a new genus of Cruciferae. Sven. Bot. Tidskr. 55 211–217. [Google Scholar]
- Madlung, A., R. W. Masuelli, B. Watson, S. H. Reynolds, J. Davison et al., 2002. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 129 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, M. B., and H. O. Smith, 1977. Specificity of Hpa II and Hae III DNA methylases. Nucleic Acids Res. 4 4211–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen, R., and V. Colot, 2001. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293 1070–1074. [DOI] [PubMed] [Google Scholar]
- Masterson, J., 1994. Stomatal size in fossil plants: evidence for polyploidy in majority of angiosperms. Science 264 421–424. [DOI] [PubMed] [Google Scholar]
- Matzke, M. A., and J. A. Birchler, 2005. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 6 24–35. [DOI] [PubMed] [Google Scholar]
- McClelland, M., M. Nelson and E. Raschke, 1994. Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res. 22 3640–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock, B., 1984. The significance of responses of the genome to challenge. Science 226 792–801. [DOI] [PubMed] [Google Scholar]
- Moser, E. B., A. M. Saxton and J. P. Geaghan, 1988. Biological applications of the SAS system: an overview. Comput. Appl. Biosci. 4 233–238. [DOI] [PubMed] [Google Scholar]
- O'Kane, S., B. Schaal and I. Al-Shehbaz, 1995. The origins of Arabidopsis suecica (Brassicaceae), as indicated by nuclear rDNA sequences, and implications for rDNA evolution. Syst. Bot. 21 559–566. [Google Scholar]
- O'Neill, R. J., M. J. O'Neill and J. A. Graves, 1998. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393 68–72. [DOI] [PubMed] [Google Scholar]
- Osborn, T. C., J. C. Pires, J. A. Birchler, D. L. Auger, Z. J. Chen et al., 2003. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19 141–147. [DOI] [PubMed] [Google Scholar]
- Pikaard, C. S., 2000. The epigenetics of nucleolar dominance. Trends Genet. 16 495–500. [DOI] [PubMed] [Google Scholar]
- Price, R. A., J. D. Palmer and I. Al-Shehbaz, 1994. Systematic relationships of Arabidopsis: a molecular and morphological perspective, pp. 7–19 in Arabidopsis, edited by E. M. Meyerowitz and C. R. Sommerville. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Richards, E. J., and S. C. Elgin, 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108 489–500. [DOI] [PubMed] [Google Scholar]
- Roemer, I., F. Grutzner, H. Winking, T. Haaf, A. Orth et al., 1999. Genome evolution. Global methylation in eutherian hybrids. Nature 401 131–132. [DOI] [PubMed] [Google Scholar]
- Salmon, A., M. L. Ainouche and J. F. Wendel, 2005. Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 14 1163–1175. [DOI] [PubMed] [Google Scholar]
- Saze, H., O. Mittelsten Scheid and J. Paszkowski, 2003. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 34 65–69. [DOI] [PubMed] [Google Scholar]
- Shaked, H., K. Kashkush, H. Ozkan, M. Feldman and A. A. Levy, 2001. Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen, T., F. A. Steiner, K. L. Thijssen and R. H. Plasterk, 2007. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science 315 244–247. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E., P. S. Soltis and J. A. Tate, 2003. Advances in the study of polyploidy since plant speciation. New Phytol. 161 173–191. [Google Scholar]
- Soltis, P. S., and D. E. Soltis, 2000. The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. USA 97 7051–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, K., P. Lu, K. Tang and T. C. Osborn, 1995. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. USA 92 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, T. L., and E. J. Richards, 2002. Induced instability of two Arabidopsis constitutive pathogen-response alleles. Proc. Natl. Acad. Sci. USA 99 7792–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, T. L., B. N. Kunkel and E. J. Richards, 2002. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 16 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa, R., C. M. McCallum, J. Delrow, J. G. Henikoff, B. van Steensel et al., 2002. Genome-wide profiling of DNA methylation reveals transposon targets of CHROMOMETHYLASE3. Curr. Biol. 12 65–68. [DOI] [PubMed] [Google Scholar]
- Vongs, A., T. Kakutani, R. A. Martienssen and E. J. Richards, 1993. Arabidopsis thaliana DNA methylation mutants. Science 260 1926–1928. [DOI] [PubMed] [Google Scholar]
- Vrana, P. B., J. A. Fossella, P. Matteson, T. del Rio, M. J. O'Neill et al., 2000. Genetic and epigenetic incompatibilities underlie hybrid dysgenesis in Peromyscus. Nat. Genet. 25 120–124. [DOI] [PubMed] [Google Scholar]
- Wang, J., L. Tian, A. Madlung, H. S. Lee, M. Chen et al., 2004. Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167 1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., J. J. Lee, L. Tian, H. S. Lee, M. Chen et al., 2005. Methods for genome-wide analysis of gene expression changes in polyploids. Methods Enzymol. 395 570–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., L. Tian, H. S. Lee, N. E. Wei, H. Jiang et al., 2006. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J. F., 2000. Genome evolution in polyploids. Plant Mol. Biol. 42 225–249. [PubMed] [Google Scholar]
- Wolfe, K. H., 2001. Yesterday's polyploidization and the mystery of diploidization. Nat. Rev. Genet. 2 333–341. [DOI] [PubMed] [Google Scholar]
- Xiao, W., M. Gehring, Y. Choi, L. Margossian, H. Pu et al., 2003. Imprinting of the MEA Polycomb gene is controlled by antagonism between MET1 methyltransferase and DME glycosylase. Dev. Cell 5 891–901. [DOI] [PubMed] [Google Scholar]
- Yoder, J. A., and T. H. Bestor, 1996. Genetic analysis of genomic methylation patterns in plants and mammals. Biol. Chem. 377 605–610. [PubMed] [Google Scholar]
- Yoder, J. A., C. P. Walsh and T. H. Bestor, 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13 335–340. [DOI] [PubMed] [Google Scholar]
- Zhang, X., J. Yazaki, A. Sundaresan, S. Cokus, S. W. Chan et al., 2006. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zilberman, D., M. Gehring, R. K. Tran, T. Ballinger and S. Henikoff, 2007. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39 61–69. [DOI] [PubMed] [Google Scholar]