

Abstract
It has been established in a number of studies that the alkaline-denatured state of pepsin (the IP state) is composed of a compact C-terminal lobe and a largely unstructured N-terminal lobe. In the present study, we have investigated the residual structure in the IP state in more detail, using limited proteolysis to isolate and characterize a tightly folded core region from this partially denatured pepsin. The isolated core region corresponds to the 141 C-terminal residues of the pepsin molecule, which in the fully native state forms one of the two lobes of the structure. A comparative study using NMR and CD spectroscopy has revealed, however, that the N-terminal lobe contributes a substantial amount of additional residual structure to the IP state of pepsin. CD spectra indicate in addition that significant nonnative α-helical structure is present in the C-terminal lobe of the structure when the N-terminal lobe of pepsin is either unfolded or removed by proteolysis. This study demonstrates that the structure of pepsin in the IP state is significantly more complex than that of a fully folded C-terminal lobe connected to an unstructured N-terminal lobe.
Keywords: Pepsin, zymogen, denaturation, partially folded state, limited proteolysis
Extensive structural studies have been carried out recently on denatured and other nonnative states of proteins (Dill and Shortle 1991; Kamatari et al. 1996, 1999; Smith et al. 1996; Plaxco and Gross 1997; Prusiner et al. 1998; Dyson and Wright 2001; Khurana et al. 2001; Zurdo et al. 2001). These investigators were largely motivated by a desire to probe the mechanisms of protein folding and aggregation or to understand the physiological roles of nonnative structures An important group of nonnative states of proteins includes those of several zymogen-derived enzymes, which unfold irreversibly and become trapped in partially denatured states (Fruton 1960; Ikemura et al. 1987; Zhu et al. 1989; Baker et al. 1993; Eder and Fersht 1995). Recently, the zymogen-derived serine protease αLP was found to require its prosequence for folding; without the prosequence it is trapped in a partially denatured state separated from the native state by a very high kinetic barrier (Sohl et al. 1998). It has also been found that mutations introduced into αLP can reduce the free energy of the transition state of the protein and allow it to fold rapidly in the absence of the prosequence (Derman and Agard 2000). A partially structured denatured state has also been identified and characterized for low-molecular-weight urokinase-type plasminogen activator; this enzyme under mildly denaturing conditions possesses native-like structure only in the N-terminal lobe of its structure (Nowak et al. 1994). There is, however, relatively little detailed information about the structures of the denatured states of these and other zymogen-derived proteins. Defining the structural characteristics of such proteins should give insights not only into the functional properties of this very important family of enzymes involved in proteolysis, but also into the general factors defining protein structure, folding, and activity (Eder and Fersht 1995; Cunningham et al. 1999).
The gastric aspartic proteinase pepsin (porcine pepsin, molecular weight = 34,623) is a zymogen-derived protein that has been the subject of extensive study (Chen et al. 1992; Richter et al. 1998; Fruton 2002). X-ray diffraction analysis shows that the substrate-binding cleft is located between two homologous portions of the structure, the N-terminal lobe (residues 1–172) and the C-terminal lobe (residues 173–327) (Fig. 1B ▶ and Cooper et al. 1990; Sielecki et al. 1990). Pepsin undergoes a conformational transition from the native (at acidic pH) to the denatured (at alkaline pH) state in a narrow pH range (between 6 and 7). This alkaline denaturation process appears to be almost completely irreversible (Fruton 1960; Lin et al. 1993), although the unfolding of the zymogen pepsinogen is reversible under carefully controlled conditions (Ahmad and McPhie 1978a). Recently, refolding of an immobilized form of the denatured pepsin was achieved without the prosequence (Kurimoto et al. 2001), but its refolding mechanism is still unsolved. Structural knowledge of the alkaline-denatured state of pepsin (designated “the IP state” for convenience) is essential not only for understanding the folding behavior of pepsin, but also for elucidating the mechanisms that govern the observed strong interaction of the IP state of pepsin with species such as molecular chaperones (Aoki et al. 1997) or amyloid fibrils (Konno 2001).
Figure 1.
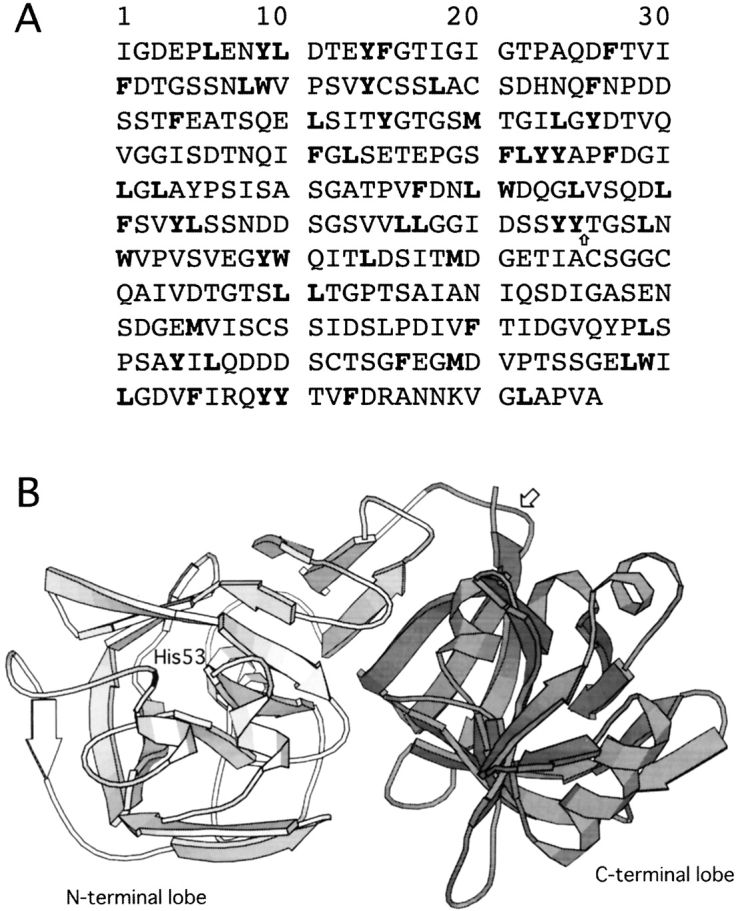
(A) Amino acid sequence of pepsin. All potential cleavage sites digested by α-chymotrypsin are shown in bold. The cleavage site to produce the C fragment is shown by an arrow. (B) Main-chain trace of native pepsin (structure 4pep) showing the N-terminal lobe of the protein (residues 1–172) in light gray, the C-terminal lobe (residues 173–326) in dark gray, and His53 in black. The cleavage site to produce the C fragment is shown by an arrow. This figure was generated with MOLSCRIPT (Kraulis 1991).
Privalov et al. (1981) isolated the C-terminal lobe using limited proteolytic digestion of native pepsin and showed that the C-terminal lobe has higher stability than does the N-terminal lobe in the native state. Lin et al. (1993) demonstrated regeneration of the enzymatic activity of alkaline-inactivated pepsin by addition of the recombinant N-terminal lobe but not by addition of the C-terminal lobe. Both lines of evidence indicate that the C-terminal lobe has higher stability than does the N-terminal lobe and that the IP state has a folded C-terminal lobe and a largely unstructured N-terminal lobe. A theoretical calculation by Lin et al. (1993) and a recent mutational experiment by Tanaka and Yada (2001) have resulted in a similar conclusion. However, in a previous study, we demonstrated that a histidine residue located in the N-terminal lobe of the pepsin molecule is located near the folded region of the IP molecule. This observation indicates that there is a significant contribution by residues in the N-terminal lobe to the residual structure of the IP state and that the structure of pepsin in the IP state could be more complex than that of a natively folded C-terminal lobe connected to an unstructured N-terminal lobe. In order to obtain more detailed structural information on the IP state of pepsin, we have carried out experiments designed to isolate the folded core of the molecule by purifying the material obtained from limited proteolysis of the alkaline-denatured protein. We have characterized the structure of this folded core spectroscopically, and we compare it with that of the full-length IP state of pepsin.
Results
Proteolysis and isolation of the C fragment
Because a polypeptide chain buried within a folded protein core is generally resistant to attack by a proteinase, limited proteolysis has often been a successful method for identifying tightly folded regions of proteins (Fontana et al. 1997; Tanaka et al. 2000). We employed this method using α-chymotrypsin to isolate the folded part of pepsin in the alkaline-denatured (IP) state. Our purpose is to study the structure of pepsin in the denatured state rather than in the folded form, in contrast to the proteolytic experiment described by Privalov et al. (1981), in which diazoacetyl-inhibited pepsin was digested in its natively folded state.
Complete digestion of pepsin by α-chymotrypsin, which mainly cleaves peptide bonds adjacent to aromatic amino acids and large hydrophobic side chain residues (Tyr, Trp, Phe, Met, and Leu, not before Pro), is expected to give 63 fragments, each with a molecular mass of <2.3 kD. All potential cleavage sites are shown in Figure 1A ▶. Digestion of the IP state of pepsin for 1–2 h under our experimental conditions by α-chymotrypsin, however, resulted in the transient accumulation of a fragment with a molecular mass of ~14 kD (Fig 2 ▶). This result indicates that this fragment represents a relatively stable region of the pepsin structure in the IP state. Mass spectroscopic and N-terminal sequencing analyses of the fragment revealed that its mass is 15,895 ± 5 Da and its N-terminal amino acid sequence is TGSLNWVPVS; this information shows that the fragment corresponds to residues 176–327 of the protein, representing the C-terminal lobe of the overall structure (and henceforth is designated the “C fragment”). Note that the sequence “TGSLNWVPVS” is unique in the pepsin sequence and that the expected molecular mass of a fragment corresponding to residues 176–327 of the protein is 15,901.67 Da. Residue 175 is Tyr, so the peptide bond between residues 175 and 176 is susceptible to digestion by α-chymotrypsin. This cleavage site is shown by an arrow on the pepsin amino acid sequence (Fig. 1A ▶) and structure (Fig. 1B ▶). It is also worth noting that there are 23 potential sites for chymotrypsin digestion within this C fragment, implying protection conferred by a stably folded structure.
Figure 2.

Limited proteolysis of pepsin with α-chymotrypsin monitored by SDS-PAGE; see the text for digestion conditions. Lanes 1 and 9 show undigested pepsin and the purified C fragment, respectively. The digestion periods were 5 min (lane 2), 10 min (lane 3), 34 min (lane 4), 1 h (lane 5), 2 h (lane 6), 4 h (lane 7), and 8 h (lane 8).
Comparative spectroscopic analysis
Figure 3 ▶ shows a 1H NMR spectrum of the C fragment at pH 8.0 in comparison with spectra of the full-length protein in the IP state at pH 8.0, the native state at pH 5.6, and the urea-denatured state at pH 8.1. The upfield-shifted signals between −0.8 and 0.6 ppm in the C fragment spectrum are clear evidence for the presence of a tightly folded structure typical of that of a native protein (Fig. 3B ▶). Peaks representing a highly denatured polypeptide conformation (for example, those at 0.9 ppm attributable to the signals of methyl groups) are not evident in the spectrum of the fragment, indicating that it is essentially completely folded (Fig. 3B ▶). Molten globule-like compact denatured states that are loosely packed show NMR signals significantly broader (Baum et al. 1989; Kamatari et al. 1996, 1999) than those observed for the C fragment or the IP state. This result demonstrates that α-chymotrypsin digestion has effectively removed the flexible part of the N-terminal lobe of the IP state that gives rise to intense signals characteristic of a denatured polypeptide chain. Most of the resonances of the C fragment in the upfield region of the spectrum appear to be in similar positions to those found in the IP state of pepsin (Fig. 3B,C ▶). In particular, a single well-resolved resonance at −0.6 ppm is evident in both spectra, as are clusters of signals at −0.2 ppm and +0.2 ppm. Some differences between the two spectra can, however, also be observed (Fig. 3B,C ▶). As well as differences in the detailed structure of the two clusters of resonances discussed earlier, a well-resolved resonance at −0.5ppm is present in the spectrum of the C-fragment but not the IP state. Although specific assignments cannot be made at this stage, these differences between the NMR spectra of the IP state and C fragment are clear evidence of contributions of the N-terminal lobe, which is lacking in the C fragment, to the residual structure specific to the IP state.
Figure 3.

750 MHz 1H NMR spectra of the native state of pepsin at pH 5.6 in H2O (A), the C fragment at pH 8.0 (B), the IP state at pH 8.0 (C), and the urea-denatured state in 4 M urea at pH 8.1 (D). The signals labeled with an asterisk and an “x” are those of the reference molecule DSS and an impurity, respectively.
In addition, the similarity between the tertiary structure of the IP state and the C fragment is further strongly supported by the appearance of the near-UV CD spectra of the two species (Fig. 4A ▶). The far-UV CD spectrum of the C fragment has a shape typical of a folded protein with a large amount of secondary structure (Fig. 4B ▶). Unlike the spectrum of the IP state, there is little evidence for substantial unstructured regions that are characterized by strong negative ellipticity at ~204–205 nm. The spectrum of the C fragment is not, however, identical to that of pepsin in its native state. The spectrum of the latter has a single minimum at ~210–215 nm, and the shape of the spectrum is typical of that for a very highly β-sheet-rich protein (Fig. 4B ▶, thick broken line). The spectrum of the C fragment, on the other hand, has two rather broad negative peaks centered at ~207 and ~220 nm. The appearance of these peaks, particularly the latter, is suggestive of significant α-helical content (Fig. 4B ▶, thick solid line).
Figure 4.
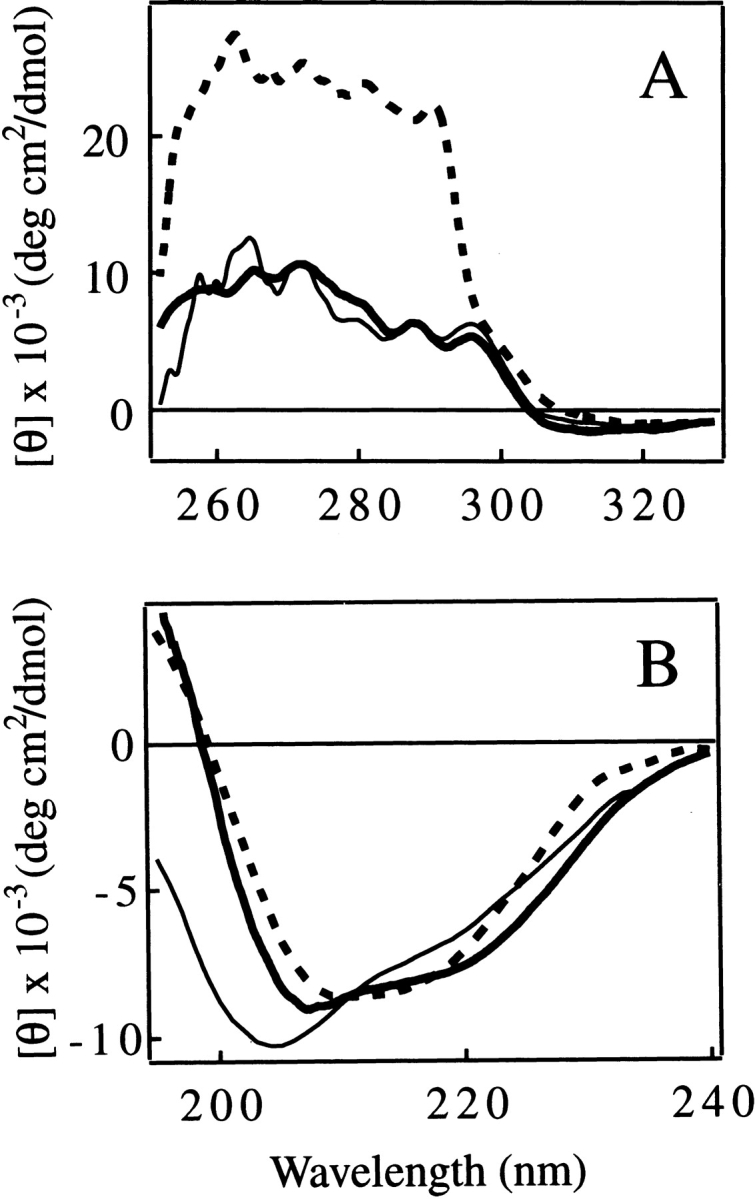
Near-UV (A) and far-UV (B) CD spectra of the C fragment (thick solid line), the IP state (pH 8.0; thin solid line), and the native state (pH 6.5; thick broken line) of pepsin. The unit of ellipticity is per protein concentration for A and per residue for B.
Numerical estimation of the secondary-structure content from the CD spectra using three widely used programs (CONTINLL, SELCOM3, and CDSSTR [Sreerama and Woody 2000]; see also the legend to Table 1) was performed to further investigate this general conclusion. Theoretical far-UV CD spectra of pepsin reconstructed from secondary structure estimates give good fits to the experimental spectra (Fig. 5 ▶), indicating that the prediction programs work well in the present case. The average ratio of the α-helical to the β-sheet content (Rα/β) from this analysis for the native state of pepsin (0.33 ± 0.06) is very close to that found in the crystal structure of pepsin (Rα/β = 0.36; calculated using the DSSP program [Kasch and Sander 1983] and five pepsin coordinates [Cooper et al. 1990]). The Rα/β value calculated for pepsinogen using its spectrum at pH 8.0 (Rα/β = 0.57 ± 0.05) also shows good agreement with the value from the crystal structure (Rα/β = 0.51; calculated using three pepsinogen coordinates). These calculations also support the reliability of the numerical analysis. Analysis of the CD spectra demonstrates that the Rα/β value for the C fragment is higher than that of the native state by a factor of 2.3 (Table 1). Because the secondary structure content calculated from the crystallographic structure of pepsin is almost identical for each lobe of the molecule (Rα/β = 0.36 in each case), the higher helical conformation observed in the C fragment is suggestive of the presence of nonnative helical structure in the C-terminal lobe of the molecule after the removal of the N-terminal lobe. A slightly larger Rα/β value than that characteristic of the native state of pepsin was also found for the IP state of pepsin (Rα/β = 0.44 ± 0.05; Table 1). The secondary structural analysis therefore indicates that either partial unfolding, or the removal of the N-terminal lobe by proteolysis, results in the conversion of part of the native conformation of the C-terminal lobe of pepsin to one containing a degree of nonnative α-helical structure.
Table 1.
Secondary structure content of pepsin and the C fragment estimated from the CD spectra
| α-Helix | β-Sheet | Turn | Random | Rα/βa | |
| Native pepsin, (pH 5.6, 25°C) | 11.3 ± 2.6 | 33.9 ± 2.7 | 23.2 ± 1.1 | 32.3 ± 2.6 | 0.33 ± 0.06 |
| IP state (pH 8.0, 25°C) | 9.6 ± 1.9 | 21.9 ± 2.0 | 18.6 ± 2.1 | 50.6 ± 3.4 | 0.44 ± 0.05 |
| C fragment (pH 8.0, 25°C) | 19.0 ± 0.8 | 24.8 ± 1.2 | 20.5 ± 1.2 | 35.5 ± 3.0 | 0.77 ± 0.05 |
| Pepsinogen (pH 8.0, 25°C) | 16.5 ± 0.5 | 29.4 ± 2.7 | 21.5 ± 0.4 | 32.7 ± 2.1 | 0.57 ± 0.05 |
Each value is the average of six independent calculations, based on the CONTINLL, SELCOM3, and CDSSTR programs and two reference data sets supplied with the CDpro package (Sreerama and Woody 2000). Two independent runs for each of the three programs were performed using the two different database sets. Thus, six independent predictions were averaged to obtain the values in this table. The error values are those obtained in this averaging process. The spectral data used in this analysis ranged in wavelength from 187 to 240 nm.
aRα/β = (α-helical content)/(β-sheet content).
Figure 5.

Reconstituted far-UV CD spectra of the native state (A), the C fragment (B), and the IP state (C) of pepsin. Solid lines are experimental spectra. Open circles represent spectra reconstituted from the secondary structure predictions generated using the programs CONTINLL, SELCOM3, and CDSSTR. Two independent runs for each of the three programs were performed using two different database sets. Thus, six independent predictions were averaged to obtain the reconstituted spectra in this figure.
Thermal denaturation of the C fragment and the IP state
The thermal stability of the C fragment was measured using CD spectroscopy and compared with that of the IP state of pepsin under the same solution conditions (Fig. 6 ▶). The heat denaturation curves of the C fragment monitored by ellipticity at 220 nm and 272 nm are identical to those of the IP state at temperatures above 25°C (Fig. 6A,B ▶; see figure legend for the data normalization method). These data indicate that the unfolding of the structure of the C-terminal lobe of pepsin under these conditions is highly cooperative, involving the concomitant loss of both secondary and tertiary structure. The midpoint of the denaturation is 52.4 ± 1.3°C. The heat denaturation curves monitored by the anisotropy (r; Perrin 1926) of tryptophan fluorescence also indicate that denaturation takes place over a similar temperature range for both protein species (Fig. 6C ▶). The cooperative increase in 1/r at 50°C–60°C probably originates from an increase in the local mobility of the three tryptophan residues in the C-terminal lobe of pepsin when this part of the structure unfolds at 52.0 ± 0.5°C. Smaller but significant changes in 1/r values for the IP state of pepsin relative to those found for the C fragment at temperatures below 40°C can be attributed to contributions to the signal from the two tryptophan residues in the relatively disordered N-terminal lobe of the IP state.
Figure 6.
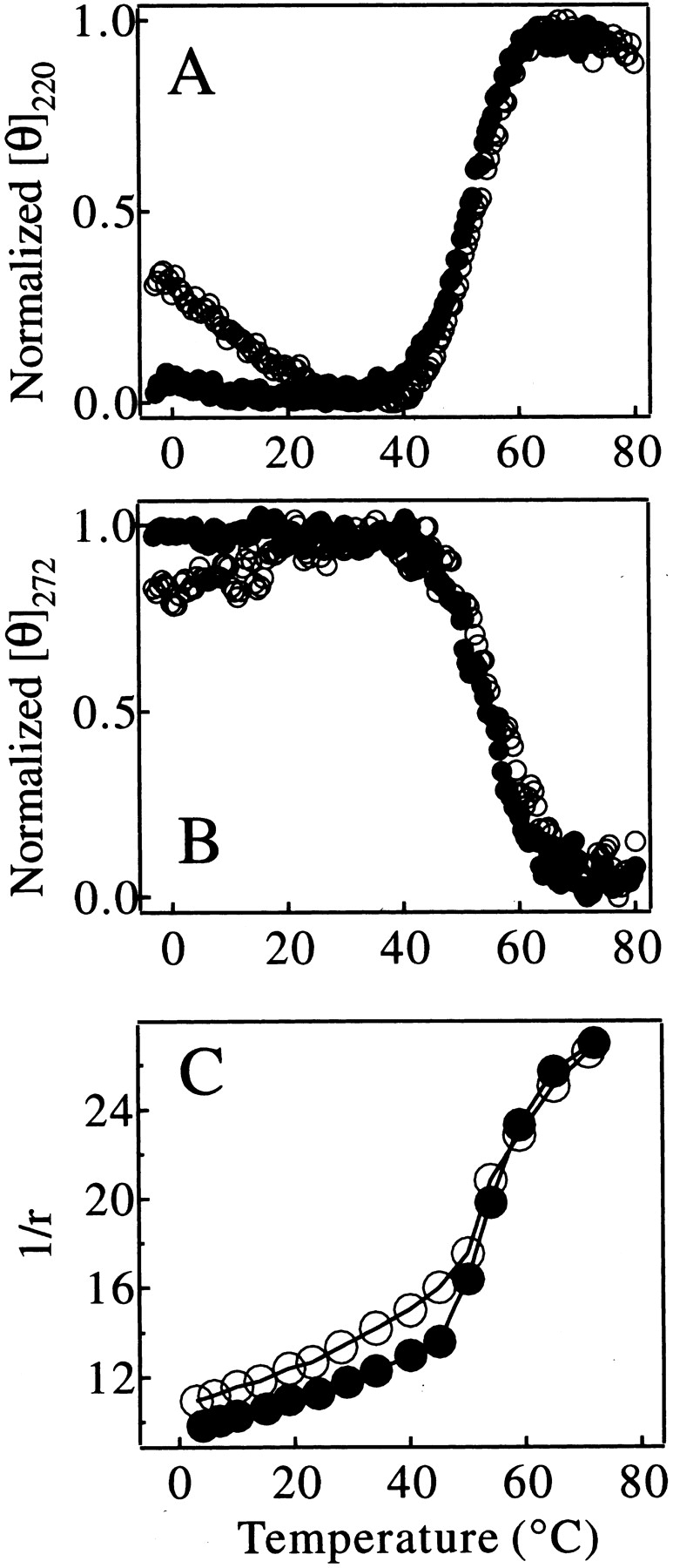
Thermal denaturation curves of the C fragment (filled circles) and the IP state (open circles) of pepsin monitored by [θ]220nm (A), [θ]272nm (B), and 1/r of tryptophan fluorescence (C). The ellipticity in A and B was normalized to the maximum ellipticity change in the temperature range shown in the figures.
The results presented in Figure 6 ▶ therefore indicate that the stability of the C fragment is closely similar to that of the structured region of the IP state. This finding is consistent with the conclusion that the folded structure of the IP state corresponds primarily to that of the C-terminal lobe of intact pepsin (Fig. 1B ▶). At temperatures below ca. 25°C, however, the temperature-dependent curve of [θ]220 for the C fragment is very different from that of the IP state of pepsin. The ellipticity value for the IP state decreases substantially in magnitude on lowering the temperature below 20°C, whereas the value for the C fragment is unchanged as the temperature is reduced (Fig. 6A ▶). Comparison of the far-UV CD spectra at 0°C with those at 25°C also shows a clear decrease in the magnitude of the ellipticity at 220 nm and loss of secondary structure with decreasing temperature, but only for the full-length IP state (Fig. 7 ▶). This type of behavior for IP at lower temperature can also be seen to a lesser extent in the curves for [θ]272 (Fig. 6B ▶). These observations indicate that the N-terminal lobe of pepsin in the IP state has some degree of nonrandom structure that experiences cold denaturation, and may also indicate the importance of hydrophobic interactions in this region. These results indicate that the N-terminal lobe, which is lacking in the C fragment, nonetheless contributes significantly to the residual structure of the IP state.
Figure 7.
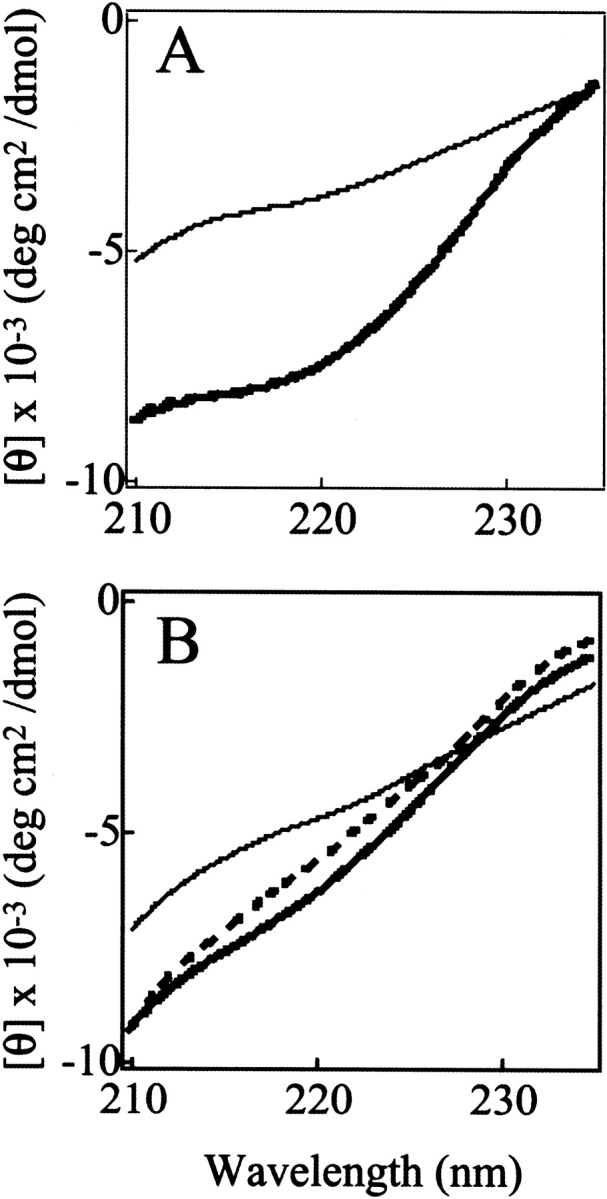
Far-UV CD spectra of the IP state (A) and the C fragment (B) of pepsin at pH 8.0. The temperature of the protein solutions is 0°C (thick broken lines), 25°C (thick solid lines), or 70°C (thin solid lines).
Discussion
The folded structure of the IP state corresponds primarily to the C-terminal lobe
The application of limited proteolysis to investigate the alkaline-denatured state of pepsin resulted in the isolation of a tightly folded fragment of this IP state, a result that strongly supports our previous biophysical studies that indicated the IP state has a structure consisting of both fully folded and highly denatured regions (Konno et al. 2000). The proteolytic experiment reveals definitively that the tightly folded fragment corresponds to the C-terminal lobe of pepsin. This finding, together with the similarity of the thermal denaturation curve above 25°C for the IP state and the C fragment monitored by far- and near-UV CD and fluorescence spectroscopy (Fig. 6 ▶), indicates that the folded cooperative core of the IP state corresponds primarily to that of the C-terminal lobe of intact pepsin. These results confirm the previous suggestion by Lin et al. (1993) and also agree with the conclusion that the C-terminal lobe of pepsin is more stable against heat or proteolytic digestion than is the N-terminal lobe in the native state (Privalov et al. 1981).
Contributions of the N-terminal part to the residual structures of the IP state and nonnative structures in the IP state
Although the folded structure of the IP state corresponds primarily to the C-terminal lobe as shown earlier, evidence from the experiments described in this and our previous paper indicate that the structure in the IP state does not correspond simply to the structure of the C-terminal lobe of the native protein in all its features. The differences in the ellipticity change at low temperatures (<25°C) between the C fragment and the IP state of pepsin indicate a degree of nonrandom conformation in the N-terminal lobe of the IP state (Figs. 6A ▶, 7 ▶). Moreover, the NMR spectra show significant differences between the tertiary structure of the C fragment and the residual structure present in the IP state (Fig. 3 ▶). There is also evidence from our previous work that additional residual structure exists within the N-terminal lobe, including residues in the vicinity of His53 (Konno et al. 2000).
There is also some evidence that the residual structure in the IP state and in the C fragment contains structure not present in the native state. Analysis of the far-UV CD spectra indicates that the IP state and the C fragment have a larger α-helical and a smaller β-sheet content than that observed in the native state of pepsin (Fig. 4B ▶ and Table 1). Moreover, our previous NMR study demonstrating involvement of the residues in the vicinity of His53 in the residual structure of the IP state is also further evidence for nonnative structure because His53 is far from the C-terminal lobe (Konno et al. 2000). One possibility is that some of the β-sheet structure localized at the interface between the N- and C-terminal lobes of folded pepsin (Cooper et al. 1990; Sielecki et al. 1990) is disrupted by the removal or unfolding of the N-terminal lobe and replaced by α-helical structure.
Our study therefore indicates a relatively complex structure of the IP state of pepsin. It contains the tightly folded C-terminal lobe with a substantial amount of nonnative secondary and tertiary structures, and additional contributions to the residual structure of the IP state from the N-terminal lobe.
Implications for the folding mechanisms of pepsin
It is interesting to speculate that the observed nonnative characteristics of the structure of pepsin in the IP state could play a role in the folding of pepsin or its precursor pepsinogen. As formation of the interfacial β-sheet structure between the two structural lobes is likely to be a crucial step for the proper folding of the protein, it is possible that such structure can only be achieved efficiently when the N-terminal lobe folds in the presence of the prosequence. Alternatively, the nonnative structural elements in the partially folded IP state of pepsin at pH 8.0 could stabilize this structure relative to the native state; in other words, “misfolding” in this state could inhibit the proper refolding of the protein when returned to conditions that stabilize the native state. Whether or not such speculation is correct, the present study indicates that further investigation of the partly folded states is likely to be of substantial importance in understanding the nature of their folding of zymogen-derived proteins and the manner in which their activity is controlled and regulated.
Materials and methods
Materials
Porcine pepsin and TLCK-treated α-chymotrypsin of the highest grade of purity were purchased from Sigma. Pepsin was purified using an S-200 gel chromatography column (Pharmacia) equilibrated with the buffer required for the subsequent experiments. α-Chymotrypsin was used without further purification. Other chemicals were of reagent grade and purchased from Nacalai Tesque. The concentration of pepsin was determined using the extinction coefficient ɛ278 = 5.10 × 104 cm−1 mol−1 (Ahmad and McPhie 1978b).
Isolation and identification of the C fragment
Solutions of pepsin and α-chymotrypsin were prepared by dissolving the lyophilized samples in 20 mM sodium phosphate buffer (pH 8.2); the protein concentrations were 10 mg/mL (pepsin) and 2 mg/mL (α-chymotrypsin). The proteolytic reaction was initiated by mixing 5 μL of the α-chymotrypsin solution with 1 mL of the pepsin solution at 25°C (enzyme:substrate ratio of 1:730), and monitored by the SDS-PAGE method (Laemmli 1970). A fragment designated the “C fragment” was chromatographically purified from pepsin solutions after digestion for between 1 and 2 h. The sample was passed through a BioCAD HPLC system installed with an anion-exchange column using POROS HQ/M resin from PerSeptive Biosystems in 50 mM Tris (pH 8.0) and a salt gradient from 0 to 1 M NaCl. The chromatogram was monitored by absorption at 280 nm, and the main protein-containing peak was collected. The purity of the fragment was then checked by SDS-PAGE analysis, which indicated the content of impurities was <5% (Fig. 2 ▶, lane 9). N-terminal protein sequencing was carried out by automatic Edman degradation using an Applied Biosystems 494A Procise protein sequencer (Applied Biosystems) in the Protein Sequencing Service of the Oxford Centre for Molecular Sciences (MRC, Immunochemistry Unit, University of Oxford). The molecular mass of the fragment was determined by nanoflow electrospray mass spectrometry using a Q-ToF mass spectrometer (Micromass) operated in negative ion mode. The extinction coefficient of the C fragment was determined by the Edelhoch’s method (Edelhoch 1967; Gill and von Hipple 1989), which gave ɛ280 = 2.43 × 104 cm−1 mol−1.
Spectroscopic measurements
1H NMR measurements were performed at 25°C on a home-built 750 MHz NMR spectrometer belonging to the Oxford Centre for Molecular Sciences. The concentration of pepsin for the NMR measurements was 10 mg/mL, and the protein was dissolved in 20 mM sodium phosphate or glycine buffer prepared with 95% H2O/5% D2O DSS was added as an internal chemical shift reference. The sample pH was adjusted using NaOH or HCl. All the NMR experiments were performed using the water-gate pulse sequence (Piotto et al. 1992) for water suppression.
CD spectra were measured with a Jasco J-720 WI spectropolarimeter (JASCO), using quartz cells with pathlengths of 0.1 and 2 mm for the far- and near-UV CD measurements, respectively. The protein concentration was maintained at 30 and 60 μM for the far- and near-UV CD measurements, respectively. The temperature of the solutions was controlled by a JASCO thermal controller. Fluorescence anisotropy experiments were performed using an F2500 fluorimeter (Hitachi) equipped for the measurement of anisotropies using a quartz cell with a light path of 10 mm. The temperature of the solutions was controlled by means of a thermostatically controlled water bath. The excitation wavelength was 295 nm and the bandwidths for excitation and emission light were both 5 nm. The protein concentration in each case was maintained at 6 μM. The anisotropy (r) at each temperature was taken as the average of the values in the wavelength range of 340–380 nm. Solutions for the CD and fluorescence measurements contained 20 mM MOPS (pH 8.0). The sample pH was adjusted using NaOH or HCl.
Secondary-structure analysis of far-UV CD spectra
Secondary-structure estimation from the far-UV CD spectra was performed using three popular programs: CONTINLL, SELCOM3, and CDSSTR. These are included in the CDPro package (Sreerama and Woody 2000) available at http://lamar.colostate.edu/~sreeram/CDPro/. The spectral data used in this analysis ranged in wavelength from 187 to 240 nm at 1-nm intervals. Two different reference data sets supplied with the package were used for the analysis. Each of the three programs was run using these reference sets, and six independent estimates were obtained for each experimental spectrum. The values in Table 1 are the averages and the standard errors of the six.
Acknowledgments
We thank Dr. Helena Hernandez and Prof. Carol V. Robinson for providing the mass spectrometric data and for general advice. We also thank Dr. K. Hun Mok for his advice on α-chymotrypsin digestion experiments. We are also indebted to Prof. Masao Miki and Dr. Masashi Unno for their helpful suggestions in the final stage of this work. We also thank Dr. Cait E. MacPhee for constructive comments on this manuscript. Y.O.K. was supported by an HFSP fellowship. This work is in part a contribution from the Oxford Centre for Molecular Sciences, which is supported by the UK Engineering and Physical Sciences Research Council, the Biotechnology and Biological Sciences Research Council, and the Medical Research Council. The research of C.M.D. is also supported in part by the Wellcome Trust and by an International Research Scholars award from the Howard Hughes Medical Research Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
UV, ultraviolet
NMR, nuclear magnetic resonance
ppm, parts per million
DSS, 2,2-dimethyl-2-silapentane-5-sulfonic acid
IP, the alkaline-denatured state of pepsin at pH 8.0 and 25°C
C fragment, the pepsin fragment designated by α-chymotrypsin and chromatographically purified
αLP, α-lytic protease
TLCK, N-α-p-tosyl-L-lysine chloromethyl ketone hydrochloride
SDS, sodium dodecylsulfate
PAGE, polyacrylamide gel electrophoresis
HPLC, high performance liquid chromatography
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0219903.
References
- Ahmad, F. and McPhie, P. 1978a. Thermodynamics of the denaturation of pepsinogen by urea. Biochemistry 17 241–246. [DOI] [PubMed] [Google Scholar]
- ———. 1978b. The denaturation of covalently inhibited swine pepsin. Int. J. Pept. Protein Res. 12 155–163. [DOI] [PubMed] [Google Scholar]
- Aoki, K., Taguchi, H., Shindo, Y., Yoshida, M., Ogasahara, K., Yutani, K., and Tanaka, N. 1997. Calorimetric observation of a GroEL-protein binding reaction with little contribution of hydrophobic interaction. J. Biol. Chem. 272 32158–32162. [DOI] [PubMed] [Google Scholar]
- Baker, D., Shiau, A.K., and Agard, D.A. 1993. The role of pro regions in protein folding. Curr. Opin. Cell Biol. 5 966–970. [DOI] [PubMed] [Google Scholar]
- Baum, J., Dobson, C.M., Evans, P.A. and Hanley, C. 1989. Characterization of a partly folded protein by NMR methods: Studies on the molten globule state of guinea pig α-lactalbumin. Biochemistry 28 7–13. [DOI] [PubMed] [Google Scholar]
- Chen, L., Erickson, J.W., Rydel, T.J., Park, C.H., Neidhart, D., Luly, J., and Abad-Zapatero, C. 1992. Structure of a pepsin/renin inhibitor complex reveals a novel crystal packing induced by minor chemical alterations in the inhibitor. Acta. Crystallogr. B 48 476–488. [DOI] [PubMed] [Google Scholar]
- Cooper, J.B., Khan, G., Taylor, G., Tickle, I.J., and Blundell, T.L. 1990. X-ray analyses of aspartic proteinases. II. Three-dimensional structure of the hexagonal crystal form of porcine pepsin at 2.3 Å resolution. J. Mol. Biol. 214 199–222. [DOI] [PubMed] [Google Scholar]
- Cunningham, E.L., Jaswal, S.S., Sohl, J.L., and Agard, D.A. 1999. Kinetic stability as a mechanism for protease longevity. Proc. Natl. Acad. Sci. 96 11008–11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman, A.I. and Agard, D.A. 2000. Two energetically disparate folding pathways of α-lytic protease share a single transition state. Nat. Struct. Biol. 7 394–397. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. and Shortle, D. 1991. Denatured states of proteins. Annu. Rev. Biochem. 60 795–825. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2001. Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol. 339 258–270. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Eder, J. and Fersht, A.R. 1995. Pro-sequence-assisted protein folding. Mol. Microbiol. 16 609–614. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Zambonin, M., Polverino de Laureto, P., De Filippis, V., Cleminti, A., and Scramella, E. 1997. Probing the conformational state of apomyoglobin by limited proteolysis. J. Mol. Biol. 266 223–230. [DOI] [PubMed] [Google Scholar]
- Fruton, J.S. 1960. Pepsin. In The enzymes III. (ed. P.D. Boyer), pp. 119–164. Academic Press, New York.
- ———. 2002. A history of pepsin and related enzymes. Q. Rev. Biol. 77 127–147. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hipple, P. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Ikemura, H., Takagi, H., and Inoue, M. 1987. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J. Biol. Chem. 262 7859–7864. [PubMed] [Google Scholar]
- Kamatari, Y.O., Konno, T., Kataoka, M., and Akasaka, K. 1996. The methanol-induced globular and expanded denatured states of cytochrome c: A study by CD fluorescence, NMR and small-angle X-ray scattering. J. Mol. Biol. 259 512–523. [DOI] [PubMed] [Google Scholar]
- Kamatari, Y.O., Ohji, S., Konno, T., Seki, Y., Soda, K., Kataoka, M., and Akasaka, K. 1999. The compact and expanded denatured conformations of apomyoglobin in the methanol-water solvent. Protein Sci. 8 873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasch, W. and Sander, C. 1983. Dictionary of protein secondary structure. Pattern recognition of hydrogen bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Khurana, R., Gillespie, J.R., Talapatra, A., Minert, L.J., Ionescu-Zanetti, C., Millett, I., and Fink, A.L. 2001. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry 40 3525–3535. [DOI] [PubMed] [Google Scholar]
- Konno, T. 2001. Amyloid-induced aggregation and precipitation of soluble proteins: An electrostatic contribution of the Alzheimer’s β(25–35) amyloid fibril. Biochemistry 40 2148–2154. [DOI] [PubMed] [Google Scholar]
- Konno, T., Kamatari, Y.O., Tanaka, N., Kamikubo, H., Dobson, C.M., and Nagayama, K. 2000. A partially unfolded structure of the alkaline-denatured state of pepsin and its implication for stability of the zymogen-derived protein. Biochemistry 39 4182–4190. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kurimoto, E., Harada, T., Akiyama, A., Sakai, T., and Kato, K. 2001. In vitro refolding of porcine pepsin immobilized on agarose beads. J. Biochem. (Tokyo). 130 295–297. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lin, X., Loy, J.A., Sussman, F., and Tang, J. 1993. Conformational instability of the N- and C-terminal lobes of porcine pepsin in neutral and alkaline solutions. Protein Sci. 2 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak, U.K., Cooper, A., Saunders, D., Smith, R.A., and Dobson, C.M. 1994. Unfolding studies of the protease domain of urokinase-type plasminogen activator: The existence of partly folded states and stable subdomains. Biochemistry 33 2951–2960. [DOI] [PubMed] [Google Scholar]
- Perrin, F. 1926. J. Phys. Radium. 7 390–401. [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W. and Gross, M. 1997. The importance of being unfolded. Nature 386 657–659. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L., Mateo, P.L., Khechinashvili, N.N., Stepanov, V.M., and Revina, L.P. 1981. Comparative thermodynamic study of pepsinogen and pepsin structure. J. Mol. Biol. 152 445–464. [DOI] [PubMed] [Google Scholar]
- Prusiner, S.B., Scott, M.R., DeArmond, S.J., and Cohen, F.E. 1998. Prion protein biology. Cell 93 337–348. [DOI] [PubMed] [Google Scholar]
- Richter, C., Tanaka, T. and Yada, R.Y. 1998. Mechanism of activation of the gastric aspartic proteinases: Pepsinogen, progastricsin and prochymosin. Biochem. J. 335 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sielecki, A.R., Fedorov, A.A., Boodhoo, A., Andreeva, N.S., and James, N.G. 1990. Molecular and crystal structures of monoclinic porcine pepsin refined at 1.8 Å resolution. J. Mol. Biol. 214 143–170. [DOI] [PubMed] [Google Scholar]
- Smith, L.J., Fiebig, K.M., Schwalbe, H., and Dobson, C.M. 1996. The concept of a random coil. Residual structure in peptides and denatured proteins. Fold Des. 1 R95–R106. [DOI] [PubMed] [Google Scholar]
- Sohl, J.L., Jaswal, S.S., and Agard, D.A. 1998. Unfolded conformations of α-lytic protease are more stable than its native state. Nature 395 817–819. [DOI] [PubMed] [Google Scholar]
- Sreerama, N. and Woody, R.W. 2000. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem 287 252–260. [DOI] [PubMed] [Google Scholar]
- Tanaka, T. and Yada, R.Y. 2001. N-terminal portion acts as an initiator of the inactivation of pepsin at neutral pH. Protein Eng. 14 669–674. [DOI] [PubMed] [Google Scholar]
- Tanaka, N., Ikeda, C., Kanaori, K., Hiraga, K., Konno, T., and Kunigi, S. 2000. Pressure effect on the conformational fluctuation of apomyoglobin in the native state. Biochemistry 39 12063–12068. [DOI] [PubMed] [Google Scholar]
- Zhu, X.L., Ohta, Y., Jordan, F., and Inouye, M. 1989. Pro-sequence of subtilisin can guide the refolding of denatured subtilisin in an intermolecular process. Nature 339 483–484. [DOI] [PubMed] [Google Scholar]
- Zurdo, J., Guijarro, J.I., Jimenez, J.L., Saibil, H.R., and Dobson, C.M. 2001. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J. Mol. Biol. 311 325–340. [DOI] [PubMed] [Google Scholar]