

Abstract
Fertility quantitative trait loci (QTL) are of high interest in dairy cattle since insemination failure has dramatically increased in some breeds such as Holstein. High-throughput SNP analysis and SNP microarrays give the opportunity to genotype many animals for hundreds SNPs per chromosome. In this study, due to these techniques a dense SNP marker map was used to fine map a QTL underlying nonreturn rate measured 90 days after artificial insemination previously detected with a low-density microsatellite marker map. A granddaughter design with 17 Holstein half-sib families (926 offspring) was genotyped for a set of 437 SNPs mapping to BTA3. Linkage analysis was performed by both regression and variance components analysis. An additional analysis combining both linkage analysis and linkage-disequilibrium information was applied. This method first estimated identity-by-descent probabilities among base haplotypes. These probabilities were then used to group the base haplotypes in different clusters. A QTL explaining 14% of the genetic variance was found with high significance (P < 0.001) at position 19 cM with the linkage analysis and four sires were estimated to be heterozygous (P < 0.05). Addition of linkage-disequilibrium information refined the QTL position to a set of narrow peaks. The use of the haplotypes of heterozygous sires offered the possibility to give confidence in some peaks while others could be discarded. Two peaks with high likelihood-ratio test values in the region of which heterozygous sires shared a common haplotype appeared particularly interesting. Despite the fact that the analysis did not fine map the QTL in a unique narrow region, the method proved to be able to handle efficiently and automatically a large amount of information and to refine the QTL position to a small set of narrow intervals. In addition, the QTL identified was confirmed to have a large effect (explaining 13.8% of the genetic variance) on dairy cow fertility as estimated by nonreturn rate at 90 days.
FOLLOWING the development of molecular tools in the beginning of the 1990s and the first experiment reported more than 10 years ago (Georges et al. 1995), detection of quantitative trait loci (QTL) in dairy cattle has been performed in most large dairy cattle populations. Hence, a large number of QTL affecting production, functional, and conformation traits have been detected (Khatkar et al. 2004). Although performed on enlarged pedigrees, the mapping resolution of most of these experiments was primarily limited by the low marker density available for the genome scan. Thus confidence intervals for QTL location spread most often over >10 cM.
In some cases, fine-mapping studies were carried out to reduce these confidence intervals (Meuwissen et al. 2002; Olsen et al. 2005; Gautier et al. 2006), leading in some instances to the identification of the underlying causal mutation (Grisart et al. 2002; Blott et al. 2003; Cohen-Zinder et al. 2005). These fine-mapping studies were mostly based on addition of new sire families, additional markers, and the use of statistical methods combining linkage analysis (LA) and linkage-disequilibrium (LD) analysis (LDLA). In general, the marker density was slightly increased by the addition of a few tens of new markers (microsatellite markers or SNPs) identified within the QTL region or in some candidate genes.
High-throughput SNP analysis and SNP microarrays now give one the opportunity to genotype many animals for hundreds of SNPs per chromosome (Khatkar et al. 2006, 2007; Gautier et al. 2007). Due to these techniques, marker density is no longer a limiting factor in QTL fine-mapping studies, which can be speeded up or even performed without prior knowledge of segregating QTL. However, these dense marker maps require efficient statistical methods that work fast and efficiently with large numbers of markers.
In France, fine mapping of QTL related to female fertility is of utmost importance. Fertility has been decreasing in the Holstein breed despite its economical importance. The heritability of this trait is particularly low and the efficiency of traditional selection remains limited. Therefore identification of genetic variants involved in the fertility decline of Holstein cows might be particularly beneficial to improve selection through marker assistance. As a consequence, it was decided to evaluate the efficiency of dense SNP maps for QTL fine mapping considering a QTL underlying nonreturn rate estimated 90 days after artificial insemination (NRR90) previously identified on bovine chromosome 3 (BTA03) and segregating in the French Holstein population using a sparse 16-microsatellite map as a model (Guillaume et al. 2007).
MATERIALS AND METHODS
Animal material:
A granddaughter design (GDD) with 17 Holstein half-sib families was analyzed in this study. In total 926 sons with known phenotypes were genotyped. Family size ranged from 20 to 112 sons (54 sons per sire on average). For the variance components method analysis, relatives of these genotyped animals (which were not further genotyped) were added to the pedigree file, which contained in total 2265 animals.
Phenotypes were equivalent to twice the daughter yield deviation (DYD) (Vanraden and Wiggans 1991) estimated on NRR90 as described previously (Guillaume et al. 2007). These represent the average performance of the daughters of a sire, corrected for the environmental effects and the genetic value of the mates.
Genotypic data:
The methods for SNP detection, selection, and genotyping were presented previously (Gautier et al. 2007). Briefly, SNPs were detected in silico on the basis of the sequences available in public databases. In total, 1373 SNPs were chosen to cover the whole BTA03 on the basis of comparative mapping results and included in a 1536-SNP GoldenGate assay provided by Illumina (http://www.illumina.com; Illumina, San Diego) to be genotyped at the Centre National de Génotypage (Evry, France). A dense linkage map of BTA03 containing 460 SNPs could then be constructed using available pedigrees and comparative mapping information after rejecting uninformative SNPs (Gautier et al. 2007). The BTA03 genetic map was estimated to span 125 Mb (and 127 cM) with an average marker spacing of 280 kb (from <500 bp to 3 Mb). For this study, 437 SNPs belonging to this dense BTA03 map and with a minor allele frequency (MAF) >0.05 in the Holstein breed were considered for further fine-mapping analysis. Genotypes for these 437 chosen SNPs were available for the 926 sons of the Holstein GDD, with few missing data or inconsistency with familial information. On average each son was genotyped for 99.7% of the SNPs (from 79.4 to 100%).
Haplotype reconstruction:
A program was developed for rapid haplotype reconstruction working with dense marker maps. The most likely genotypes of the different sires were first determined. For each SNP, the likelihood of each of the three possible sire genotypes (all SNPs in the study being biallelic) was equal to the product of the contribution of all its sons. The contribution of each son corresponded to its genotype probability given the sire genotype considered and the probability of inheriting the observed maternal allele estimated by its population frequency. A probability of genotyping error was set to 0.001 in the computation of the likelihood.
Paternally and maternally inherited haplotypes for the different animals were then calculated using the following algorithm:
Alleles from homozygous SNPs are assigned to both haplotypes of a given animal.
For offspring for which allele origin (maternal or paternal) can be determined unambiguously (conditionally on sire genotype), alleles are assigned accordingly to the corresponding haplotype.
- Within family, the most likely sire haplotypes are then sequentially constructed with marker alleles in offspring for which allele origin was already determined (in step 2):
- 3.1. For each marker, search the closest informative flanking markers (informative markers must already be assigned to a haplotype for both offspring and parent and be heterozygous in the parent).
- 3.2. Given genetic distances and offspring genotypic data, compute the probability for each sire marker allele that it belongs to the first (k = 1) and second (k = 2) sire haplotype. According to the closest informative markers upstream, this probability can be computed as
 (Windig and Meuwissen 2004), where y (x) is the number of offspring with a marker phase in agreement (disagreement) with the tested sire haplotype configuration and Θl is the recombination rate between the tested marker and the closest informative marker for offspring l. Similarly,
(Windig and Meuwissen 2004), where y (x) is the number of offspring with a marker phase in agreement (disagreement) with the tested sire haplotype configuration and Θl is the recombination rate between the tested marker and the closest informative marker for offspring l. Similarly,  is computed with the closest informative markers downstream. If
is computed with the closest informative markers downstream. If  , then the probability that the first marker allele of the sire belongs to the haplotype k is
, then the probability that the first marker allele of the sire belongs to the haplotype k is  (Windig and Meuwissen 2004).
(Windig and Meuwissen 2004). - 3.3. If
 > 0.95, the corresponding marker allele is assigned to the sire haplotype k. Thus, the other sire allele is then assigned to the sire haplotype 3 − k.
> 0.95, the corresponding marker allele is assigned to the sire haplotype k. Thus, the other sire allele is then assigned to the sire haplotype 3 − k.
- Unassigned markers in offspring are determined with the help of neighboring markers already assigned and parental haplotypes (Qian and Beckmann 2002):
- 4.1. Search the two closest informative flanking markers.
- 4.2. Check for these two flanking markers, whether or not the marker alleles of the paternal haplotype of the offspring originate from the same haplotype of the sire (no recombination is observed).
- 4.3. If the probability of double recombination between the informative flanking markers is <0.05, assign the marker allele of the corresponding haplotype of the sire to the son's paternal haplotype and the other allele to the maternal one.
QTL mapping method:
First, half-sib linear regression (Knott et al. 1996) was performed,
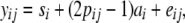 |
where yij is twice the DYD for NRR90 of son j of sire i, si is the fixed effect of sire i, pij is the probability of inheriting the first allele from sire i for son j, ai is half of the substitution effect of the QTL carried by the sire i, and eij is the residual. The residual variance was estimated within the sire family and assumed to be heterogeneous to account for the amount of information in the progeny: the residual variance was equal to 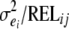 , where
, where 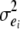 is the residual variance in sire family i and RELij is the reliability of the proof of son j of sire i based on progeny information only. The model was tested for each marker interval. Marker informativity for each interval was computed as the mean
is the residual variance in sire family i and RELij is the reliability of the proof of son j of sire i based on progeny information only. The model was tested for each marker interval. Marker informativity for each interval was computed as the mean 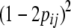 . Chromosomewide significance thresholds were estimated with 30,000 within-family permutations (Churchill and Doerge 1994).
. Chromosomewide significance thresholds were estimated with 30,000 within-family permutations (Churchill and Doerge 1994).
At the location of the maximum likelihood-ratio test (LRT), heterozygous status of the sires was estimated with a t-test on the basis of the value of the substitution effect, residual variance in the sire family, and the number of sons. A 90% confidence interval was estimated using the Lod drop-off approach and corresponds to the positions around the peak (with a LRT value of LRTp) for which the LA curve is >(LRTp − 3.84).
In addition to the half-sib regression analysis, a variance component (VC)-based linkage analysis (George et al. 2000) was performed with the model
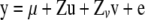 |
(Fernando and Grossman 1989), where y is a vector containing twice the DYD for NRR90 for bulls, μ is the mean, u is a vector of random polygenic effects, v is a vector of random gametic effects, and e is a vector of random residual terms. X, Z, and Zv are known design matrices relating results to fixed, random polygenic, and gametic effects, respectively.
The (co)variance structure was
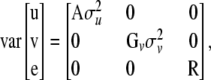 |
where A is the additive relationship matrix and 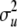 is the polygenic variance. Gv is the relationship matrix among QTL allelic effects estimated due to relationships and marker information (Fernando and Grossman 1989) and
is the polygenic variance. Gv is the relationship matrix among QTL allelic effects estimated due to relationships and marker information (Fernando and Grossman 1989) and 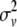 is the gametic variance. As in Pong-Wong et al. (2001), the method for calculating the gametic matrix used the closest informative bracket (as defined in 3.1 above) instead of estimating probabilities-of-descent of a gamete (PDQ) from parent to offspring by integration over all possible haplotypes. Rules to compute the PDQ using the closest informative bracket can be found in Table 1 in Pong-Wong et al. (2001). In summary, the probability to receive the QTL allele from the first haplotype of the sire is equal to
is the gametic variance. As in Pong-Wong et al. (2001), the method for calculating the gametic matrix used the closest informative bracket (as defined in 3.1 above) instead of estimating probabilities-of-descent of a gamete (PDQ) from parent to offspring by integration over all possible haplotypes. Rules to compute the PDQ using the closest informative bracket can be found in Table 1 in Pong-Wong et al. (2001). In summary, the probability to receive the QTL allele from the first haplotype of the sire is equal to 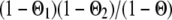 ,
, 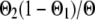 ,
, 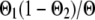 , or
, or 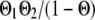 if both, only the first, only the second, or none of the marker alleles of the closest informative markers are equal in the paternal haplotype of the offspring and the first haplotype of the sire (where Θ, Θ1, and Θ2 are the recombination rates between the two closest informative markers, between the first informative marker and the QTL location, and between the second informative marker and the QTL location, respectively). The variances of paternal and maternal alleles were assumed to be equal and a single parameter was estimated (
if both, only the first, only the second, or none of the marker alleles of the closest informative markers are equal in the paternal haplotype of the offspring and the first haplotype of the sire (where Θ, Θ1, and Θ2 are the recombination rates between the two closest informative markers, between the first informative marker and the QTL location, and between the second informative marker and the QTL location, respectively). The variances of paternal and maternal alleles were assumed to be equal and a single parameter was estimated (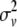 ) as previously described (Grignola et al. 1996a). Then, variance associated with the QTL (QTL allelic variance) was twice
) as previously described (Grignola et al. 1996a). Then, variance associated with the QTL (QTL allelic variance) was twice 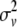 . The proportion of total genetic variance due to the QTL was
. The proportion of total genetic variance due to the QTL was
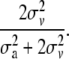 |
R is a diagonal matrix containing the residual variance (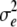 ) divided by the weight of the corresponding DYD for NRR90. These weights were daughter equivalent and were corrected for the number of inseminations and cows in each herd (Vanraden and Wiggans 1991).
) divided by the weight of the corresponding DYD for NRR90. These weights were daughter equivalent and were corrected for the number of inseminations and cows in each herd (Vanraden and Wiggans 1991).
TABLE 1.
Half-allelic substitution effect (in percentage of NRR90) and probability of heterozygosity for the 17 sires
| Sire no. | No. of sons | Half-substitution effect | pr(t < T) |
|---|---|---|---|
| 6 | 44 | −0.32 | 0.7732 |
| 25 | 20 | 0.78 | 0.6578 |
| 41a | 91 | −0.98 | 0.0871 |
| 74 | 34 | −0.18 | 0.8505 |
| 99 | 111 | −0.04 | 0.9602 |
| 102 | 37 | 0.66 | 0.5655 |
| 103 | 61 | 0.36 | 0.6833 |
| 305 | 51 | −0.72 | 0.5059 |
| 307b | 68 | 2.14 | 0.0060 |
| 310a | 43 | −2.24 | 0.0579 |
| 311b | 60 | 2.02 | 0.0075 |
| 314 | 51 | −0.66 | 0.4934 |
| 633b | 49 | −2.70 | 0.0001 |
| 974 | 62 | −0.58 | 0.4682 |
| 975 | 45 | 1.22 | 0.2215 |
| 976b | 54 | −2.22 | 0.0121 |
| 977 | 27 | −0.64 | 0.6213 |
Sires with a significant t-test (P < 0.10).
Sires with a significant t-test (P < 0.05).
LDLA:
QTL fine mapping was based on an approach similar to the one previously described (Kim and Georges 2002; Blott et al. 2003) and derived from the original method proposed by Meuwissen and Goddard (2000). It consists of a VC mapping method that includes information from linkage disequilibrium (LD) between base haplotypes in the construction of the relationship matrix among QTL allelic effects estimated (see above). Chromosomes were grouped in different categories: sire chromosomes (SC) and paternally and maternally inherited chromosomes (PC and MC) of the sons. SCs and MCs were considered as base haplotypes. At each tested position the following procedure is applied:
Probability of transmission (pij) is computed to determine to which SC a PC corresponds. These probabilities are the same as those computed in the linkage analysis method.
Identity-by-descent (IBD) probabilities (φp) were estimated among each pair of base haplotypes conditionally on the identity-by-state (IBS) status of the neighboring markers using windows of 10 flanking markers (Meuwissen and Goddard 2001).
Base haplotypes were grouped with a clustering algorithm with SAS proc CLUST, using (1 − φp) as a distance measure. Base haplotypes were grouped if φp > 0.50 (Ytournel et al. 2007). PCs were also grouped within the clusters if (i) the two SCs of a sire are grouped in the same cluster (the PCs of all his sons are then grouped in this cluster) or (ii) a PC can be associated with a base haplotype with a probability >0.95 (it is grouped to the corresponding cluster).
A model similar to the linkage analysis model is then applied,
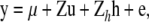 |
where h is a vector of random QTL effects corresponding to the haplotype clusters and Zh is a design matrix relating phenotypes to corresponding haplotype clusters. IBD10 is the notation for this model. Information on the structure of the groups was indicated by the percentage of base haplotypes grouped in the 20 largest clusters (N20) and the repartition of the haplotypes of n heterozygous sires (P < 0.10) in the groups was also described.
In addition, a similar model with the following new rules was applied: (1) all chromosomes, even PCs, were considered as base haplotypes; (2) smaller marker windows were used (three markers); and (3) haplotype groups were no longer constructed on the basis of IBD probabilities but on the basis of IBS status (if haplotypes were IBS for all markers they were grouped together). This method searches if an effect can be associated with a small haplotype covering a small region. Small marker windows were preferred to obtain a small number of groups. HAP3 is used to refer to this model.
For the three different models (VC linkage analysis, IBD10, and HAP3), genetic parameters were estimated after maximizing likelihoods with an average information–restricted maximum-likelihood (AI–REML) approach (Jensen et al. 1996). The BLUPF90 software (Misztal et al. 2002) was modified to incorporate relationship matrices among QTL allelic effects.
The likelihood-ratio test statistic considered variance components as parameters and was used to confirm whether there was a QTL present at the studied position,
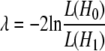 |
(George et al. 2000), where L(H0) and L(H1) are the maximum values of the likelihood functions estimated by REML under the polygenic model with no QTL fitted and with one of the tested models (LA or LDLA model), respectively. The distribution of the test is not known but was previously shown to be intermediate between the 1- and the 2-d.f. chi-square distribution (Grignola et al. 1996b). In this study, the 2-d.f. chi-square distribution was used. Consequently, the test was conservative for the assumption of no existing QTL.
RESULTS
Linkage analysis:
Respectively 90 and 67% of the 437 SNPs used in our study had a MAF > 0.1 and >0.2. Along BTA03, the mean informativity was 0.96 (with a minimum of 0.85) and >0.95 for each sire family (the minimum always remaining >0.70). Two sire families departed from these rules. The first sire (305) was homozygous for the first 93 markers (26 cM) while the second (310) was homozygous from marker 293 until marker 430 (over 54 cM). This homozygosity is probably a result of the high level of inbreeding in the Holstein breed. Indeed, the sires of our study are strongly inbred; for instance, the inbreeding coefficient for sires 305 and 310, computed only over a four-generation pedigree, is ∼5% (data not shown). As a consequence, for these two sire families, informativity dropped to 0.35 and 0.40. Although this might affect power of LA, in the case that the corresponding sires are found heterozygous at the QTL, assuming the two sire haplotypes are IBD at the corresponding (large) homozygous positions and the causal mutation(s) are relatively old, the QTL could be excluded from this part of the chromosome. The LDLA approach uses to a broader extent this kind of IBD information.
The QTL was confirmed by both regression and the VC linkage analysis (Figure 1). The maximum LRT location was found in neighboring intervals with both methods (positions 19.03 and 18.98 cM with the regression analysis and the VC approach, respectively). With the regression analysis the QTL was significant (P < 0.001) while with the VC approach, the likelihood-ratio test (−2(ln(H0) − ln(H1)) = 9.92 (P < 0.01). The confidence interval spanned 9 cM on the map: from position 15.85 to 24.82 cM.
Figure 1.—

Linkage analysis curves obtained for NRR90 on chromosome 3. Significance threshold P < 0.001. - - -, LA curve obtained with the VC approach; ——, LA curve obtained with the regression analysis ( ).
).
The estimated part of genetic variance explained by the QTL was 13.8% at the maximum LRT location. According to the t-test performed at the peak position, four sires were heterozygous for the QTL at the 5% threshold (Table 1). In addition, two sires (41 and 310) were also heterozygous if the threshold for significance was reduced (P < 0.10). As mentioned above, the complete homozygosity of sire 310 over 54 cM from SNP 293 to SNP 430 is in perfect agreement with the confidence-interval positions at the beginning of BTA03. The average allelic substitution effect for twice the DYD of the four heterozygous sires is 4.54% NRR90 units (from 4.04 to 5.40). This effect is close to one genetic standard deviation. Among the six sires possibly heterozygous, two pairs of them have a common grandsire.
Linkage disequilibrium and linkage analysis:
These analyses were performed on the 150 first marker intervals (until position 34.96 cM) corresponding to a region larger than the 90% QTL confidence interval as determined by the Lod drop off (Figure 1).
The LRT curves presented five and six peaks >6.0 with models IBD10 and HAP3, respectively (Figure 2 and Table 2). For most of the described peaks, the LRT curve obtained with LDLA models exceeded clearly the LA LRT curve while outside these regions it was below it and often reduced to zero. As shown in Table 2, only peaks 1, 2, and 4 were significant for both LDLA models. For both models, the maximum was located in the second peak (at position 16.9 cM on the map). The first peak (at position 11.7 cM on the map) also presented particularly high LRT values with both models.
Figure 2.—

LA and LDLA curves obtained for NRR90 on chromosome 3. LA curve (·····), LDLA curve with model IBD10 ( ), and LDLA curve with model HAP3 (----) are shown.
), and LDLA curve with model HAP3 (----) are shown.
TABLE 2.
Position, LRT, and percentage of base haplotypes grouped in the 20 largest clusters for LRT peaks (LRT > 6.0) obtained with models IBD10 and HAP3
| Peak no. | Position (in cM) | LRT with model
|
||
|---|---|---|---|---|
| IBD10 | HAP3 | N20a | ||
| 1 | 11.68 | +8.93 | +11.02 | 0.93 |
| 2 | 16.87 | +10.05 | +12.69 | 0.87 |
| 3 | 19.68 | +5.25 | +11.84 | 0.90 |
| 4 | 23.12 | +6.37 | +7.00 | 0.99 |
| 5 | 25.55 | +7.49 | +4.73 | 0.75 |
| 6 | 26.93 | +2.96 | +9.06 | 0.97 |
| 7 | 28.03 | +8.55 | +5.40 | 0.94 |
| 8 | 34.86 | +1.26 | +6.25 | 0.99 |
Percentage of base haplotypes grouped in the 20 largest clusters.
The N20 value (see materials and methods) varied from 0.59 to 0.99 (see Table 2) and was >0.75 for all the identified peaks (i.e., at least 700 base haplotypes were grouped within 20 clusters). For most intervals, <20 clusters contained more than one haplotype and all the grouping was done in the 20 largest clusters. The remaining haplotypes were ungrouped and corresponded most often to haplotypes with missing alleles.
In addition to N20, the haplotypes of the six heterozygous sires (P < 0.10) were studied. As expected, for all the positions, more base haplotypes were grouped with model HAP3 than with the IBD10 model. However, this was also true for haplotypes with opposite effects. At the first and fifth peaks, all the positive haplotypes of the heterozygous sires were identical for three markers and were grouped together with the HAP3 model (Table 3). The HAP3 model grouped at least five haplotypes of six in one cluster at peaks 1, 2, 4, 5, and 6 (see haplotypes of sires in Table 3). The peaks 3, 7, and 8 were locations where base haplotype clusters contained less than four haplotypes of heterozygous sires together.
TABLE 3.
Haplotypes of heterozygous sires (P < 0.10) at five peak locations
| Favorable (+) or unfavorable (–) QTL allele | Haplotypes
|
||||||
|---|---|---|---|---|---|---|---|
| Sire | t-test | Peak 1 | Peak 2 | Peak 4 | Peak 5 | Peak 6 | |
| 633 | + | 0.0001 | ATCCAATGTA | CGGATACGGA | ATCGCAATTG | TTGTAATGAA | CCCCATGCAG |
| 307 | + | 0.0085 | ACTCAACGCG | CCGACGGGGA | ATCGCAGTTA | TTATAATGAA | CGCAATGCAG |
| 311 | + | 0.0105 | ACTCAACGCG | CGGATACGGA | ATCGCAGTTA | TTATAATGAA | CCCAATACGA |
| 976 | + | 0.0117 | TTCCAACCCG | CGGGCAGGAA | ATCGCGACTG | CTGTAAAGAA | CGCAATACAG |
| 310 | + | 0.0697 | TTCCAATGCG | CGAATAGGAA | ATCGCGACTA | CTATAATGAG | CCTAATGTAA |
| 41 | + | 0.0852 | ATCCAATGTA | CGGATACGGA | ATCGCAGCTA | CTATAATGGA | CGCAATGCGG |
| 633 | − | 0.0001 | TTCCAACGCG | TGGATGGAGA | GTCGCGGTTA | TTATAATGAA | CGCAATGCAG |
| 307 | − | 0.0085 | ACTCAGCGCA | CGAATGGGGG | ATCGCGGCTG | CTGTGAAGAA | CCCCCTACAG |
| 311 | − | 0.0105 | ACTCAGCGCG | CGGATGGAAA | ACCGCGGTTA | TTACAATGAG | CCTACTGCAG |
| 976 | − | 0.0117 | ACTTTACCCG | CGGATGGGGA | ATCGCGATTA | TTATAAAGAA | TCCAACACAG |
| 310 | − | 0.0697 | ACTTTACCCG | CGGATGGGGA | ATCGCGATTA | TTATAAAGAA | TCCACTACAG |
| 41 | − | 0.0852 | ACTTTATGTG | CGAATAGGAA | ATCGCAGCCG | CCGTGATAAA | CCCACTGCAA |
With both models, the grouping was identical at the fourth peak. The haplotypes of the six sires are grouped in only two clusters (Table 3) but both groups contain haplotypes of opposite effects.
With the IBD10 model, four haplotypes with positive effects were grouped in one cluster and the two remaining haplotypes were grouped in another cluster at the first position. At other positions, three or fewer haplotypes of heterozygous sires were clustered together with the IBD10 model.
At peaks 5 and 7, LRT is high with IBD10 while low with HAP3. For these positions, there is little grouping with the IBD10 model (the haplotypes of the heterozygous sires are not grouped together) while there is more grouping with the HAP3 model. However, haplotypes with opposite effects are also grouped together with the HAP3 model. Therefore, the LRT curve dropped with model HAP3 for these positions.
DISCUSSION
Haplotype reconstruction:
With dense marker maps, methods evaluating all possible haplotypes or using a search algorithm to find the most probable haplotype are no longer feasible. Our former haplotype reconstruction program estimated the probability of 2n−1 possible haplotype pairs (where n is the number of markers). With >25 or 30 markers, this program was no longer working and a new program had to be developed. The new algorithm is described in materials and methods and is similar to a method proposed by Windig and Meuwissen (2004). Construction of haplotypes of 500 markers for 1000 animals was no longer a problem and the program performed this step in only a few seconds. In addition, use of haplotypes of parents to determine unassigned markers in offspring (step 4 of the algorithm) strongly improved haplotype reconstruction in comparison to the former program. In this study, while on average sons were genotyped on 99.7% of the SNPs considered, phase could be deduced on average for 97.7% of the SNPs (from 74.1 to 100%). With hundreds of markers, missing alleles in haplotype reconstruction are not very important for linkage analysis methods since there is always a neighboring informative marker that can be used to compute the probability of transmission of QTL alleles from sire to progeny. However, for methods based on linkage disequilibrium, reconstruction of the haplotype for all markers is crucial. In addition, missing information can also lead to computational problems, for instance, assuming three haplotypes are identical for a 10-marker window except at one position for which the allele is different in the first two haplotypes and missing in the third one. The IBD probability between the first two haplotypes will then be low. However, if the missing allele is ignored to compute φp between haplotypes 1 and 3, this probability will be high since all the nine remaining alleles are identical alleles. The same high φp will be computed between haplotypes 2 and 3, which is inconsistent with the first φp estimated. To avoid these problems, minimal φp were used in this study: if a marker allele was missing in a haplotype, it was supposed different from another haplotype one for φp computation. This has consequences on the LDLA since not all the LD information is used and haplotypes containing missing markers will have low φp with other base haplotypes and will not be clustered. Therefore, it is very important to reconstruct as much as possible the base haplotypes. For SCs, the haplotype reconstruction program leaves few missing markers but for MCs, there are still some missing markers for ungenotyped markers, in low-informativity regions and in regions displaying high recombination rates. The haplotype reconstruction program can still be improved to reduce the number of missing alleles by using the LD between markers.
Linkage analysis:
The QTL was confirmed with respect to the previously published study (Guillaume et al. 2007) based on a larger half-sib design comprising 26 families. The LRT peak was within the 95% confidence interval of this latter one and the obtained significance was higher with fewer families. The use of a high-density marker map resulted in almost optimal genetic information along the whole chromosome. In consequence, a sharper and higher LRT curve was obtained. With this density of markers, QTL transmission is followed more precisely and locations of recombinations are determined within smaller intervals, allowing an almost perfect achievement of the pedigree linkage-mapping resolution. Four sires were heterozygous (P < 0.05), which represents <25% of the tested families. This proportion is close to the one previously obtained (Guillaume et al. 2007), where 6 of 26 sires (23%) were determined as heterozygous. This corresponds approximately to allelic frequencies of 0.15 and 0.85 for the two alternate QTL alleles, assuming a biallelic QTL and that QTL genotypes are distributed according to Hardy–Weinberg proportions in the sire population. The average allelic substitution effect was 4.6% of NRR90 and represents 0.7 standard genetic deviation, which is higher than the effect (3% corresponding to 0.45 standard genetic deviation) previously reported (Guillaume et al. 2007). Nevertheless, these estimated effects have a rather large impact on the fertility and would represent a large part of the genetic variation. Indeed, the variance explained by the QTL can be approximated as 2pqα2 (where p and q are the allelic frequencies and α is the average allelic substitution effect) and this would result in 12% of the genetic variance in our study. However, these are rough approximations since both average substitution effects and allelic frequencies are estimated approximately. For instance, some old sire families had low reliabilities (because recording of fertility traits started when their daughters were already old) while younger ones might also get more records in the coming years.
With the VC approach, the location of the peak was nearly identical to the one found with the regression method although different hypotheses and information were used (complex pedigree, numerous QTL alleles). The significance of the QTL was also high (P < 0.01). The variance associated with the QTL represented 14% of the genetic variance. This value is close to the variance approximated through the average allelic substitution effect and the allelic frequencies. The part of variance associated with this QTL and the allelic substitution effect are rather large and the use of this QTL in selection would help breeders to improve fertility of dairy cows, which is dramatically decreasing.
Linkage disequilibrium and linkage analysis:
The method used for LDLA was based on LDLA methods proposed recently (Meuwissen and Goddard 2000; Kim and Georges 2002; Blott et al. 2003). Meuwissen et al. (2002) proposed to use φp between base haplotypes, which generates a very dense relationship matrix among haplotypes that is often nonpositive and requires bending strategies, reducing IBD relationships between base haplotypes. In addition, inversion of this dense matrix might be computationally demanding. Due to the clustering approach, the method by Kim and Georges (2002) uses a diagonal matrix associated with the random base haplotype effects since base haplotypes are assigned to a given cluster according to LD information and there is no relationship between different clusters. The model and the results are easier to interpret since pairs of base haplotypes can be described as equal or as different.
Kim and Georges (2002) and Blott et al. (2003) tested all possible distances from 0 to 1 to cluster the base haplotypes. In this study, only one grouping was tested at each position to reduce computational time and because multiplication of the test might lead to large amounts of results that are difficult to interpret. At the chosen 0.5 maximum distance, it was shown in a simulation study (Ytournel et al. 2007) that most of the base haplotype pairs were actually IBD while in 75% of the cases where two QTL alleles were non-IBD, the probability was <0.10. The distance was also chosen to obtain a small number of groups with many base haplotypes. Indeed, if there is little grouping, the groups are correct but little LD information (only from common ancestors from recent generations) is used whereas if there is too much grouping, the grouping is incorrect.
From 58 to 99% of the base haplotypes were grouped within 20 clusters. All the LD information was not used since some base haplotypes were not grouped. This is due to haplotypes containing missing alleles. However, already a large part of LD is used since generally 75% or more of the base haplotypes are grouped and information of SCs is used. Indeed, these haplotypes are known with more precision and are therefore more important. Fortunately, these haplotypes are also reconstructed with more precision and therefore contain only a few missing markers. Most PCs were grouped directly in the clusters because with the marker density the probability of transmission of the paternal allele was >0.95 at most locations. These probabilities were <0.95 if there was a recombination or if the sire was noninformative in that region. In the latter case, the sire was homozygous for all the markers in the region and therefore, both SCs were grouped in the same cluster. In consequence, the corresponding PC was also grouped in that cluster.
Despite the fact that the LDLA did not result in a single peak, it improved strongly the information on the QTL location with respect to the LA. Indeed, many regions were discarded according to the LDLA because QTL alleles of opposite effects were grouped in the same cluster. The LDLA discarded regions where heterozygous sires did not share common haplotypes. As a consequence, the possible location of the QTL is confined to a few small intervals.
Additional information at the peaks such as grouping of haplotypes of sires estimated to be heterozygous helps discard some peaks. This information is similar to that used in haplotype-sharing methods (Riquet et al. 1999; Nezer et al. 2003): positive or negative alleles of heterozygous sires should be surrounded by small identical haplotypes. Peaks 1, 2, 4, 5, and 8 were the positions where the four heterozygous sires (P < 0.05) shared a common haplotype of three markers for their favorable or unfavorable haplotypes. At peak 2, the only negative haplotype not grouped with the others was the haplotype of sire 41 that had the less significant t-test (P < 0.10). Peaks 1 and 2 seem to be the most interesting since at other peaks, the common haplotype is also associated with several haplotypes of opposite effect. These three-marker haplotypes cover regions of 0.1 and 2.04 cM for peaks 1 and 2, respectively. Flanking markers are at a distance of −0.05 and +2.04 cM and −0.70 and +0.15 cM, respectively.
In contrast, there was little grouping at some positions even with the HAP3 model: haplotypes with favorable (or unfavorable) effects do not share a common three-marker haplotype. This was found at peaks 3, 6, and 7. Therefore, confidence in these peaks is lower than in the first two peaks.
In addition, peaks where some heterozygous sires at the QTL appear homozygous at the markers are less likely to contain the QTL. For instance, for sire 633, with the strongest t-test, φp between his two haplotypes was 0.84 at peak 1. Similarly, when favorable and unfavorable haplotypes of heterozygous sires are grouped together, there is less probability that the QTL is located at that peak. The negative haplotype of sire 633 is identical to the positive haplotypes of sire 307 and 311 for a region spanning from marker 83 to marker 101. Therefore, φp for these haplotypes is >0.90 at peak 5. At peak 6, this is still true for the haplotypes of sires 633 and 307. Similarly, peaks 4 and 8 are less likely to contain the QTL because several haplotypes with opposite effects have high φp and are clustered together.
In summary, the first two peaks appear to be the most likely intervals for the QTL location because LRT values are high with both methods, and most heterozygous sires share a common haplotype, while at other peaks there is little grouping of haplotypes of heterozygous sires or haplotypes with opposite effects are grouped together.
These conclusions are based on a model assuming a single QTL with two alleles. This is the most parsimonious model and it appears to be in agreement with the data since studies in linkage analysis did not show evidence for multiple QTL, the same sires are heterozygous at different positions, and heterozygous sires have a common haplotype (Table 3). For several peaks, the grouping of the sire haplotypes is relatively similar, indicating that these peaks are likely to reflect the same QTL. In this context, the multiple-peak profile would then be explained by the heterogeneous LD structure within the QTL region. This might be increased by possible local inconsistencies in the map order, which was based on draft assembly or on comparative map information. Moreover, the method and the data structure might not allow the discarding of some regions even though they do not harbor the QTL. In addition, NRR90 is a complex biological trait resulting from several distinct unobserved events such as fertilization or embryo survival (Guillaume et al. 2007). More discriminating phenotypes would then enhance accuracy of the QTL fine mapping. However, such phenotypes are difficult and costly to obtain. Hence, several (possibly linked) genes might thus be expected to underlie the genetic variability of different biological related events of such a broad trait. Nevertheless the method should be relatively robust if there are several QTL alleles, each possibly embodied in a different cluster, since the model does not assume only two alleles. Alternatively, if several QTL located between positions 10 and 35 cM on our map are affecting NRR90, fine mapping of these linked QTL would require a more complex method such as multi-QTL fine-mapping methods (Meuwissen and Goddard 2004; Olsen et al. 2005). Larger designs might be advised to clearly and correctly separate several QTL in a 25-cM region.
The models HAP3 and IBD10 have some complementary properties. First, HAP3 searches for small informative regions of three markers that have a quantitative effect. Therefore, these regions must be in LD with the QTL. Noninformative regions are not detected if all QTL alleles (favorable and unfavorable) are grouped together. If all (un)favorable alleles of a QTL are associated with one haplotype, the HAP3 method should detect that region unless the opposite allele is associated with the same haplotype (noninformative haplotype with very high frequency). The method automatically performs the search for haplotype sharing for a three-marker haplotype. It is expected that within the QTL region, the QTL will be detected but also that false positive results might be found. Indeed, by chance, four or six haplotypes of heterozygous sires might have a common haplotype over three markers, especially if these three markers have low MAF. For instance, according to the allelic frequencies, the probability for a haplotype to be CAA was 0.417 and to be ATG was 0.370 at peaks 1 and 2, respectively. Therefore, the probability to observe that the six positive haplotypes of heterozygous sires are CAA was 0.005 and the probability to observe that the five negative haplotypes of the five sires with the highest t-test are ATG was 0.007. Since 150 marker intervals were tested, it is possible to find by chance regions for which haplotypes of heterozygous sires are IBS and not IBD. The IBD10 method uses IBD probabilities and uses a large marker window. Therefore, it helps to discard regions that were identical for three markers by chance from regions where haplotypes were grouped because they have high φp. However, IBD10 will be more sensitive to missing information or to genetic map inconsistencies. Finally, the LD information (haplotype sharing) must be confirmed by the LA information used by the method. Indeed, a tested region will present a high LRT value only if LD and LA information is high in that region.
Use of two distinct techniques, one based on the regression on a small number of markers and one based on φp obtained from larger haplotypes, is in agreement with recent findings (Grapes et al. 2006; Zhao et al. 2007) showing that a method based on single-marker regression can be as efficient as the IBD method (Meuwissen and Goddard 2000). In addition, according to one of these studies (Grapes et al. 2006), 4 or 6 markers must be used in the latter method, which represents the optimal compromise between power (increasing with the number of markers) and discrimination between successive tested positions (decreasing with the number markers). In our study, a larger number of markers (namely 10) are used but our method is different since φp's are used to cluster haplotypes together. Therefore, the number of effects considered in the model is smaller. Most of the haplotypes are grouped in 20 clusters, which corresponds more to the number of haplotypes obtained with 4–6 markers than with 10 markers. Finally, differences between likelihood values at successive tested positions indicate that the method discriminates sufficiently between these positions.
Conclusions:
The fertility QTL on BTA3 was highly significant (P < 0.001) and has large effects on insemination results. Indeed, the average allelic substitution effect was 0.7 genetic standard deviation and the QTL explained 14% of the genetic variance. The program implemented for this study successfully handled the large amount of data for haplotype construction, LA, and LDLA. The QTL was not fine mapped to a small single region. However, the analysis, combined with the analysis of haplotypes of heterozygous sires, refined the QTL position to a small set of narrow intervals out of which two intervals appeared to be the regions the most likely to harbor the QTL. However, additional information is required to find the causal mutations.
Acknowledgments
We thank the two anonymous reviewers for their helpful suggestions and corrections.
References
- Blott, S., J. J. Kim, S. Moisio, A. Schmidt-Kuntzel, A. Cornet et al., 2003. Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics 163 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold value for quantitative trait mapping. Genetics 138 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Zinder, M., E. Seroussi, D. M. Larkin, J. J. Loor, A. Everts-van der Wind et al., 2005. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 15 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando, R., and M. Grossman, 1989. Marked assisted selection using best linear unbiased prediction. Genet. Sel. Evol. 21 467–477. [Google Scholar]
- Gautier, M., R. R. Barcelona, S. Fritz, C. Grohs, T. Druet et al., 2006. Fine mapping and physical characterization of two linked quantitative trait loci affecting milk fat yield in dairy cattle on BTA26. Genetics 172 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier, M., T. Faraut, K. Moazami-Goudarzi, V. Navratil, M. Foglio et al., 2007. Genetic and haplotypic structure in 14 European and African cattle breeds. Genetics 177 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, A. W., P. M. Visscher and C. S. Haley, 2000. Mapping quantitative trait loci in complex pedigrees: a two-step variance component approach. Genetics 156 2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges, M., D. Nielsen, M. Mackinnon, A. Mishra, R. Okimoto et al., 1995. Mapping quantitative trait loci controlling milk production in dairy cattle by exploiting progeny testing. Genetics 139 907–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grapes, L. M., J. C. Firat, J. C. Dekkers, M. F. Rotschild and R. L. Fernando, 2006. Optimal haplotype structure for linkage disequilibrium-based fine mapping of quantitative trait loci using identity by descent. Genetics 172 1955–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignola, F. E., I. Hoeschele and B. Tier, 1996. a Mapping quantitative trait loci in outcross populations via residual maximum likelihood. I. Methodology. Genet. Sel. Evol. 28 479–490. [Google Scholar]
- Grignola, F. E., I. Hoeschele, Q. Zhang and G. Thaller, 1996. b Mapping quantitative trait loci in outcross populations via residual maximum likelihood. II. A simulation study. Genet. Sel. Evol. 28 491–504. [Google Scholar]
- Grisart, B., W. Coppieters, F. Farnir, L. Karim, C. Ford et al., 2002. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 12 222–231. [DOI] [PubMed] [Google Scholar]
- Guillaume, F., M. Gautier, S. Ben Jemaa, S. Fritz, A. Eggen et al., 2007. Refinement of two female fertility QTL using alternative phenotypes in French Holstein dairy cattle. Anim. Genet. 38 72–74. [DOI] [PubMed] [Google Scholar]
- Jensen, J., E. A. Mantysaari, P. Madsen and R. Thompson, 1996. Residual maximum likelihood estimation of (co)variance components in multivariate mixed linear models using average information. J. Ind. Soc. Agric. Stat. 49 215–236. [Google Scholar]
- Khatkar, M. S., P. C. Thomson, I. Tammen and H. W. Raadsma, 2004. Quantitative trait loci mapping in dairy cattle: review and meta-analysis. Genet. Sel. Evol. 36 163–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatkar, M. S., P. C. Thomson, I. Tammen, J. A. Cavanagh, F. W. Nicholas et al., 2006. Linkage disequilibrium on chromosome 6 in Australian Holstein-Friesian cattle. Genet. Sel. Evol. 38 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatkar, M. S., K. R. Zenger, M. Hobbs, R. J. Hawken, J. A. Cavanagh et al., 2007. A primary assembly of a bovine haplotype block map based on a 15,036-single-nucleotide polymorphism panel genotyped in Holstein–Friesian cattle. Genetics 176 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. J., and M. Georges, 2002. Evaluation of a new fine-mapping method exploiting linkage disequilibrium: a case study analysing a QTL with major effect on milk composition on bovine chromosome 14. Asian-Aust. J. Anim. Sci. 15 1250–1256. [Google Scholar]
- Knott, S. A., J. M. Elsen and C. S. Haley, 1996. Methods for multiple marker mapping of quantitative trait loci in half-sib populations. Theor. Appl. Genet. 93 71–80. [DOI] [PubMed] [Google Scholar]
- Meuwissen, T. H., and M. E. Goddard, 2000. Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics 155 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H., and M. E. Goddard, 2001. Prediction of identity by descent probabilities from marker-haplotypes. Genet. Sel. Evol. 33 605–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H., and M. E. Goddard, 2004. Mapping multiple QTL using linkage disequilibrium and linkage analysis information and multitrait data. Genet. Sel. Evol. 36 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H., A. Karlsen, S. Lien, I. Olsaker and M. E. Goddard, 2002. Fine mapping of a quantitative trait locus for twinning rate using combined linkage and linkage disequilibrium mapping. Genetics 161 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misztal, I., T. Tsuruta, T. Strabel, B. Auvray, T. Druet, 2002. BLUPF90 and related programs (BGF90). 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, Communication no. 28–07.
- Nezer, C., C. Collette, L. Moreau, B. Brouwers, J. J. Kim et al., 2003. Haplotype sharing refines the location of an imprinted quantitative trait locus with major effect on muscle mass to a 250-kb chromosome segment containing the porcine IGF2 gene. Genetics 165 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, H. G., S. Lien, M. Gautier, H. Nilsen, A. Roseth et al., 2005. Mapping of a milk production quantitative trait locus to a 420-kb region on bovine chromosome 6. Genetics 169 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong-Wong, R., A. W. George, J. A. Woolliams and C. S. Haley, 2001. A simple and rapid method for calculating identity-by-descent matrices using multiple markers. Genet. Sel. Evol. 33 453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, D., and L. Beckmann, 2002. Minimum-recombinant haplotyping in pedigrees. Am. J. Hum. Genet. 70 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquet, J., W. Coppieters, N. Cambisano, J. J. Arranz, P. Berzi et al., 1999. Fine-mapping of quantitative trait loci by identity by descent in outbred populations: application to milk production in dairy cattle. Proc. Natl. Acad. Sci. USA 96 9252–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRaden, P. M., and G. R. Wiggans, 1991. Derivation, calculation, and use of national animal model information. J. Dairy Sci. 74 2737–2746. [DOI] [PubMed] [Google Scholar]
- Windig, J. J., and T. H. E. Meuwissen, 2004. Rapid haplotype reconstruction in pedigrees with dense marker maps. J. Anim. Breed. Genet. 121 26–39. [Google Scholar]
- Ytournel, F., H. Gilbert and D. Boichard, 2007. Concordance between IBD probabilities and linkage disequilibrium. 58th Annual Meeting of the European Association of Animal Production, Dublin, Ireland.
- Zhao, H. H., R. L. Fernando and J. C. Dekkers, 2007. Power and precision of alternate methods for linkage disequilibrium mapping of quantitative trait loci. Genetics 175 1975–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]