

Abstract
We report the effects of peptide binding on the 15N relaxation rates and chemical shifts of the C-SH3 of Sem-5. 15N spin-lattice relaxation time (T1), spin-spin relaxation time (T2), and {1H}-15N NOE were obtained from heteronuclear 2D NMR experiments. These parameters were then analyzed using the Lipari-Szabo model free formalism to obtain parameters that describe the internal motions of the protein. High-order parameters (S2 > 0.8) are found in elements of regular secondary structure, whereas some residues in the loop regions show relatively low-order parameters, notably the RT loop. Peptide binding is characterized by a significant decrease in the 15N relaxation in the RT loop. Concomitant with the change in dynamics is a cooperative change in chemical shifts. The agreement between the binding constants calculated from chemical shift differences and that obtained from ITC indicates that the binding of Sem-5 C-SH3 to its putative peptide ligand is coupled to a cooperative conformational change in which a portion of the binding site undergoes a significant reduction in conformational heterogeneity.
Keywords: Sem-5, SH3, 15N relaxation, NMR, backbone dynamics, order parameter, polyproline peptide, ligand binding
Molecular recognition remains one of the most challenging problems in structural biology, and a complete understanding of this phenomenon can only come by elucidating the complex interplay between structure, dynamics, and energetics (Weber 1992). Recently we solved the solution structure of the unliganded C-SH3 of Sem-5 (Ferreon et al. 2003), which when compared with the previously solved liganded structure (Lim et al. 1994b) revealed several significant differences in the RT loop, a region that makes direct contact with the ligand. In addition, as with other SH3 domains (Wang et al. 2001), 15N relaxation and hydrogen deuterium exchange reveal that the RT loop possesses significant conformational heterogeneity, indicating that consideration of the static structural differences alone may not be sufficient to provide a detailed accounting of the energetic contributions to molecular recognition.
In order to determine the quantitative role of dynamics in the recognition process, it is essential to determine not only the average structure of the bound and free forms of the protein, but also the magnitude and extent of the conformational fluctuations for each state. In ensemble terms, a quantitative characterization of the binding process requires knowledge of the structure and energetics of each of the microscopic states within the macroscopic bound and unbound ensembles.
Motions on a variety of timescales from femtoseconds to minutes have been shown to be important in enzyme catalysis, binding specificity, and regulation (Collins et al. 1995; Nicholson et al. 1995; Wagner 1995; Stivers et al. 1996; Gagné et al. 1998; Kay et al. 1998; Feher and Cavanagh 1999; Stock 1999), and NMR spectroscopy has emerged as a powerful tool to characterize these motions. Of particular importance is the use of 15N relaxation rate measurements to obtain information about picosecond to nanosecond dynamics (Kay et al. 1989; Clore et al. 1990; Palmer III et al. 1991). On application of the appropriate motional models (Lipari and Szabo 1982a,b; Mandel et al. 1995), relaxation measurements provide access to residue-specific order parameters, which can in turn be related to the entropy changes in the system (Akke et al. 1993; Yang and Kay 1996).
It has been shown that ligand-induced changes in backbone dynamics can be used to obtain unique insights into the role of dynamics in binding affinity and specificity (Nicholson et al. 1995; Kay et al. 1996, 1998; Spyracopoulos et al. 1998; Zidek et al. 1999; Wang et al. 2001). Several studies have shown an increase in backbone rigidity on ligand binding (Akke et al. 1993; Hodsdon and Cistola 1997; Olejniczak et al. 1997; Alexandrescu et al. 1998; Kristensen et al. 2000), whereas others have shown an increase in disorder on binding (Farrow et al. 1994; Stivers et al. 1996; Zidek et al. 1999). Here we investigate the changes in residue-specific 15N relaxation rates in Sem-5 C-SH3 domain on ligand binding. We correlate these changes with the observed cooperativity of the binding process. We show that consideration of the location, magnitude, and cooperativity of the dynamics changes provides unique insights into the thermodynamic origins of the dynamic contributions.
The model system used in these studies is the C-terminal SH3 of the protein Sem-5. SH3 domains are adaptor proteins that recognize proline-rich sequences and are found in many signal transduction and cytoskeletal proteins (Pawson 1995; Dalgarno et al. 1997; Buday 1999). In particular, the C-SH3 of Sem-5 from Caenorhabditis elegans and its mammalian and Drosophila homologs, Grb2 and Drk, are involved in linking protein tyrosine kinase activity to Ras. Sem-5 activates Ras signaling by binding to the PPPXPPR motifs found in the guanine nucleotide exchange factor Sos (Clark et al. 1992; Lowenstein et al. 1992; Egan et al. 1993; Olivier et al. 1993).
Results
Identification of the residues affected by ligand binding
Previously, we have determined the chemical shifts for the unliganded Sem-5 C-SH3 domain (Ferreon et al. 2003). Backbone amide 15N and 1H were assigned using various homonuclear and heteronuclear NMR techniques. In the past, changes in chemical shifts observed on ligand binding have been used to identify residues that are involved in the binding interface for different SH3 domains (Booker et al. 1993; Wittekind et al. 1994; Horita et al. 1998). For Sem-5 C-SH3, chemical shifts in the 1H-15N HSQC spectra were obtained in various saturating and subsaturating concentrations (15 concentrations) of the putative peptide ligand. Figure 1 ▶ shows the 1H-15N HSQC spectra of both unbound (green) and bound Sem-5 C-SH3 domain (red). Although many resonances are not affected, residues near the binding interface (Yu et al. 1992; Booker et al. 1993; Lim et al. 1994b) show significant deviation (>0.1 ppm for 1H and >0.5 ppm for the 15N dimension; Fig. 2A,B ▶). These residues are found primarily in the conserved 310 helix and the n-Src and RT loops. The line widths of the amide resonances do not change significantly throughout the titration, indicating that the complex is in fast exchange on the chemical shift NMR timescale (Lian and Roberts 1993). Such conditions enable calculation of the dissociation constant (Kd) assuming two states, unbound and bound (Lian and Roberts 1993; Horita et al. 1998). A nonlinear regression analysis (see Materials and Methods) of chemical shift changes as a function of peptide concentration yields Kd values for all residues showing significant chemical shifts (Fig. 2C,D ▶). The residues considered are F163, N166, E169, S170, L173, F175, W191, W192, F203, S205, and N206. The Kd values range from 286–430 μM with the average and standard deviation of 377 ± 38 μM. The associated error from the regression analysis is 5%–13%. The similarity in calculated equilibrium constants obtained from different chemical shift perturbations indicates that each of the perturbed residues is monitoring the same equilibrium and that the binding can be approximated as a two-state process. It should be noted and will be discussed below that several residues in the RT loop show significant chemical shift differences despite the fact that they are not directly involved in binding.
Figure 1.

Assigned 1H-15N HSQC spectra of Sem-5 C-SH3 without peptide (green) and with peptide (red), taken using a 750-MHz Varian spectrometer at 25°C.
Figure 2.
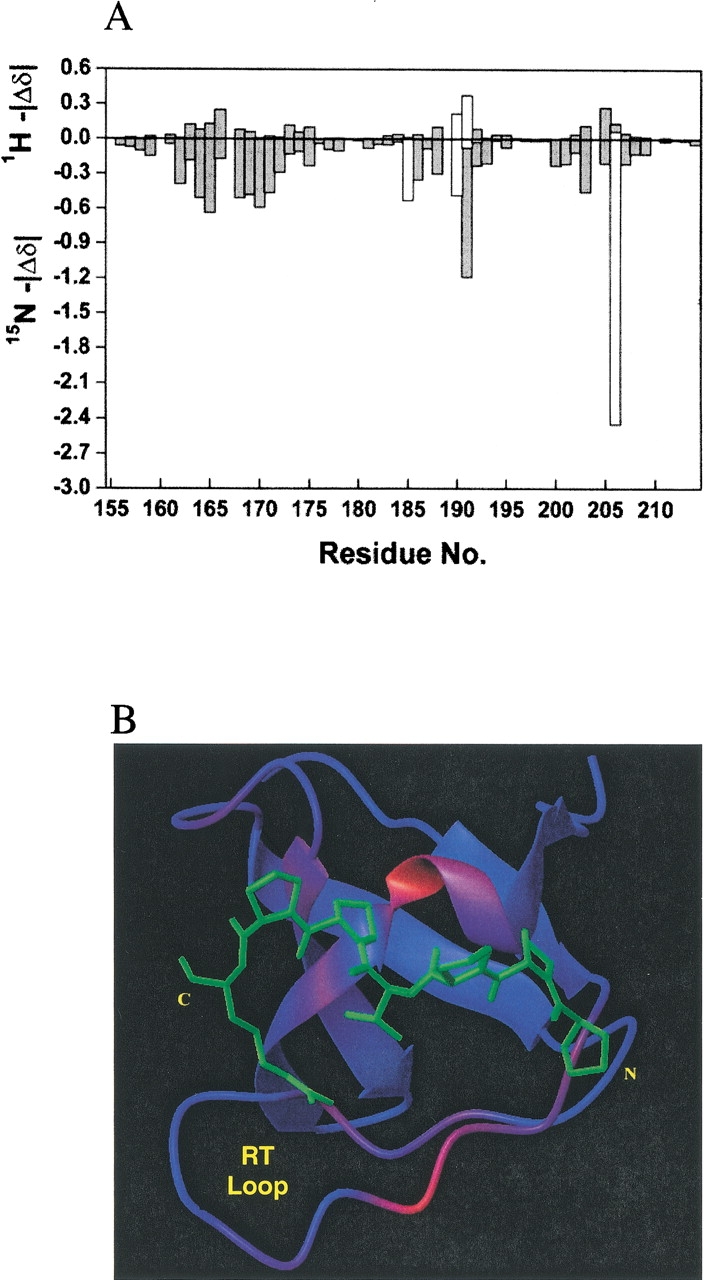
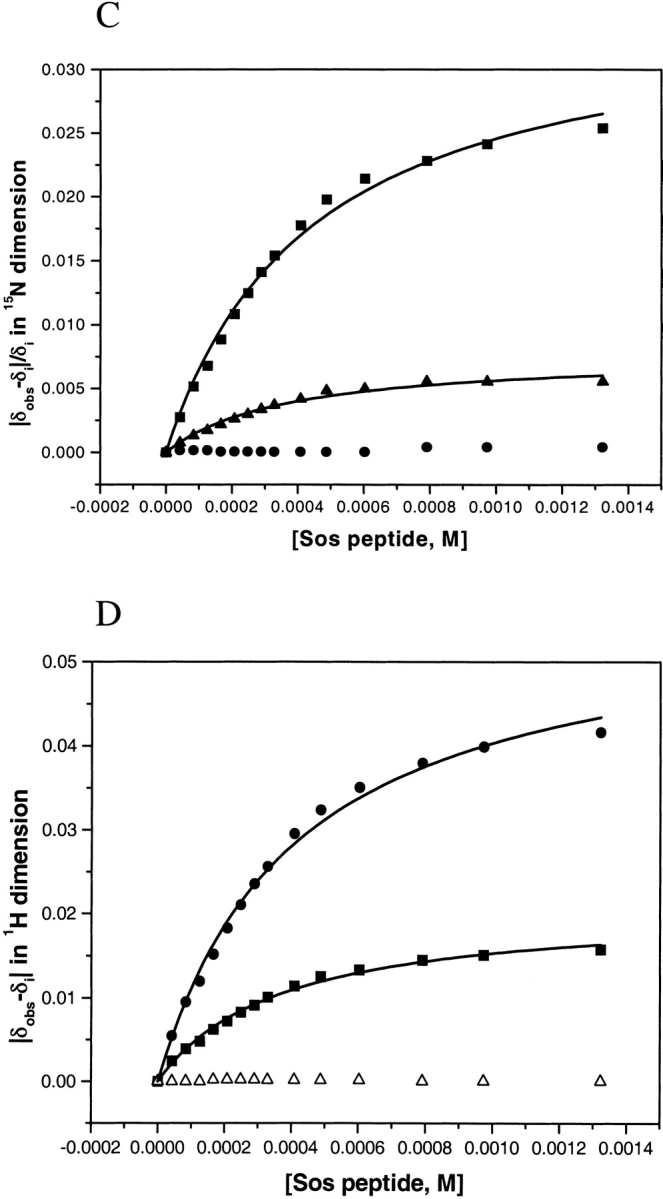
(A) The magnitude of differences in 1H (top graph) and 15N (bottom) chemical shifts for each residue between the free and complexed state. (B) Color mapping of the 1H chemical shift difference in the NMR minimized average structure with the peptide overlayed in the minus orientation and the same position as seen in the X-ray structure. (C and D) Binding curves of chemical shift changes for sample of residues (Ser170, 362 ± 46 μM and Asn206H2Ne, 427 ± 53 μM in 15N dimension; Ser205, 397 ± 36 μM and Leu173, 355 ± 34 μM in 1H dimension) as a function of peptide concentration. Lines are drawn as the best-fitted lines as calculated (see text). T156 and Q160 are residues not involved in binding and do not show chemical shift changes during the titration of the ligand.
Energetics of binding
The binding affinity of Sem-5 C-SH3 domain to polyproline peptide was also assessed using ITC under similar conditions as those used for the chemical shift experiments (Fig. 3 ▶). Analysis of the fitted parameters reveals the binding between Sem-5 C-SH3 and the Sos peptide occurs in a 1:1 stoichiometry with a favorable enthalpy of interaction (ΔH°~−4.9 kcal mole−1), a slightly unfavorable entropy of interaction (ΔS°~−0.33 cal deg−1 mole−1), and a dissociation constant of 337 μM at pH 4.8. This value is in agreement with that obtained from NMR analysis of the chemical shifts (Fig. 2 ▶) and is approximately 10-fold less than the binding constant obtained at pH 7.3 (Lim et al. 1994a) and 7.5 (data not shown). The agreement (within experimental error) between the calorimetrically obtained binding affinity and that obtained from the chemical shifts data indicates that both techniques are monitoring the same cooperative two-state process.
Figure 3.

Calorimetric titration of Sem-5 C-SH3 domain with the Sos peptide (Ac-VPPPVPPRRR-NH2) in 50 mM NaOAc and 100 mM NaCl, and 10 mM CaCl2 (pH 4.8) at 25°C.
Relaxation data analysis
15N relaxation data were obtained at 25°C and 400 MHz field strength, both for the free and complex C-SH3 domain. All of 15N−T1 and 15N−T2 were adequately fit to single exponential two-parameter decays. Figure 4 ▶ shows the representative best and worst 15N−T1 and T2 fits for the free C-SH3 domain. Best fits have standard errors <1% whereas worst fits have standard errors <2%. Forty-seven and 52 resonances (of 58 with 4 prolines) were determined for the unliganded and liganded forms, respectively. Data were unavailable for the remaining residues because resonances were either highly overlapped or too weak for reliable analysis. The average value of R1 is 2.84 ± 0.29 s−1, R2 is 6.30 ± 0.86 s−1, and NOE is 0.53 ± 0.39 for the free C-SH3, and for the complex C-SH3 the average value of R1 is 2.85 ± 0.27 s−1, R2 is 6.50 ± 0.84 s−1, and NOE is 0.65 ± 0.30. Figure 5 ▶ shows the relaxation rates R1 and R2, and NOE profiles for both the free and the bound C-SH3 domain. In both cases, the R1 profiles show similar distributions, with significantly lower than average R1 values in the loop regions (RT loop, n-Src, and distal loop), as well as residues in the termini. Slight increases in R1 are observed for most residues on binding. Although several residues show a slight decrease in R1, these changes are generally within error. The R2 and NOE distribution also show similar profiles, with most residues in the loop regions exhibiting lower values. Significant increases occur for almost all residues in R2 and NOE on ligand binding.
Figure 4.
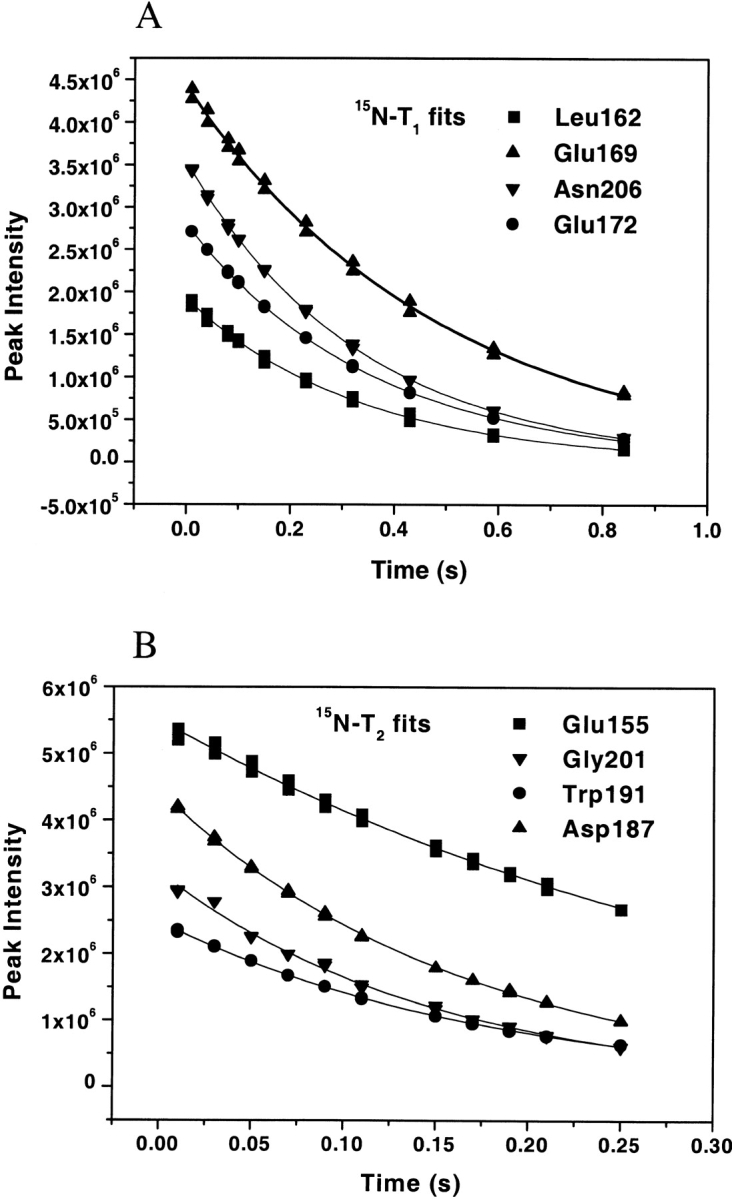
Representatives of the worst (L162, E169, E155, G201) and the best (N206, E172, W191, D187) fits of 15N−T1 (A) and T2 (B). Fitting errors are <2% and <1% for the worst and best fits, respectively. Duplicate points are taken for each time delay and shown in the same graph.
Figure 5.

15N-relaxation data for Sem-5 C-SH3 domain both in the unliganded (open circles) and liganded state (filled circles). Elements of secondary structure are the same for both states. β-Sheets are represented as arrows, 310 helix by a cylinder, loops by straight black lines, and turns by bent lines.
Isotropic diffusion tensor
The degree of anisotropy is important in choosing the appropriate spectral density functions, as a slight degree of anisotropy could introduce potential errors in describing the appropriate motional model for each amide (Tjandra et al. 1995). Using the average NMR solution structure of the unbound state (Ferreon et al. 2003), the principal axes of the inertia tensor were found to be 1.00:0.91:0.67 using the program pdbinertia (Dr. Arthur Palmer, Columbia University). For the bound state, the crystal structure was used and the relative moments are 1:00:0.94:0.75. The r2r1_diffusion v.1.11 program (Dr. Arthur Palmer, Columbia University) was used to analyze the degree of rotational diffusion anisotropy.
Table 1 shows the summary of the rotational diffusion anisotropy. The diffusion tensor was also fitted to two models, isotropic and axially symmetric. Under the axial symmetric model, D|/D⊥ (D| and D⊥ are the principal components of the diffusion tensor along and perpendicular to, respectively, the long axis of rotation) is 0.89 and 0.96 (oblate) for free and bound states, respectively. A statistical test shows that Sem-5 C-SH3 domain can be approximated by an isotropic diffusion tensor (Tjandra et al. 1995). Initial estimates of the overall rotational correlation time (τm) were obtained by two methods (see Materials and Methods). Both gave the same result of ~5 nsec. Initial τm obtained from tmest program was chosen to be 5.091 ns for the unbound and 5.198 ns for the bound SH3 domain based on the procedure by Kay et al. (1989), wherein the initial estimate was obtained from the trimmed R1/R2 ratios. Residues that show conformational exchange contributions to R2, and which have internal motions faster than 100 psec, are not included in obtaining the initial estimate of τm.
Table 1.
Rotational diffusion anisotropy of Sem-5 C-SH3 domain
| C-SH3 (free)a | C-SH3: Sos complex (bound)b | |||
| Parameter | Isotropic | Axially symmetric | Isotropic | Axially symmetric |
| θc | 1.62 | −0.526 | ||
| φc | 6.53 | 2.10 | ||
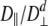 |
1 | 0.891 | 1 | 0.957 |
| χ2e | 102.70 | 64.61 | 426.50 | 398.05 |
| χ2 (red)f | 3.67 | 2.58 | 10.66 | 10.76 |
| Fg | 4.91 | 0.88 | ||
a NMR minimized average structure.
b Crystal structure (PDB entry 1 SEM).
c Orientation angles of the diffusion tensor.
d Defined as 2 Dz/(Dx + Dy).
e χ-square value (χ2 = Σ[R1e/R2e − R1c/R2c]2/σ2R1/R2).
f Reduced χ-square value (χ2 [red] = χ2/[N-m]).
gF-test to test the significance of addition of extra variables to the fit.
A recent study has shown that removing the extreme values can bias the results toward isotropic tumbling (Pawley et al. 2001). Using the filtering procedure presented in that work resulted in the elimination of similar residues from the analysis to those eliminated as described earlier, confirming that our data can be approximated by an isotropic tumbling analysis. Final optimization yielded τm of 5.019 and 5.091 nsec for the free and bound states, respectively. It is noteworthy that the difference in τm observed here (~0.1 nsec) is less than the value determined for other SH3 domains (0.4 nsec; Pawley et al. 2001). Although we have no unequivocal explanation for this difference, it is likely that the spherical approximation of the model is a contributing factor. Nonetheless, iterative optimization using different values for τm did not qualitatively change the results.
Model-free analysis
Using the Lipari-Szabo model-free formalism, backbone amides were fitted to either one of the five models using the modeling strategy described in Materials and Methods. Table 2 shows the summary of the number of residues that fit to each of the five spectral density models for both the free and bound states. For both forms of the protein, the majority of residues were satisfactorily fit with the simplest model, either with S2 (model 1) or S2 and τe (model 2). Figure 6A ▶ illustrates the generalized order parameters for both ligation states of the protein. The average S2 for the free C-SH3 domain is 0.79 ± 0.13 and 0.79 ± 0.12 for the bound state. These values are only slightly lower than the average found for the 20 proteins from the Indiana Dynamics database (0.84 ± 0.11; Ye et al. 1999).
Table 2.
Summary of spectral density models used to fit R1, R2, and NOE data
| Parameters optimized | Uncomplexed C-SH3 | Complexed C-SH3 |
| Model 1: S2, τm | 12 | 21 |
| Model 2: S2, τm, τe | 20 | 6 |
| Model 3: S2, τm, Rex | 4 | 13 |
| Model 4: S2, τm, τe, Rex | 6 | 4 |
Model 5: 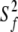 , ,  , τm, τs , τm, τs
|
3 | 4 |
| Not fit | 2 | 4 |
| Proline residues | 4 | 4 |
| Overlap | 6 | 0 |
| Weak peak | 1 | 2 |
Figure 6.

(A) Order parameters (S2) profile both for the free (open circles) and complexed (filled circles) C-SH3 domain with Sos peptide. (B) Difference between the order parameters of the free and complexed C-SH3 domain.
The S2 profiles are similar for both ligation states, with higher-than-average S2 (>0.8) found in β strands I-V and the 310 helix and lower-than-average S2 values found in the terminal residues and residues in the loop regions (i.e., RT, n-Src, and distal loop). Of particular note is the fact that the S2 values in the RT loop (N166–S171) are lowest in both the bound and free forms. Residue E169 was found to be the most mobile in both states (S2 of 0.52 and 0.59 for free and bound state, respectively). Interestingly, the backbone amide of this residue forms H-bond interactions with the carboxylate group of E172 in the bound state. Despite the high mobility observed in many RT-loop residues, the conserved diverging type II β-turn of all SH3 domains shows S2 of >0.83 in both states, a result that is consistent with other SH3 domains.
Most terminal residues were fitted with a distinct two-timescale motion, which includes slower motions (τs<2 nsec), with τe < τs < τm. Clore et al. (1990) described this two-timescale model as involving fast motions or local oscillations such as diffusion in a cone and slow motions originating from transition jumps between two distinct states. Hydrogen-deuterium exchange data (Ferreon et al. 2003) also show that amides in the secondary structural elements have slower exchange, whereas residues in the loops have faster exchange rates (i.e, beyond the detection limit). In the case of Sem-5 C-SH3 domain, there is a strong correlation between the high-order parameters and the location of secondary structure elements.
Although the S2 profile is similar in both ligation states, significant differences in magnitude exist (Fig. 6B ▶). For example, residues in the RT loop show significant increases in rigidity on complex formation, the trend persisting over the entire length of the loop. Residues W191 and R199 of the distal loop, and residues comprising the 310 helix, also showed an increase in order parameters on binding. All residues exhibiting significant increases in order parameters (aside from R199 in the distal loop) are either in direct contact with the peptide or are proximal to the binding interface in the RT loop. This observation, coupled with the quality of the fits of the various motional models to the data, indicates that the observed differences are real and reflect decreases in disorder on binding. It is also important to note that the observed increase in S2 is consistent with changes in the relaxation data.
Additionally, as shown in Table 3, most of the significant changes in S2 arise for residues that are fitted using the same model in the bound and free states. For example, the relaxation data for E169 in the RT loop, which showed the largest change (ΔS2 = 0.06), was fit to model 4, both in the unliganded and the liganded states. Moreover, data analyses showed that, regardless of models used, the S2 values for the unliganded state are consistently lower than those in the liganded state for most residues in the RT loop.
Table 3.
Differential effects on the Sem-5 C-SH3 residues coupled by ligand binding
| Res. No. | ASA changea | Chemical shiftb | Structural changec | S2d | Model freee | Model bounde | ΔS2i | |
| Leu 162 | + | |||||||
| Phe 163 | + | + | + | 2 | 1 | + | ||
| Asp 164 | R | + | ++ | + | n.a.f | 1 | n.a.f | |
| Phe 165 | T | + | ++ | + | + | n.a.f | 2 | n.a.f |
| Asn 166 | + | + | + | ++ | 4 | n.a.f | n.a.f | |
| Pro 167 | N.A.g | + | N.A.g | N.A.g | N.A.g | N.A.g | ||
| Gln 168 | + | ++ | + | +++ | n.a.f | 4 | n.a.f | |
| Glu 169 | L | + | ++ | + | +++ | 4 | 4 | + |
| Ser 170 | O | ++ | + | ++ | 4 | 3 | + | |
| Gly 171 | O | ++ | + | 4 | 3 | + | ||
| Glu 172 | P | + | + | + | 3 | 3 | ||
| Leu 173 | + | |||||||
| Asn 190 | + | ++ (s.ch) | ||||||
| Trp 191 | + | ++ | + | 2 | 2 | + | ||
| Pro 204 | + | N.A. | ||||||
| Asn 206 | + | ++ (s.c.h) | ||||||
| Tyr 207 | + | + |
a Accessible surface area (ASA) buried in the binding interface, (+) >0Å2.
b (+) <0.5 ppm; (++) >0.5 ppm in 15N dimension.
c (+) >1.0 Å difference in Cα (bound vs. unbound).
d (+) <0.82; (++) <0.75; (+++) <0.65 in S2.
e Models used in fitting (see Materials and Methods), either for the free or the bound states.
f Overlap either in the free or the complexed state.
g Proline residue.
h Effect monitored in the side-chain amide.
i (+) 70.025.
In fitting the data, 10 residues required the conformational exchange term Rex in the fitting model for the unbound state and 17 residues for the bound state. These residues are found in the RT loop, diverging type II turn (175–177), βIII strand, and include residues F203 and N206. Most of these residues are involved in binding. All the values of the Rex term, except for S170 (1.097 ± 0.07 and 1.358 ± 0.079 in the bound and unbound states, respectively) are <1 Hz. (Fig. 7B ▶). Three residues (N-terminal residues, 155–157) required a two-timescale motion model in the unbound state as well as four residues (N-terminal 155 and 157 and C-terminal 211 and 212) in the bound state. As shown in other cases (Tjandra et al. 1996), most of the terminal residues experience a two-timescale motion. The timescale of the slow motions is ~2 nsec.
Figure 7.

(A) Internal correlation times (τe) profile both for the free and complexed C-SH3 domain with Sos peptide. (B) Residues, which exhibit conformational exchange (Rex), both for the free and complexed C-SH3 domain.
Tryptophan side-chain dynamics
Using the same procedure described for the backbone amides, dynamics of the side-chain amide (HNɛ) of tryptophan was also determined. There are two tryptophans in the Sem-5 C-SH3, which are consecutive in sequence. One is buried (W192) and the other (W191) is solvent-exposed and directly intercalates with the polyproline peptide in the bound state (Lim et al. 1994b). W192 HNɛ cannot be fit to any model, whereas W191 HNɛ was satisfactorily fit to model 1 with S2 equal to 0.77 ± 0.002 and 0.85 ± 0.006 (in free and bound states, respectively).
Comparison with other SH3 domains
The general pattern of low-order parameters in the loop regions (RT, n-Src, and distal loop) has been observed in other SH3 domains (Hansson et al. 1998; Horita et al. 2000; Wang et al. 2001); however, the relative magnitude of disorder in each loop is different between different SH3 domains. In the Hck SH3 domain (Horita et al. 2000), the n-Src loop shows a higher degree of disorder compared with the other loops. In the Btk SH3 domain (Hansson et al. 1998), the RT and n-Src loop show similar disorder, which are both higher than in the distal loop. In the Sem-5 SH3 domain, the RT loop shows more disorder than the n-Src and the distal loop. Although we have no explanation for the relative differences, we note that the Hck n-Src loop, which has more disorder than the n-Src loop in Sem-5 SH3 domain, contains two additional residues (Horita et al. 2000).
The results presented here can be compared with a recent study focused on the binding of the peptide (RALPPLPRY) to the c-Src SH3 domain (Wang et al. 2001). As is the case with Sem-5, most of the regions of c-Src, which experience an increase in S2 on binding, are located in loop regions. Interestingly, residues E36 and Q49 of c-Src, corresponding to residues D188 and R199 in Sem-5, showed a decrease in motion even though these residues are >9 Å from the ligand interface. Although ΔS2 for D188 of Sem-5 could not be determined due to overlap, R199 showed a substantial decrease in S2, similar to the case in c-Src. Conversely, residues V32 and T35 in c-Src (as well as the corresponding residues I184 and D187 in Sem-5) showed an increase in motion on binding. Despite the numerous similarities, differences between the two SH3 domains are also observed. For instance, in c-Src residues T17 and D20 show an increase in motion on binding, whereas the corresponding residues in Sem-5, E169 and E172, show a decrease on ligand binding.
Discussion
Dual character of peptide binding site on Sem-5
Accessible surface area calculations on the crystal structure of the Sem-5:Sos peptide complex reveal that the following residues are in direct contact with the peptide: F163, D164, F165, N166, Q168, E169, E172, N190, W191, P204, N206, Y207 (Table 3). Residues that also show a significant change in chemical shift in the 1H-15N HSQC spectra on ligand titration (difference of 0.1 and 0.5 ppm in 1H and 15N dimension, respectively) include those stated earlier, as well as residues S170, G171, L173, F175, W192, S205. In addition, of the total number of residues that show chemical shift differences, only a subset show structural (Ferreon et al. 2003) and dynamic changes on binding of ligand (Table 3). A significant observation can be made from these results. Namely, the binding site of Sem-5 can be viewed as having a dual character (Fig. 8 ▶).
Figure 8.

Space-filling model representation of the crystal structure determined (Lim et al. 1994b) for Sem-5 C-SH3 (gray) with and without the Sos peptide (yellow). Residues belonging to the binding interface but showing no dynamic changes in the picosecond timescale on binding are shown in green, and residues in red are those showing dynamic changes. Residues shown in blue (S170 and G171) are those that do not interact with the peptide in the complex, but which nonetheless experience significant changes in chemical shift and dynamics on binding.
Residues E172, N190, P204, N206, and Y207, although burying significant surface area on binding, do not undergo a change in either the structure or the dynamics. In essence, the binding in this part of the molecule can be considered a rigid body association, the structural determinants of the binding being well approximated by the high-resolution structure. Residues F163, D164, F165, N166, Q168, E169, and W191, on the other hand, show significant changes in chemical shifts and dynamics. These differences occur in the regions of Sem-5 C-SH3 that show the highest structural differences between the free and complexed protein (Ferreon et al. 2003). These results indicate that binding is coupled to a conformational change in the RT loop. The fact that residues 170 and 171, which do not directly interact with ligand, also show both chemical shift and dynamic differences between bound and unbound states, further supports this conclusion.
Two key features of the observed conformational change in the RT loop are important. First, as determined from chemical shift perturbation and ITC experiments, the binding reaction is two state, and thus the conformational change is cooperative. Second, the dynamics measurements clearly show that the binding reaction is coupled to the decrease in the flexibility in the RT loop, as discussed earlier. In ensemble terms, the results indicate that the unbound state consists of a conformational manifold, wherein the RT loop has considerably greater conformational variation than the rest of the Sem-5 C-SH3 molecule. On binding of the ligand, that conformational manifold is significantly reduced in a cooperative fashion, with the average structure of the bound and unbound states differing considerably. For this region of the molecule, it is likely that the thermodynamics of binding cannot be deduced simply by considering the differences in structure between the canonical bound and unbound conformation. Instead, elucidation of the thermodynamics requires insight into the thermodynamic consequences of redistributing (i.e., reducing the size of) the ensemble.
The spatial partitioning of the two portions of the binding site, as seen in Figure 8 ▶, is intriguing, as both regions are generally contiguous groupings of residues. In essence, the binding site has a rigid structural scaffold on one side and a flexible surface on the other. It should be noted that the dynamic character of the RT loop, although decreased on binding, does not acquire a level of rigidity consistent with the remainder of Sem-5 C-SH3 (Fig. 6 ▶). Instead, the bound form also maintains conformational flexibility in this region. This leads to the conclusion that the bound state, as well as the unbound, is more accurately represented as an ensemble of states rather than as a single discrete conformation. Although the current studies do not provide information about the determinants of affinity and specificity in Sem-5 C-SH3, the observed dual character of the binding site offers interesting possibilities in this regard. Conformation variability of the RT loop in the bound state may indicate that specificity for different peptide ligands may be a function of the number of accessible protein conformations that can also facilitate low-energy contacts with the peptide. The role of dynamics in determining the thermodynamics of binding is currently under investigation.
Conformational entropy contributions
The results reported here have important thermodynamic consequences pertaining to the relationship between order parameters and conformational entropy. As shown by Yang and Kay (1996), NMR-derived order parameters are related to the conformational entropy, as both terms contain the same description of the orientational probability distributions of the amide vectors. Assuming diffusion-in-a-cone for the motions of the amide vectors, the conformational entropy has been expressed as:
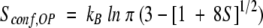 |
(1) |
where Sconf,OP is the conformational entropy determined from order parameters, S is the order parameter from picosecond and nanosecond contributions, and kB is the Boltzmann constant. Although the precise relationship between the conformational entropy determined by equation 1 and the experimentally obtained entropy remains ambiguous, the qualitative implications are straightforward. Specifically, the increase in order parameters in the RT loop of Sem-5 C-SH3 on ligand binding is indicative of a decrease in conformational entropy.
The observation that Sem-5 C-SH3 undergoes a cooperative conformational transition on binding and that the transition results in a decrease in conformational entropy means that the two processes are linked functions (Wyman 1964). This observation has several noteworthy implications. First, the ΔSconf,OP values determined for each residue from equation 1 are monitoring the same process. As such, the overall conformational entropy for the binding process cannot be represented as a sum of the individual residue-specific contributions.
Second, the conformational entropy changes observed on binding do not provide direct access to the magnitude of the conformational variations of the various states in the ensemble. This is evident from the binding constant expression of a system that undergoes a cooperative decrease in the conformational manifold on binding:
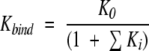 |
(2) |
where K0 is the intrinsic association constant in the absence of conformational fluctuations, and ΣKi is the sum of statistical weights of all binding-incompetent states (i.e., the conformational variants). In essence, the denominator is the conformational partition function for the protein. As indicated by equation 2, the magnitude of the effect of conformational redistribution on the observed binding energetics will depend on the statistical weight of the binding-incompetent states (i.e., if ΣKi ≫ 1, the effect will be large; if ΣKi ≪ 1, the effect will be small). In other words, the magnitude of the change in conformational entropy on binding, as determined from equation 1, does not provide details of the conformational entropy of the individual states, even if the relationship between order parameters and conformational entropy are known precisely. Instead, the magnitude of the observed conformational entropy change (or any other observable sensitive to the equilibrium) will depend on where the equilibrium is poised, the precise effect being determined by the linkage relationship in equation 2.
Third, the cooperative redistribution of the ensemble on binding will likely have enthalpic as well as entropic consequences, as the free energy of redistributing the ensemble, ΔGi (=−RTln ΣKi) will, under most circumstances, have both enthalpic and entropic contributions (ΔGi = ΔHi − T * ΔSi). Equation 2 indicates that if the binding-incompetent states differ enthalpically from the binding-competent species, redistributing the ensemble will reflect these differences in the apparent enthalpy (Eftink et al. 1983). For Sem-5 C-SH3, it is possible, indeed probable, that the observed dynamics changes are coupled to enthalpy changes, although direct experimental validation awaits further study. Nevertheless, it is clear that enthalpy changes coupled to dynamics would be difficult to rationalize and impossible to predict in the context of a static structural model of the protein.
The results presented here show that Sem-5 C-SH3 undergoes a cooperative two-state binding process, wherein binding is coupled to a conformational transition. This cooperative transition is associated with a decrease in conformational entropy in the protein, and that decrease is restricted to a relatively contiguous group of binding site residue. Despite these experimental insights, deciphering the energetic consequences of dynamics in the Sem-5:Sos binding process is not straightforward. Because it is not possible to resolve the structural and energetic attributes of the component states in the ensemble from relaxation data (using the current experimental protocol), it is not possible to elucidate the structural determinants of the observed energetics of binding. As equation 2 reveals that such uncertainties will likely affect the enthalpy as well as the entropy, it is clear that an accurate molecular-level description of binding is predicated on resolving the structure and energy of the relevant states in both the bound and unbound ensembles.
Materials and methods
Sample preparation
The C terminus of SH3 domain Sem-5 was cloned, overexpressed, and purified as described (Lim et al. 1994a,b). This SH3 domain has a mutation on residue 209, from cysteine to alanine, to prevent possible oxidation and intermolecular crosslinking. The purity of the eluted protein (>95%) was checked by electrospray ionization mass spectrometry, reverse-phase high-performance liquid chromatography (HPLC), and SDS-PAGE electrophoresis. 15N-labeled protein was obtained from cells grown in M9 minimal medium containing 15NH4Cl (Isotec Inc.). The purified protein was concentrated to ~0.7 mM in 50 mM NaOAc, 100 mM NaCl, and 10 mM CaCl2 (pH 4.8; 90% H20/10% D20). Because of cloning, there are two extra residues both at the N and C terminus of the SH3 domain that are not present in the crystal structure. These resonances are also weak and were also excluded from the relaxation analysis.
The HN and 15N chemical shifts for the unliganded form were assigned using various 3D and 2D heteronuclear NMR experiments (Ferreon et al. 2003). For the bound SH3 domain, unlabeled peptide (Ac-PPPVPPRRR-amide) was titrated to 15N-labeled SH3 domain and chemical shift perturbations were monitored via 1H-15N HSQC spectra. The peptide used in the studies was synthesized and purified by the Peptide Synthesis Laboratory (UTMB). The purity of the peptide (>95%) was checked by mass spectrometry and reverse-phase HPLC.
NMR spectroscopy
All NMR experiments were collected at 25°C on a Varian UnityPlus 400-MHz spectrometer using a triple resonance probe equipped with a pulsed field z gradient. 15N−T1, T2, and {1H}-15N NOE NMR experiments were done for both the unliganded and liganded forms of C-SH3 using the sensitivity-enhanced pulse field gradient sequences derived from the Varian ProteinPack, which are based on the pulse sequences described previously (Farrow et al. 1994). R1 (T1−1) data were acquired with the following 10 relaxation delays: 10, 40, 80, 100, 150, 230, 320, 430, 590, 840 msec, and R2 (T2−1) data were obtained with the following 11 relaxation delays: 10, 30, 50, 70, 90, 110, 150, 170, 190, 210, 250 msec. Both R1 and R2 experiments use 1-sec recycle delays. The {1H}-15N NOEs were measured by recording HSQC spectra with and without proton saturation. The former spectrum uses 3-sec proton saturation and 2-sec delays, whereas the latter only has the 5-sec delay. Duplicates were done for all spectra acquired.
Data processing and analysis
All spectra were processed using the FELIX v.98 software (Biosym Technologies, Inc.) on Silicon Graphics Indy workstation. The spectra were applied with a 90° sine bell shift window function and zerofilled to a 256 × 1024 matrix. Peak height intensities were measured by the automated routine in the software. The 15N−T1 and T2 time constants were obtained by fitting the data to a two-parameter single exponential decay function using software obtained from Neil A. Farrow. The uncertainties of T1 and T2 values were estimated from the nonlinear least squares fit. The relaxation rates R1 and R2 were obtained from the inverse of the T1 and T2 and the uncertainties were obtained by using the formula σ/T1,22 where σ is the uncertainty in the T1 or T2 value. We have also tested this by fitting the data to the R1 or R2 parameter and we obtain the same results both for the rates and the uncertainties. The NOE values were obtained from the ratio of the volumes with and without proton saturation and averaged for the duplicate spectra. Uncertainties in the NOEs were estimated from the base plane noise of the spectrum (Farrow et al. 1994).
Model-free formalism
The theoretical basis of the 15N relaxation has been discussed in a number of papers. Briefly, 15N relaxation is primarily dominated by dipolar and chemical shift anisotropy and is related to the spectral density functions at the 1H and 15N frequency combinations (ω; Abragam 1961). The transverse relaxation rate (R2) can also have, aside from contributions from dipolar and chemical shift anisotropy, conformational exchange contributions in the microsecond-to-millisecond timescale (Rex) by the following relation:
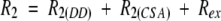 |
The spectral density function is then approximated by the following equation, which is the model-free formalism developed by Lipari and Szabo (1982a,b):
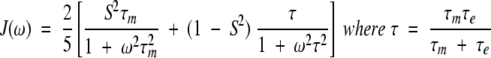 |
This model-free approach characterizes the spectral density function as composed of the overall tumbling of the molecule (τm) as well as the internal motions (τe) of a given 15N−1H vector. This equation simplifies to the following spectral density function:
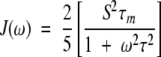 |
if all internal motions are very fast (τe < 10 psec). Using this Lipari-Szabo approach, relaxation parameters (R1, R2, and NOE) were fitted to the different models with an isotropic diffusion tensor using Modelfree 4.01 (Mandel et al. 1995,Mandel et al. 1996) to obtain parameters that describe the internal motions of the molecule. The five models are summarized as follows:
Model 1: S2
Model 2: S2 and τe
Model 3: S2 and Rex
Model 4: S2, τe, and Rex
Model 5:
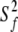 ,
, 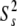 , and τs
, and τs
For models 1–4 → S2 = 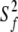 where S2 =
where S2 = 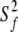 *
* 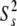 and
and 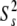 = 1
= 1
The last model uses a two-timescale model to characterize both the fast motions (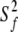 ) and slow motions (
) and slow motions (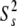 , τs; Clore et al. 1990). As described elsewhere (Mandel et al. 1995,Mandel et al. 1996), model fitting involves three steps: initial estimation of the rotational correlation time (τm) or the diffusion tensor, model selection, and final optimization.
, τs; Clore et al. 1990). As described elsewhere (Mandel et al. 1995,Mandel et al. 1996), model fitting involves three steps: initial estimation of the rotational correlation time (τm) or the diffusion tensor, model selection, and final optimization.
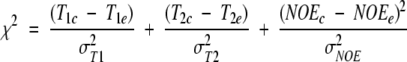 |
Initial estimate of τm
Programs (which include the Modelfree 4.01) obtained from Dr. A.G. Palmer III (Columbia University, http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer) were used for obtaining the inertia tensor, statistical tests of anisotropy, and initial estimate of τm. The program pdbinertia was used to calculate the principal moments of the inertia tensor using the recently solved NMR solution structure for the unbound state and the crystal structure (wherein hydrogens are added) for the complexed state. MOLMOL v. 2K.1 (Koradi et al. 1996) was used to generate the pdb coordinates of the crystal structure with the hydrogens. The program r2r1_diffusion determines the significance of fitting to an isotropic versus an axially symmetric model. This uses the approach of Tjandra et al. (1995) to determine the diffusion tensors for spherical and axially symmetric motional models from the experimental relaxation data. An initial estimate of τm was obtained by two methods. The first is through the r2r1_diffusion, which also gives the τm for the isotropic model from the structural coordinates. The second uses the program tmest and is based on the approach of Kay et al. (1989). Residues having an R1/R2 ratio above or below one standard deviation from the mean and NOE <0.55 are not included in the analysis (Kay et al. 1989; Clore et al. 1990; Tjandra et al. 1995). Model selection is based on the procedure described in Mandel et al. (1995).
Model selection and optimization
Model-free parameters were calculated from the measured relaxation parameters using the program Modelfree 4.01 (Palmer III et al. 1991; Mandel et al. 1995). The process of analyzing the relaxation data starts with an estimate of the overall correlation time. The selection of the appropriate model for amide vector follows the same strategy outlined by Palmer III and coworkers (Mandel et al. 1995). Five hundred Monte Carlo simulations were generated to determine the cumulative probability distribution and to compare the χ2 value with the critical value at α = 0.05 or 0.10. Models 2 and 3 were preferred over model 1 by evaluating the F-statistic at α = 0.20. Because we only have three relaxation parameters, model 4 and model 5 were resorted to only if the sum of the squares error was zero and if the residues were not modeled adequately by models 1–3. Model optimization was then done by Powell minimization to obtain the final τm and the optimized parameters. Uncertainties in the dynamics parameters were determined using the Monte Carlo simulations carried out by the Modelfree program, as described earlier. Residues were considered adequately fit if they followed the criteria for Farrow et al. (1994): (a) all three experimental relaxation parameters are reproduced within 95% confidence limits, i.e., 1.96 times the experimental uncertainty and (b) fitting parameters must be greater than their error. The tryptophan side chains were fitted in a similar procedure, except the value used for the difference between the parallel and perpendicular components of the chemical shift tensor for the indole amides was taken to be −89 ppm (Cross and Opella 1983; Stone et al. 1993), as opposed to the value for backbone amide groups (−160 ppm; Hiyama et al. 1988).
Chemical shift perturbation
The peptide Ac-PPPVPPRRR-amide was synthesized and purified by the Peptide Synthesis Laboratory (UTMB). The purity of the peptide (>95%) was checked by mass spectrometry (Louisiana State University) and reverse-phase HPLC. Peptide concentration was determined from quantitative amino acid analysis. The 15N-labeled sample (0.66 mM) was prepared in 50 mM NaOAc, 100 mM NaCl, and 10 mM CaCl2 (pH 4.8; 90% H20/10% D20). 1H-15N HSQC spectra were collected for each titration point using a 750-MHz Varian UnityPlus spectrometer. A total of 15 titration points were collected and, at the final point, the peptide concentration was approximately twice that of Sem-5 C-SH3 protein. The binding curves were analyzed using a nonlinear regression analysis following the procedure of Lian and Roberts (1993), using the following equation to monitor the chemical shifts in the labeled protein resonance:
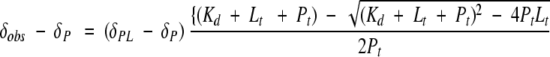 |
where Pt, Lt, and δobs are the total protein, ligand concentrations, and the observed change in chemical shift on ligand titration, respectively, which are known, and δPL and Kd are to be determined by the regression analysis, where Kd is the dissociation constant and δPL is the chemical shift of the complex.
Isothermal titration calorimetry (ITC)
Titration experiments were performed using the VP-ITC system at 25°C as described elsewhere (Wiseman et al. 1989; Gómez and Freire 1995; Renzoni et al. 1996). The unlabeled protein was extensively dialyzed against 50 mM NaOAc, 100 mM NaCl, and 10 mM CaCl2 (pH 4.8). The peptide (Ac-PPPVPPRRR-amide) was lyophilized from pure water and dissolved with the buffer from the final dialysis. Protein and peptide were then centrifuged to remove particulates and degassed, and the pH was adjusted to the final pH of 4.8. The protein (0.269 mM) was loaded into the 1.3-mL sample cell and the peptide (4.22 mM) into the injector. An initial 2-μL injection and 31 × 4μ-L injections were made, with a 6-min equilibration period between injections. A control experiment (peptide titrated into buffer alone) was performed to evaluate the heats of dilution, which were subtracted from the experimental titration results. Data were then fitted using a nonlinear least squares program in Origin for ITC v. 5.0 (Microcal), varying the stoichiometry (N), binding constant (Kb), and binding enthalpy (ΔH°). The Kb determined from the best-fit curve was used to determine the free energy (ΔG°) and the entropy (ΔS°) involved in binding using the following thermodynamic relation:
 |
where R is the gas constant; T is the absolute temperature; and ΔG°, ΔH°, and ΔS° are the standard free energy, enthalpy, and entropy for the binding.
Acknowledgments
We thank Drs. David Volk, Shanmin Zhang, David Gorenstein, and Krishna Rajarathnam for NMR assistance and helpful discussions. We also thank Allan Chris M. Ferreon for ITC technical assistance and NMR analyses of binding isotherms. We are also indebted to Drs. Neil A. Farrow, Lewis Kay, and Andrew Lee for valuable advice and useful scripts in analyzing relaxation data, and Dr. Arthur Palmer III for Modelfree 4.0 software. We also thank Dr. Judah Rosenblatt for help in statistical analysis. Supported by National Science Foundation grant MCB-9875689, National Institutes of Health RO1-GM13747, and Welch Award H-1461.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
SH3, src-homology domain 3
C-SH3, C-terminal SH3 domain
Sos, Son of Sevenless
NMR, nuclear magnetic resonance
2D, two-dimensional
NOE, nuclear Overhauser effect
ITC, isothermal titration calorimetry
HSQC, heteronuclear single quantum coherence
ppm, parts per million
PDB, Protein Data Bank
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0238003.
References
- Abragam, A. 1961. Principles of nuclear magnetism. Clarendon Press, Oxford, UK.
- Akke, M., Bruschweiler, R., and Palmer III, A.G. 1993. NMR order parameters and free-energy: An analytical approach and its application to cooperative Ca2+ binding by calbindin-d(9k). J. Am. Chem. Soc. 115 9832–9833. [Google Scholar]
- Alexandrescu, A.T., Rathgeb-Szabo, K., Rumpel, K., Jahnke, W., Schulthess, T., and Kammerer, R.A. 1998. 15N backbone dynamics of the S-peptide from ribonuclease A in its free and S-protein bound forms: Toward a site-specific analysis of entropy changes upon folding. Protein Sci. 7 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker, G.W., Gout, I., Downing, A.K., Driscoll, P.C., Boyd, J., Waterfield, M.D., and Campbell, I.D. 1993. Solution structure and ligand-binding site of the SH3 domain of the p85α subunit of phosphatidylinositol 3-kinase. Cell 73 813–822. [DOI] [PubMed] [Google Scholar]
- Buday, L. 1999. Membrane-targeting of signalling molecules by SH2/SH3 domain-containing adaptor proteins. Biochim. Biophys. Acta 1422 187–204. [DOI] [PubMed] [Google Scholar]
- Clark, S.G., Stern, M.J., and Horvitz, H.R. 1992. C. elegans cell-signalling gene sem-5 encodes a protein with SH2 and SH3 domains. Nature 356 340–344. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Driscoll, P.C., Wingfield, P.T., and Gronenborn, A.M. 1990. Analysis of the backbone dynamics of interleukin-1 β using two-dimensional inverse detected heteronuclear 15N−1H NMR spectroscopy. Biochemistry 29 7387–7401. [DOI] [PubMed] [Google Scholar]
- Collins, J.R., Burt, S.K., and Erickson, J.W. 1995. Flap opening in HIV-1 protease simulated by ‘activated’ molecular dynamics. Nat. Struct. Biol. 2 334–338. [DOI] [PubMed] [Google Scholar]
- Cross, T.A. and Opella, S.J. 1983. Protein-structure by solid-state NMR. J. Am. Chem. Soc. 105 306–308. [Google Scholar]
- Dalgarno, D.C., Botfield, M.C., and Rickles, R.J. 1997. SH3 domains and drug design: Ligands, structure, and biological function. Biopolymers 43 383–400. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R., Anusiem, A.C., and Biltonen, R.L. 1983. Enthalpy-entropy compensation and heat capacity changes for protein-ligand interactions: General thermodynamic models and data for the binding of nucleotides to ribonuclease A. Biochemistry 22 3884–3896. [DOI] [PubMed] [Google Scholar]
- Egan, S.E., Giddings, B.W., Brooks, M.W., Buday, L., Sizeland, A.M., and Weinberg, R.A. 1993. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 363 45–51. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Feher, V.A. and Cavanagh, J. 1999. Millisecond-timescale motions contribute to the function of the bacterial response regulator protein Spo0F. Nature 400 289–293. [DOI] [PubMed] [Google Scholar]
- Ferreon, J.C., Volk, D.E., Luxon, B.A., Gorrenstein, D.G., and Hilser, V.J. 2003. Solution structure, dynamics and thermodynamics of the native state ensemble of the Sem-5 C-terminal SH3 domain. Biochemistry (in press). [DOI] [PubMed]
- Gagné, S.M., Tsuda, S., Spyracopoulos, L., Kay, L.E., and Sykes, B.D. 1998. Backbone and methyl dynamics of the regulatory domain of troponin C: Anisotropic rotational diffusion and contribution of conformational entropy to calcium affinity. J. Mol. Biol. 278 667–686. [DOI] [PubMed] [Google Scholar]
- Gómez, J. and Freire, E. 1995. Thermodynamic mapping of the inhibitor site of the aspartic protease endothiapepsin. J. Mol. Biol. 252 337–350. [DOI] [PubMed] [Google Scholar]
- Hansson, H., Mattsson, P.T., Allard, P., Haapaniemi, P., Vihinen, M., Smith, C.I.E., and Härd, T. 1998. Solution structure of the SH3 domain from Bruton’s tyrosine kinase. Biochemistry 37 2912–2924. [DOI] [PubMed] [Google Scholar]
- Hiyama, Y., Niu, C.-H., Silverton, J.V., Bavoso, A., and Torchia, D.A. 1988. Determination of N-15 chemical-shift tensor via N-15-H-2 dipolar coupling in BOC-glycylglycyl[N-15]glycine benzyl ester. J. Am. Chem. Soc. 110 2378–2383. [Google Scholar]
- Hodsdon, M.E. and Cistola, D.P. 1997. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry 36 2278–2290. [DOI] [PubMed] [Google Scholar]
- Horita, D.A., Baldisseri, D.M., Zhang, W., Altieri, A.S., Smithgall, T.E., Gmeimer, W.H., and Byrd, R.A. 1998. Solution structure of the human Hck SH3 domain and identification of its ligand binding site. J. Mol. Biol. 278 253–265. [DOI] [PubMed] [Google Scholar]
- Horita, D.A., Zhang, W., Smithgall, T.E., Gmeiner, W.H., and Byrd, R.A. 2000. Dynamics of the Hck-SH3 domain: Comparison of experiment with multiple molecular dynamics simulations. Protein Sci. 9 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, L.E., Torchia, D.A., and Bax, A. 1989. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: Application to staphylococcal nuclease. Biochemistry 28 8972–8979. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Muhandiram, D.R., Farrow, N.A., Aubin, Y., and Forman-Kay, J.D. 1996. Correlation between dynamics and high affinity binding in an SH2 domain interaction. Biochemistry 35 361–368. [DOI] [PubMed] [Google Scholar]
- Kay, L.E., Muhandiram, D.R., Wolf, G., Shoelson, S.E., and Forman-Kay, J.D. 1998. Correlation between binding and dynamics at SH2 domain interfaces. Nat. Struct. Biol. 5 156–163. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of molecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kristensen, S.M., Siegal, G., Sankar, A., and Driscoll, P.C. 2000. Backbone dynamics of the C-terminal SH2 domain of the p85α subunit of phosphoinositide 3-kinase: Effect of phosphotyrosine-peptide binding and characterization of slow conformational exchange processes. J. Mol. Biol. 299 771–788. [DOI] [PubMed] [Google Scholar]
- Lian, L. and Roberts, G.C.K. 1993. Effects of chemical exchange on NMR spectra. In NMR of macromolecules. A practical approach (ed. G.C.K. Roberts), pp. 153–182. Oxford University Press, Oxford, UK.
- Lim, W.A., Fox, R.O., and Richards, F.M. 1994a. Stability and peptide binding affinity of an SH3 domain from the Caenorhabditis elegans signaling protein Sem-5. Protein Sci. 3 1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, W.A., Richards, F.M., and Fox, R.O. 1994b. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372 375–379. [DOI] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104 4559–4570. [Google Scholar]
- ———. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- Lowenstein, E.J., Daly, R.J., Batzer, A.G., Li, W., Margolis, B., Lammers, R., Ullrich, A., Skolnik, E.Y., Bar-Sagi, D., and Schlessinger, J. 1992. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70 431–442. [DOI] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer III, A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246 144–163. [DOI] [PubMed] [Google Scholar]
- ———. 1996. Dynamics of ribonuclease H: Temperature dependence of motions on multiple time scales. Biochemistry 35 16009–16023. [DOI] [PubMed] [Google Scholar]
- Nicholson, L.K., Yamazaki, T., Torchia, D.A., Grzesiek, S., Bax, A., Stahl, S.J., Kaufman, J.D., Wingfield, P.T., Lam, P.Y.S., Jadhav, P.K., et al. 1995. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 2 274–279. [DOI] [PubMed] [Google Scholar]
- Olejniczak, E.T., Zhou, M.-M., and Fesik, S.W. 1997. Changes in the NMR-derived motional parameters of the insulin receptor substrate 1 phosphotyrosine binding domain upon binding to an interleukin 4 receptor phosphopeptide. Biochemistry 36 4118–4124. [DOI] [PubMed] [Google Scholar]
- Olivier, J.P., Raabe, R., Henkemeyer, M., Dickson, B., Mbamalu, G., Margolis, B., Schlessinger, J., Hafen, E., and Pawson, T. 1993. A Drosophila SH2-SH3 adaptor protein implicated in coupling the sevenless tyrosine kinase to an activator of Ras guanine nucleotide exchange, Sos. Cell 73 179–191. [DOI] [PubMed] [Google Scholar]
- Palmer III, A.G., Rance, M., and Wright, P.E. 1991. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J. Am. Chem. Soc. 113 4371–4380. [Google Scholar]
- Pawley, N.H., Wang, C., Koide, S., and Nicholson, L.K. 2001. An improved method for distinguishing between anisotropic tumbling and chemical exchange in analysis of 15N relaxation parameters. J. Biomol. NMR 20 149–165. [DOI] [PubMed] [Google Scholar]
- Pawson, T. 1995. Protein modules and signalling networks. Nature 373 575–579. [DOI] [PubMed] [Google Scholar]
- Renzoni, D.A., Pugh, D.J.R., Siligardi, G., Das, P., Rossi, C., Waterfield, M.D., Campbell, I.D., and Ladbury, J.E. 1996. Structural and thermodynamic characterization of the interaction of the SH3 domain from Fyn with the proline-rich binding site on the p85 subunit of P13-kinase. Biochemistry 35 15646–15653. [DOI] [PubMed] [Google Scholar]
- Spyracopoulos, L., Gagné, S.M., Li, M.X., and Sykes, B.D. 1998. Dynamics and thermodynamics of the regulatory domain of human cardiac troponin C in the apo- and calcium-saturated states. Biochemistry 37 18032–18044. [DOI] [PubMed] [Google Scholar]
- Stivers, J.T., Abeygunawardana, C., and Mildvan, A.S. 1996. 15N NMR relaxation studies of free and inhibitor-bound 4-oxalocrotonate tautomerase: Backbone dynamics and entropy changes of an enzyme upon inhibitor binding. Biochemistry 35 16036–16047. [DOI] [PubMed] [Google Scholar]
- Stock, A. 1999. Relating dynamics and function. Nature 400 221–222. [DOI] [PubMed] [Google Scholar]
- Stone, M.J., Chandrasekhar, K., Holmgren, A., Wright, P.E., and Dyson, H.J. 1993. Comparison of backbone and tryptophan side-chain dynamics of reduced and oxidized Escherichia coli thioredoxin using 15N NMR relaxation measurements. Biochemistry 32 426–435. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Feller, S.E., Pastor, R.W., and Bax, A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117 12562–12566. [Google Scholar]
- Tjandra, N., Szabo, A., and Bax, A. 1996. Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J. Am. Chem. Soc. 118 6986–6991. [Google Scholar]
- Wagner, G. 1995. The importance of being floppy. Nat. Struct. Biol. 2 255–257. [DOI] [PubMed] [Google Scholar]
- Wang, C., Pawley, N.H., and Nicholson, L.K. 2001. The role of backbone motions in ligand binding to the c-Src SH3 domain. J. Mol. Biol. 313 873–887. [DOI] [PubMed] [Google Scholar]
- Weber, G. 1992. Protein interactions. Chapman and Hall, London.
- Wiseman, T., Williston, S., Brandts, J.F., and Lin, L. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179 131–137. [DOI] [PubMed] [Google Scholar]
- Wittekind, M., Mapelli, C., Farmer II, B.T., Suen, K., Goldfarb, V., Tsao, J., Lavoie, T., Barbacid, M., Meyers, C.A., and Mueller, L. 1994. Orientation of peptide fragments from Sos proteins bound to the N-terminal SH3 domain of Grb2 determined by NMR spectroscopy. Biochemistry 33 13531–13539. [DOI] [PubMed] [Google Scholar]
- Wyman, J. 1964. Linked functions and reciprocal effects in hemoglobin: A second look. Adv. Protein Chem. 19 223–286. [DOI] [PubMed] [Google Scholar]
- Yang, D.W. and Kay, L.E. 1996. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: Application to protein folding. J. Mol. Biol. 263 369–382. [DOI] [PubMed] [Google Scholar]
- Ye, J., Mayer, K.L., and Stone, M.J. 1999. Backbone dynamics of the human CC-chemokine eotaxin. J. Biomol. NMR 15 115–124. [DOI] [PubMed] [Google Scholar]
- Yu, H., Rosen, M.K., Shin, T.B., Seidel-Dugan, C., Brugge, J.S., and Schreiber, S.L. 1992. Solution structure of the SH3 domain of Src and identification of its ligand-binding site. Science 258 1665–1667. [DOI] [PubMed] [Google Scholar]
- Zidek, L., Novotny, M.V., and Stone, M.J. 1999. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat. Struct. Biol. 6 1118–1121. [DOI] [PubMed] [Google Scholar]