

Abstract
α-Hemolysin (αHL) is secreted by Staphylococcus aureus as a water-soluble monomer that assembles into a heptamer to form a transmembrane pore on a target membrane. The crystal structures of the LukF water-soluble monomer and the membrane-bound α-hemolysin heptamer show that large conformational changes occur during assembly. However, the mechanism of assembly and pore formation is still unclear, primarily because of the difficulty in obtaining structural information on assembly intermediates. Our goal is to use disulfide bonds to selectively arrest and release αHL from intermediate stages of the assembly process and to use these mutants to test mechanistic hypotheses. To accomplish this, we created four double cysteine mutants, D108C/K154C (αHL-A), M113C/K147C (αHL-B), H48C/ N121C (αHL-C), I5C/G130C (αHL-D), in which disulfide bonds may form between the pre-stem domain and the β-sandwich domain to prevent pre-stem rearrangement and membrane insertion. Among the four mutants, αHL-A is remarkably stable, is produced at a level at least 10-fold greater than that of the wild-type protein, is monomeric in aqueous solution, and has hemolytic activity that can be regulated by the presence or absence of reducing agents. Cross-linking analysis showed that αHL-A assembles on a membrane into an oligomer, which is likely to be a heptamer, in the absence of a reducing agent, suggesting that oxidized αHL-A is halted at a heptameric prepore state. Therefore, conformational rearrangements at positions 108 and 154 are critical for the completion of αHL assembly but are not essential for membrane binding or for formation of an oligomeric prepore intermediate.
Keywords: Double cysteine mutants, disulfide bonds, assembly intermediate, pore-forming toxin, Staphylococcal α-hemolysin
The molecular mechanisms of oligomeric membrane protein assembly and insertion are poorly understood to a great extent because of the difficulty in obtaining structural information on assembly intermediates. In many cases, the assembly and insertion of oligomeric membrane proteins involve complex pathways in which multiple accessory proteins, such as chaperones, play essential roles and complicate the elucidation of molecular mechanisms (Booth et al. 2001). However, because of its spontaneous and relatively simple assembly pathway, α-hemolysin (αHL) is an ideal molecule to study to determine mechanisms of membrane protein assembly and insertion (Heuck et al. 2001; Prevost et al. 2001; Montoya and Gouaux 2003).
αHL is a lytic cytotoxin secreted by Staphylococcus aureus, a common and potentially deadly human pathogen (Menestrina et al. 2001). αHL is related in amino acid sequence and biological function to a host of other pore-forming Staphylococcal toxins that include a family of bicomponent toxins requiring two subunits for assembly and function (Gouaux et al. 1997). The bicomponent toxins are γ-hemolysin (γHL), leukocidin (Luk), and Panton-Valentine leukocidin (PVL), and are composed of the subunits LukF + HγII, LukF + LukS, and PV-LukF + PV-LukS, respectively (Tomita and Kamio 1997). In addition to being similar to αHL in terms of function and amino acid sequence, LukF, LukS, and HγII are related in three-dimensional structure (Gouaux et al. 1997; Gouaux 1998). All of these Staphylococcal toxins are secreted as water-soluble monomers, and assemble on target cell membranes to form oligomeric membrane pores. αHL is a homoheptamer and assembles on erythrocyte and egg phosphatidylcholine membranes, and in deoxycholate micelles (Bhakdi et al. 1981; Gouaux et al. 1994; Song et al. 1996; Fang et al. 1997; Malghani et al. 1999; Krasilnikov et al. 2000). Under some conditions, however, Shao and colleagues contend that a hexamer may also be formed, as determined by atomic force microscopy (Czajkowsky et al. 1998). At the present time, evidence for the subunit stoichiometry of the bicomponent heteromeric Luk pores suggests that they are either heptameric (Sugawara-Tomita et al. 2002) or octameric (Miles et al. 2002).
A number of different probes have been employed to elucidate the mechanism of toxin assembly for the heptameric αHL pore, including (1) proteolytic susceptibility (Palmer et al. 1993; Walker et al. 1995), (2) site-directed mutagenesis (e.g., Walker and Bayley 1995a), and (3) fluorescence spectroscopy (Valeva et al. 1997a,b; Vécsey-Semjén et al. 1997). The most parsimonious mechanism for αHL assembly is shown below.
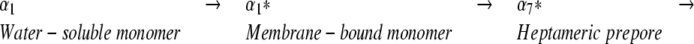 |
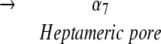 |
Structural studies of the water-soluble monomer (α1) and the heptameric pore (α7) have shown that there are significant conformational changes between α1 and α7 (Olson et al. 1999; Pédelacq et al. 1999, 2000). In α1, the amino latch and the pre-stem domain are located adjacent to the β-sandwich domain and participate in intraprotomer interactions (Walker et al. 1992; Walker and Bayley 1995a; Valeva et al. 1997b). In α7, however, the amino latch is directed toward the lumen of the pore, and makes a number of significant contacts with residues on a neighboring protomer (Song et al. 1996). The pre-stem domain has also rearranged in α7 and now forms a transmembrane β-barrel, called the stem domain. Although the atomic structure for the end point on the assembly pathway has been solved (Song et al. 1996), and a number of key residues that play essential roles in the assembly process have been found, determination of molecular mechanisms for the assembly of the heptamer and for the formation and insertion of the transmembrane β-barrel have proven elusive.
To elaborate a detailed mechanism for the assembly of αHL, we must have structural information on assembly intermediates. However, because the intermediates are inherently short-lived, one must find methods to arrest the toxin at specific points along the assembly pathway. Although a number of toxin variants have been produced that are arrested at intermediate stages of assembly (Walker and Bayley 1995b; Walker et al. 1995; Valeva et al. 2001), variants that can be turned on and off with a switch that is as stereospecific as a disulfide bond are particularly attractive for mechanistic studies (Harrison and Sternberg 1996). In this study, we created double cysteine mutants of αHL that are predicted to form disulfide bonds between the β-sandwich domain and the pre-stem domain. When the cysteine residues form a disulfide bond, the pre-stem will be prevented from rearranging, and the toxin should be inactive. In the presence of a reducing agent, however, the disulfide bond will be reduced, the pre-stem will be able to rearrange, and the toxin should have hemolytic activity. Depending on where the disulfide bond is located, we can halt specific conformational rearrangements. Here, we describe the functional behavior of a panel of four double cysteine mutants, show that rearrangement of the pre-stem is essential for lytic activity and for formation of the SDS-stable heptamer, and demonstrate that formation of the oligomeric prepore intermediate precedes and is not dependent upon rearrangement of the pre-stem.
Results
Design of the double cysteine mutants
We created four double cysteine mutants that could form disulfide bonds between the pre-stem domain and the β-sandwich domain. The design of three of the mutants was based on the crystal structure of the LukF water-soluble monomer (Olson et al. 1999) where mutation sites were selected on the basis of (1) Cα–Cα distances between the pre-stem domain and the β-sandwich domain of ~5 Å in the monomeric state, (2) appropriate orientation of the Cα–Cβ bond vectors (Harrison and Sternberg 1996) in the monomeric state, and (3) an increase in the Cα–Cα separation of the selected residues by more than 10 Å upon heptamer formation. The resulting mutants were αHL-A (D108C/K154C), αHL-B (M113C/K147C), and αHL-C (H48C/N121C), and the locations of the sites are illustrated in Figure 1 ▶. In addition, we made a fourth mutant, αHL-D (I5C/G130C), because Valeva and colleagues have shown that introduction of cysteine residues at these positions results in disulfide bond formation between the N-terminus and the β-sandwich domain, thereby preventing pore formation and cell lysis (Valeva et al. 2001).
Figure 1.
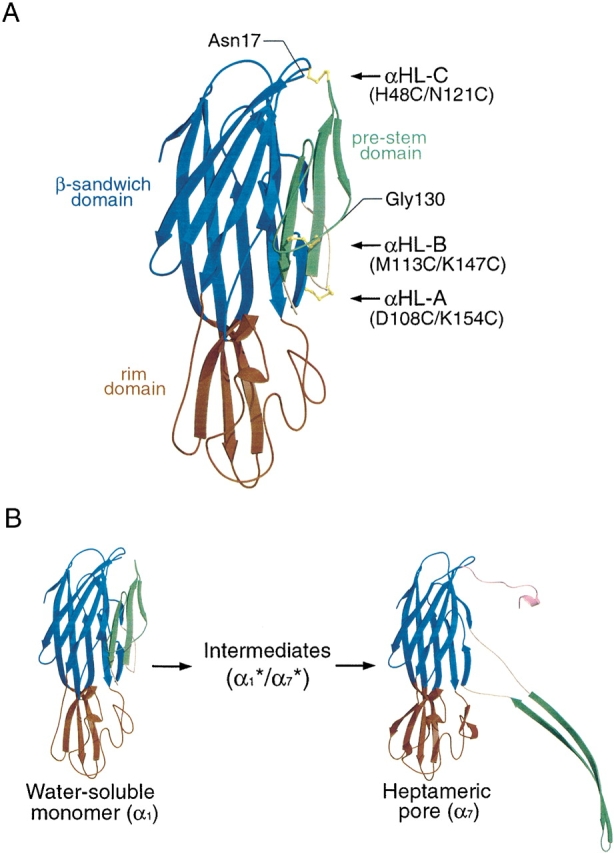
Structures of a homology model of the water-soluble αHL monomer and a protomer from the assembled heptamer. (A) Location of the double cysteine mutation sites in αHL. The model of water-soluble αHL monomer was created using Swiss-Model program (Peitsch and Tschopp 1995; Peitsch 1996; Guex and Peitsch 1997) based on the structure of water-soluble LukF monomer. Because the amino terminus of αHL is not homologous to that of LukF, the first 16 residues are not shown. Three putative disulfide bridges in the mutants αHL-A (D108C/K154C), αHL-B (M113C/K147C), and αHL-C (H48C/N121C) are depicted for illustration purposes only. For mutant αHL-D (I5C/G130C), the locations of Gly130 and the amino terminus of this model (Asn17) are illustrated. In parts (A) and (B) of this figure, the pre-stem is in green, the β-sandwich is in blue, the rim is in red, and the triangle region is in yellow. (B) A schematic presentation of the αHL conformational change during pore formation. The water-soluble monomer was modeled as indicated in (A). A protomer of the heptameric pore is depicted on the right-hand side of the panel. Here, the amino-latch is in pink.
Purification of the double cysteine mutants
The double cysteine mutations were generated by PCR using pT7[αHL-His6] as a template. The resulting mutant proteins were expressed in the cytoplasm of Escherichia coli cells (Prinz et al. 1997) and purified to homogeneity by affinity chromatography, as shown in Figure 2 ▶. The molecular masses were confirmed by mass spectrometry (calculated and predicted masses were, respectively: A, 34.832 and 34.847 kD; B, 34.816 and 34.839 kD; C, 34.824 and 34.845 kD; D, 34.905 and 34.931 kD). As illustrated in Figure 2 ▶, in the absence of a reducing agent, αHL-A, αHL-C and a fraction of αHL-D migrated faster than WT-His6, implying that disulfide bonds might have formed, resulting in a more compact protein that migrates faster on the gel. In the presence of a reducing agent, by contrast, the mutants and the wild-type protein comigrated. The mobility of αHL-B relative to the WT protein did not change in the presence or absence of a reducing agent.
Figure 2.

SDS-PAGE of purified mutant proteins in the presence and absence of a reducing agent. Recombinant proteins were purified by Ni-NTA chromatography, and 4 μg of each protein was analyzed +/− β-mercaptoethanol. The lanes are composed of wild-type, untagged αHL (WT), wild-type His-tagged αHL, and mutants αHL-A (A), αHL-B (B), αHL-C (C), and αHL-D (D).
To determine whether the cysteine residues resulted in intermolecular disulfide bond formation or in higher order aggregation, the Ni-NTA-purified proteins were subject to size-exclusion chromatography (SEC). All four mutants eluted at 16 mL from a Superose 12 10/30 column run in the absence of a reducing agent (data not shown). Comparison of the elution position of the mutants to standard proteins suggests that the molecular weight of the mutants is ~35 kD. The mutants also coeluted with the wild-type, cysteine-free protein. The mutants are, therefore, monomeric in aqueous solution under oxidizing conditions.
Hemolytic assays of the mutant and wild-type proteins
If the mutant proteins form disulfide bonds and become trapped at an intermediate stage of assembly, they will not have hemolytic activity in the absence of a reducing agent but will have hemolytic activity in the presence of a reducing agent. To test for hemolytic activity, the mutant proteins were incubated with rabbit blood cells (RBC), with and without a reducing agent, and the increase of supernatant absorbance at 475 nm due to released hemoglobin was followed (Fig. 3 ▶). In the absence of a reducing agent, αHL-A showed background hemolytic activity that was the same as that of buffer only (10%), suggesting that αHL-A forms a disulfide bond and does not assemble to a lytic pore. The relatively low hemolytic activity of αHL-C (25%) also indicates that a large proportion of αHL-C may form disulfide bonds, thus blocking pore formation. For αHL-A and αHL-C, a reducing agent rescued their hemolytic activity to levels close to that of WT-His6 (95%–100%), suggesting that the introduced cysteine residues do not substantially disrupt their structures.
Figure 3.
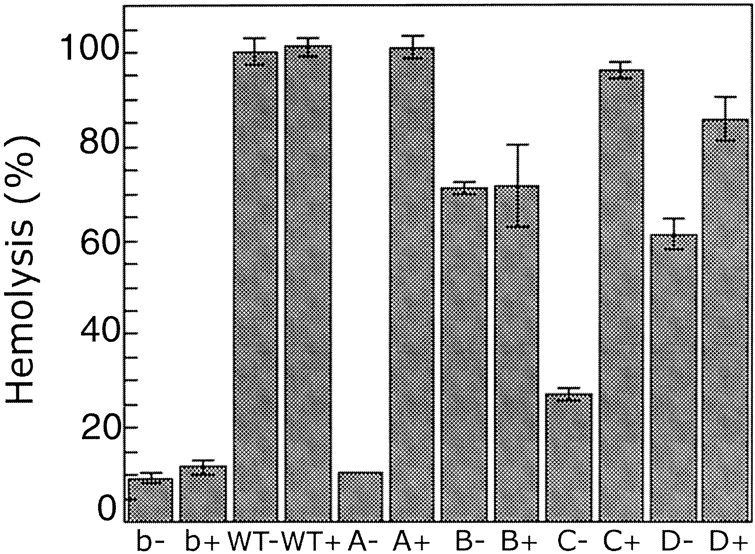
Hemolytic activity of mutant proteins, defined as in Figure 2 ▶, under reducing and nonreducing conditions. RBC in hemolysis (HL) buffer were incubated with wild-type and mutant proteins at 20°C for 20 min. The hemolytic activity was monitored by following the increase of absorbance at 475 nm. The reactions were normalized to the extent of lysis caused by diluting an equivalent volume of erythrocytes into 8 volumes of Milli Q water. The reported value is the mean and the error bars define standard deviations (n = 3). The symbol “b” indicates buffer, the symbol “WT” indicates His-tagged wild-type protein, and the symbol “+/−” indicates +/− DTT.
Although αHL-D also showed a high hemolytic activity in the presence of a reducing agent (85%), it also showed substantial activity (60%) in the absence of a reducing agent, suggesting that only a portion of the αHL-D mutant protein forms a disulfide bond. In contrast to the other mutants, αHL-B showed similar hemolytic activities in the presence or absence of a reducing agent (70%), indicating that αHL-B probably does not readily form a disulfide bond. Because αHL-A showed the most distinct hemolytic behavior in the presence and absence of a reducing agent, and because the protein expression level of αHL-A was at least 10-fold greater than any of the other proteins, including the wild-type toxin, we decided to concentrate our studies on αHL-A.
Time course of hemolysis and binding assay of αHL-A
To further explore the hemolytic potency of αHL-A, a hemolytic assay was carried out with the time course over the first 10 min. Although αHL-A did not have hemolytic activity in the absence of a reducing agent, αHL-A showed an equivalent hemolytic curve to that of the wild-type protein in the presence of a reducing agent (Fig. 4A ▶). These data strongly suggest that αHL-A retains wild-type structure under reducing conditions, resulting in essentially the same hemolytic potency as the wild-type protein. However, αHL-A might have a lower hemolytic activity but a higher affinity to rabbit blood cells (RBC), compared to that of the wild-type protein. If this were the case, αHL-A might then show hemolytic activity similar to that of the wild-type protein. Therefore, we examined the binding of αHL-A to RBC. After incubation of the wild-type toxin and αHL-A with RBC on ice, a condition in which αHL does not assemble but does bind to RBC (Hildebrand et al. 1991), the supernatant and pellet fractions were analyzed by Western blotting. As shown in Figure 4B ▶, there was no substantial difference between αHL-A and wild-type toxin in binding to RBC. Moreover, αHL-A binds to RBC to the same extent in the presence or absence of a reducing agent.
Figure 4.

Lysis and binding experiments using wild-type toxin and αHL-A. (A) Time course hemolytic activity of αHL-A. RBC in hemolysis (HL) buffer were incubated with wild-type and αHL-A protein either in the absence (left) or presence (right) of DTT at 20°C for 0, 2, 4, 6, 8, and 10 min. The hemolytic activity was monitored by following the increase of absorbance at 475 nm. The reactions were normalized to the extent of lysis caused by diluting an equivalent volume of erythrocytes into 8 volumes of Milli Q water. The reported value is the mean, and the error bars define one standard deviation (n = 3). (B) αHL-A binding to RBC. The recombinant proteins were incubated with RBC in the presence or absence of DTT on ice for 1 h. The supernatant and pellet fractions were separated by centrifugation, and the amount of recombinant protein was detected by Western blotting using anti-His6 antibody. The symbol “S” indicates supernatant and the symbol “P” indicates pellet.
αHL-A forms a disulfide bond
To show that αHL-A forms a disulfide bond, a limited trypsin digestion assay was performed. Because there are two major trypsin cleavage sites in αHL (Fig. 5A ▶), when the wild-type monomeric toxin is treated with a low concentration of trypsin, the resulting digestion yields four major bands including uncut material (35 kD) and three smaller fragments of 34.2, 20.5, and 13.7 kD. One cleavage site is near the amino terminus at Lys 8, and the second site is located in the middle of the sequence at Lys 131. Therefore, if αHL-A forms a disulfide bond between the residues C108 and C154 as we predict, the 20.5-kD fragment and the 13.7-kD fragment will remain linked together in the absence of a reducing agent. As shown in Figure 5B ▶, neither the 20.5-kD nor the 13.7-kD fragments are seen under conditions that maintain the integrity of a disulfide bond (lanes 3–6). Digestion is occurring because we see the appearance of the 34.2-kD fragment, as well as a slightly larger ~36-kD fragment as the trypsin concentration increases (lanes 4–6). The approximate yield of these fragments at the highest trypsin concentration (trypsin/protein ratio of 1/50) were ~30% (34.2 kD) and ~20% (~36 kD), respectively. The ~36-kD band may represent a species that is cleaved at Lys 131 but remains intact and has a less compact structure compared to the uncut protein. In the presence of a reducing agent, both the 20.5- and the 13.7-kD bands are produced (lanes 8–11) and the ~36-kD band is not seen. These results suggest that there is a disulfide bond between residues C108 and C154 in αHL-A, and that this bond covalently links the amino and carboxyl terminal fragments following cleavage at Lys 131 by trypsin. All fragment sizes were confirmed by mass spectrometry (data not shown).
Figure 5.

SDS-PAGE of trypsin-digested αHL-A protein under reducing and nonreducing conditions. (A) The two most sensitive trypsin digestion sites (K8 and K131) in αHL-A are shown. (B) The digestion mixtures were analyzed in the absence of β-mercaptoethanol (lanes 2–6) or in the presence of β-mercaptoethanol (lanes 7–11). Lanes 2–6 and 7–11 are digestion mixtures with trypsin/protein ratios of 0, 1/5000, 1/500, 1/250, 1/50 (w/w), respectively.
αHL-A forms an oligomer in the presence of lipid-like micelles
We have shown that αHL-A does not have hemolytic activity under oxidizing conditions but is hemolytic under reducing conditions. To test whether αHL-A is trapped as a monomer or oligomer in the absence of a reducing agent but in the presence of lipids, the mutant protein was incubated with DiC8PC micelles and analyzed by SDS-PAGE (Fig. 6 ▶). Like deoxycholate (Bhakdi et al. 1981), DiC8PC micelles readily promote the assembly of αHL (Southall 1997). In the presence of a reducing agent, αHL-A oligomerized like WT-His6 (Fig. 6 ▶, right side of gel). Because the oligomer band size of αHL-A comigrates with the WT-His6 oligomer band, we assert that αHL-A forms a heptamer. However, in the absence of a reducing agent but in the presence of DiC8PC, there is no oligomer band for αHL-A lane (Fig. 6 ▶, left side of gel). These results indicate that αHL-A does not assemble to form a SDS-stable oligomer in the absence of a reducing agent. Nevertheless, αHL-A may assemble on a membrane but the assembled oligomer may be SDS-sensitive.
Figure 6.

Wild-type and αHL-A toxin assembly in the absence or presence of β-mercaptoethanol. The recombinant proteins (0.5 mg/mL) were incubated with 8 mM DiC8PC at room temperature for 1 h. Oligomerization was detected by SDS-PAGE +/− 0.7 M β-mercaptoethanol. The positions of molecular weight standards are shown on the left, and the numbers are in units of kD. The symbol “WT” indicates His-tagged wild-type protein.
To determine whether αHL-A assembles into a SDS-sensitive oligomer on a membrane without a reducing agent, αHL-A was crosslinked using glutaraldehyde (Peters and Richards 1977) and analyzed by SDS-PAGE. When αHL-A was incubated with the cross-linker in the absence of DiC8PC, there was no band corresponding to the αHL-A oligomer, with or without the cross-linker. In the presence of a reducing agent and DiC8PC, αHL-A formed an oligomer and the assembly was not dependent upon the presence of a cross-linker (Fig. 7 ▶ B, lanes 8–11 and 13–15), which is consistent with our previous results as shown in Figure 6 ▶. In the absence of a reducing agent, however, the detection of an assembled αHL-A oligomer was dependent upon the presence of the cross-linker (Fig. 7A ▶, lanes 8–11, 13–16). These results suggest that αHL-A assembles into a SDS-sensitive oligomer on a membrane, even in the absence of a reducing agent. Upon assembly under reducing conditions, there are two major bands on the gel at high molecular weight (Fig. 7 ▶, lanes 8–11 and 13–15), which may correspond to either the same stoichiometry of oligomer trapped in two conformational states or to oligomers of different subunit stoichiometry.
Figure 7.

SDS-PAGE of crosslinked αHL-A mutant oligomer under reducing and nonreducing conditions. αHL-A protein (1.3 mg/mL) was incubated with 8 mM DiC8PC (lanes 8–17) or without DiC8PC (lanes 3–7) at room temperature for 1 h with DTT (B) or without DTT (A). The resulting reaction mixtures were incubated with glutaraldehyde (GA) at room temperature either for 1 h (lanes 8–12) or 24 h (lanes 3–7, 13–17). Five different GA concentrations (0, 0.25, 0.5, 2.5, and 25 μM) were used for each set of lanes 3–7, 8–12, and 13–17, respectively. The triangle depicts the increase in GA concentrations.
Discussion
Structural information on assembly intermediates of transmembrane pore complexes have proven difficult to obtain. A promising approach to isolate and stabilize assembly intermediates involves the introduction of specific disulfide bonds between portions of the structure that undergo dramatic rearrangement during the transformation from the water-soluble to the membrane-bound species (Falke and Koshland 1987; Matsumura and Matthews 1989; Rossjohn et al. 1998; Hotze et al. 2001). In the present study, we created and screened four double cysteine mutants for the purpose of isolating αHL assembly intermediates. The idea was to find double cysteine mutants that formed a stable disulfide bond between the β-sandwich and the pre-stem domains, and prevented assembly and membrane insertion under oxidizing conditions while allowing wild-type like assembly and pore formation under reducing conditions. Because disulfide bonds have specific stereochemical requirements (Harrison and Sternberg 1996), we investigated the behavior of a number of mutants in an effort to select the one(s) that possessed the most favorable properties.
Among the four mutant proteins, we focused our research on αHL-A because it is inactive under oxidizing conditions and fully active under reducing conditions. The mutation sites in αHL-A are located between the β-sandwich domain and the neck of the pre-stem domain in the α1 form (Fig. 1A ▶). The pre-stem domain is probably quite flexible in the water-soluble state, not only because it contains numerous glycine residues and is protease sensitive, but also because it undergoes a dramatic rearrangement in the transition from α1 to α7. Because αHL-A has wild-type hemolytic potency in the presence of a reducing agent, the loss of hemolytic activity does not result from the disulfide bond simply disrupting the native structure in a nonspecific manner. Therefore, we conclude that the disulfide bond in αHL-A specifically connects the β-sandwich domain and the pre-stem domain, restraining the pre-stem and preventing assembly (Fig. 8 ▶).
Figure 8.

A schematic showing the arrest and the release of αHL-A. Water-soluble αHL-A monomer (α1) binds to a target membrane through the rim-domain (α1*) followed by prepore formation (α7*). αHL-A is arrested from forming a pore at this stage by the disulfide bond formed between the β-sandwich domain and the rim domain under oxidizing conditions. Under reducing conditions, however, the disulfide bridge between residues 108 and 154 is reduced, the pre-stem can rearrange, and αHL-A forms a heptameric pore (α7). In this schematic, only four protomers are depicted in the heptameric prepore (α7*) and the heptameric pore (α7) states.
The next question to address concerns the stage at which the assembly of αHL-A is arrested. The oligomerization assay showed that there was no oligomer band seen in the αHL-A lane in the absence of a reducing agent, indicating that either αHL-A does not form an oligomer or αHL-A forms an oligomer that is SDS-sensitive. To differentiate between these two possibilities, we cross-linked αHL-A with glutaraldehyde in the presence of DiC8PC micelles, conditions that allow for the assembly of the wild-type toxin (Southall 1997). As shown in Figure 7 ▶, oligomer bands were seen in the absence of a reducing agent but in the presence of a cross-linker, suggesting that αHL-A forms an oligomer on the membrane-like micelles (Fig. 7A ▶; lanes 10–12, 15, and 16). When αHL-A was incubated with DiC8PC in the presence of a reducing agent, two major oligomer bands were seen in the presence or absence of a cross-linker (Fig. 7B ▶; lanes 8–11, 13–15). These two different migration patterns may represent different conformational states, that is, the heptameric transmembrane pore and the heptameric prepore, or they may represent different oligomerization states, that is, heptamer versus octamer, for example. In the case of the I5C/G130C double mutant, Bhakdi and colleagues also found that under oxidizing conditions the oligomer migrated more slowly than under reducing conditions (Valeva et al. 2001). In the absence of a reducing agent, on the other hand, there was only one major oligomer band, which corresponds to the slower moving band, suggesting that αHL-A does not form a membrane pore but does form an oligomeric prepore. Therefore, our results support the idea that oligomerization precedes pore formation, and that αHL-A is arrested at the heptameric prepore (α7*) stage of membrane pore formation.
Efforts to conclusively determine whether αHL-A forms a heptamer in the absence of reducing agents but in the presence of a cross-linker have not been definitive. We attempted to obtain the molecular weight of the cross-linked species by mass spectrometry, but have not been able to observe species of such high molecular weight in the spectrometer. Estimation of the size of the complex in DiC8PC micelles by SEC has also been inconclusive. Indeed, determining the molecular mass of oligomeric membrane proteins that are in the 200–300-kD range is challenging, particularly for species that have been subject to chemical cross-linking. Nevertheless, one important observation that strongly indicates a specific complex is being cross-linked is that the cross-linking gels show primarily a single major band of mass corresponding approximately to a heptamer. The gels do not show a ladder of bands that would indicate the successive cross-linking of the monomer, dimer, trimer, etc. One would expect to see such a ladder of bands if the αHL-A protein were monomeric and simply forming transient, nonspecific contacts with other toxin molecules on the surface of the DiC8PC micelles.
In conclusion, we created and screened four double cysteine mutants to isolate an assembly intermediate of αHL. The αHL-A mutant is remarkably stable, and is expressed at a level at least 10-fold greater than that of the wild-type protein. The hemolytic and trypsin digestion analysis showed that αHL-A forms a disulfide bond between C108 and C154, and this disulfide bond prevents the toxin from forming a transmembrane pore. Furthermore, the cross-linking analysis showed that αHL-A assembles on a membrane into an oligomer, which is likely a heptamer, in the absence of a reducing agent, suggesting that αHL-A is halted at a heptameric prepore (α7*) state. Therefore, conformational rearrangements at positions 108 and 154 are critical to the completion of the assembly of αHL but are not essential for membrane binding or for formation of an oligomeric prepore intermediate.
Materials and methods
Primers
All primers used for the site-directed mutagenesis were purchased from Operon and purified by HPLC. They are A1F (αHL-A 1st mutation forward), CCAAGAAATTCGATTTGCACAAAAGAG; A1R (αHL-A first mutation reverse), CTCTTTTGTGCAAATCG AATTTCTTGG; A2F, CTGATTTCTGCACAATTTTAGAGAG CCC; A2R, GGCTCTCTAAAATTGTGCAGAAATCAGG; B1F, GATACAAAAGAGTATTGCAGTACTTTAACTTATGG; B1R, CCATAAGTTAAAGTA CTGCAATACTCTTTTGTATC; B2F, GGTCATACACTGTGCTATGTTCAACCTG; B2R, CAGGTT GAACATAGCACAGTGTATGACC; C1F, CGATGATAAAA ATTGCAATAAAAAACTGC; C1 R, GC AGTTTTTTATTG CAATTTTTATCATCG; C2F, CTTATGGATTCTGCGGTAAT GTTACTGGTG; C2R, CACCAGTAACATTACCGCAGAATC CATAAG; D1F, GGCAGATTCTGATTGCAATATTAAAAC CGG; D1R, CCGGTTTTAATATTGCAATCAGAATCTGCC; D2F, CTGGTGATGATACATGCAAAATTGGCGGCC; D2R, GGCCGCCAATTTTGCATGTATCATCACCAG.
Mutant DNA construction
All four double-cysteine mutants were made by Quickchange Site-Directed Mutagenesis (Stratagene). The first PCR was carried out for 18 cycles (95°C/30 sec, 50°C/1 min, 68°C/10 min) with 10 ng of pT7[αHL-His6] (His-tagged, wild-type αHL; generously provided by Hagan Bayley) as the template, 125 ng of primers (A1F/A1R, B1F/B1R, C1F/C1R, D1F/D1R), 1 μL of dNTPs (25 mM), 5 μL of 10× buffer, 40 μL of H2O, and 1 μL of PFU polymerase. The resulting single-cysteine mutants were confirmed by DNA sequencing through the whole αHL coding region and used as the template for the second PCR. The second PCR was carried out with the second sets of primers (A2F/A2R, B2F/B2R, C2F/C2R, D2F/D2R) for 18 cycles of 95°C/30 sec, 52°C/1 min, and 68°C/10 min. The entire αHL coding region of each double-cysteine mutant was confirmed by DNA sequencing.
Mutant protein purification
Mutant plasmids and pT7[αHL-His6] were separately transformed into Origami cells (Novagen; Prinz et al. 1997) and cultured at 37°C for 16–18 h in 40 mL of Luria-Bertani (LB) media containing 15 μg/mL of kanamycin, 50 μg/mL of ampicillin, and 12.5 μg/mL of tetracycline (LB KAT). Two liters of LB KAT were inoculated with the overnight culture and grown at 37°C for about 4 h until the OD600 reached 0.5. The temperature was then shifted to 20°C, and expression was induced with 0.1 mM isopropyl-β-d-thiogalactoside (IPTG). After 6 h of induction, cells were harvested by centrifugation at 4000 rpm for 20 min and resuspended in 30 mL of ice-cold lysis buffer (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 1 mM PMSF, 0.5 mM MgSO4, 0.25 mg/mL lysozyme, 25 μg/mL DNaseI). Cells were broken using a cell disrupter (Avestin), and the soluble fraction was collected by centrifugation at 45,000 rpm at 4°C for 45 min with a Ti-45 rotor. Following filtration through a 0.2-μm filter (Millipore), the soluble fraction was loaded onto a Ni-NTA column (5 mL, Amersham Pharmacia Biotech) at 4°C. After washing with 150 mL of Buffer A (50 mM Tris-Cl pH 8.0, 200 mM NaCl, 1 mM glutamate, 5 mM methionine), recombinant proteins were eluted with a gradient of 300 mM imidazole in Buffer A. Protein containing fractions (110–140 mM imidazole) were dialyzed against size-exclusion chromatography (SEC) buffer (20 mM Tris-Cl pH 8.0, 150 mM NaCl, 1 mM EDTA) overnight, concentrated to 0.5 mg/mL (estimated from OD280, ɛ = 63,500) using a Centricon YM10 (Amicon) and stored at −80°C.
SEC analysis
Recombinant proteins were concentrated to 1 mg/mL using a Microcon YM10 (Amicon) and 200 μL were loaded onto a Superose 12 10/30 column (Amersham Pharmacia Biotech) equilibrated with SEC Buffer at 4°C. A constant flow rate (0.5 mL/min) was used, and the elution patterns were detected by the absorbance at 280 nm.
Limited trypsin digestion
Trypsin digestion reactions (100 μL) were carried out by mixing 0.5 mg/mL of recombinant protein in SEC buffer (20 mM Tris-Cl pH 8.0, 150 mM NaCl, 1 mM EDTA) with a final concentration of 11 mM CaCl2 and trypsin/protein ratios of 0, 1/5000, 1/500, 1/250, 1/50 (w/w) at room temperature (rt) for 40 min. To stop the reactions, phenylmethylsulfonyl fluoride (PMSF) and EDTA were added to final concentrations of 1.5 and 20 mM, respectively, and the reactions were incubated for another 5 min at rt. The reaction mixtures were separated into two fractions (50 μL each) and mixed with 12.5 μL of 4× SDS sample loading buffer +/− 0.7 M β-mercaptoethanol. The digestions were analyzed by SDS-PAGE.
Hemolysis assays
Recombinant proteins (0.5 mg/mL) were pretreated with 10 mM dithiothreitol (DTT) in hemolysis (HL) buffer (20 mM KH2PO4 pH 7.4, 150 mM NaCl, 1 mg/mL bovine serum albumin) or with a reducing agent-free buffer at rt for 5 min. Hemolysis reactions were then carried out by mixing 20 μL of recombinant protein and 500 μL of rabbit blood cells (RBC; Animal Technologies, Inc) diluted in HL buffer to 12.5% (v/v) with or without 10 mM DTT at 20°C for 20 min (Fig. 3 ▶) or 0, 2, 4, 6, 8, and 10 min (Fig. 4A ▶). The reaction mixture was then centrifuged at 13,200 rpm (Eppendorf centrifuge 5415D) for 5 min, and the absorbance at 475 nm of the supernatant was measured spectrophotometrically.
Binding assay
Wild-type and αHL-A recombinant protein (0.5 mg/mL) were pretreated with 10 mM DTT in HL buffer or with a reducing agent-free buffer at rt for 5 min. The protein solution (20 μL) was then mixed with 200 μL of RBC diluted in HL buffer to 10% (v/v) with or without 10 mM DTT. The reaction mixture was incubated on ice for 1 h and centrifuged at 13,200 rpm (Eppendorf centrifuge 5415D) for 5 min at 4°C to separate the supernatant and pellet fractions. The resulting supernatant fraction was mixed with 35 μL of 6× SDS sample loading buffer, and the pellet fraction was resuspended in 40 μL of 1× SDS sample loading buffer. The amount of αHL was detected by Western blotting using anti penta-His antibody.
Oligomerization assays
Oligomerization reaction mixtures (100 μL) contained 8 mM dioctanoyl phosphatidylcholine (DiC8PC) and 0.5 mg/mL recombinant protein pretreated either with 10 mM DTT in SEC buffer (20 mM Tris-Cl pH 8.0, 150 mM NaCl, 1 mM EDTA) or with a reducing agent-free buffer at rt for 5 min. The reaction mixtures were incubated at rt for 1 h with stirring and were then mixed with 40 μL of 4× SDS sample loading buffer with or without 0.7 M β-mercaptoethanol. The oligomerization reactions were detected by SDS-PAGE.
Cross-linking assays
The αHL-A mutant was dialyzed against cross-linking (XL) buffer (20 mM sodium phosphate pH 8.0, 150 mM NaCl) three times and pretreated with 10 mM DTT in XL buffer or with a reducing agent-free buffer at rt for 5 min. αHL-A (1 mg/mL) was then incubated with or without 8 mM DiC8PC at rt for 1 h to allow for oligomer formation. The resulting reaction mixture (50 μL) was incubated with 50 μL of glutaraldehyde diluted in XL buffer (final concentrations: 0, 0.25, 0.5, 2.5, and 25 μM) at rt for either 1 h or 24 h. SDS-PAGE was carried out to detect crosslinked αHL-A.
Acknowledgments
We thank Michelle Montoya, Rich Olson, and Yu Sun for helpful discussions and critical comments on the manuscript. We also thank Hagan Bayley for valuable comments and for the gift of the pT7[αHL-His6] plasmid. This work was supported by a grant from the NIH. E.G. is an assistant investigator with the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
αHL, α-Hemolysin
Luk, leukocidin
γHL, γ-hemolysin
SEC, size-exclusion chromatography
βME, β-mercaptoethanol
DTT, dithiothreitol
DiC8PC, dioctanoyl phosphatidyl choline
GA, glutaraldehyde
WT-His6, wild-type α-hemolysin with a carboxyl terminal hexa-histidine tag
RBC, rabbit blood cells
SDS-PAGE, SDS polyacrylamide gel electrophoresis
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0231203.
References
- Bhakdi, S., Füssle, R., and Tranum-Jensen, J. 1981. Staphylococcal α-toxin: Oligomerization of hydrophilic monomers to form amphiphilic hexamers induced through contact with deoxycholate detergent micelles. Proc. Natl. Acad. Sci. 78 5475–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth, P.J., Templer, R.H., Meijberg, W., Allen, S.J., Curran, A.R., and Lorch, M. 2001. In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol. Biol. 36 501–603. [DOI] [PubMed] [Google Scholar]
- Czajkowsky, D.M., Sheng, S., and Shao, Z. 1998. Staphylococcal α-hemolysin can form hexamers in phospholipid bilayers. J. Mol. Biol. 276 325–330. [DOI] [PubMed] [Google Scholar]
- Falke, J.J. and Koshland, D.E. 1987. Global flexibility in a sensory receptor: A site-directed cross-linking approach. Science 237 1596–1600. [DOI] [PubMed] [Google Scholar]
- Fang, Y., Cheley, S., Bayley, H., and Yang, J. 1997. The heptameric prepore of a Staphylococcal α-hemolysin mutant in lipid bilayers imaged by atomic force microscopy. Biochemistry 36 9518–9522. [DOI] [PubMed] [Google Scholar]
- Gouaux, E. 1998. α-Hemolysin from Staphylococcus aureus: An archetype of β-barrel, channel-forming toxins. J. Struct. Biol. 121 110–122. [DOI] [PubMed] [Google Scholar]
- Gouaux, E., Hobaugh, M.R., and Song, L. 1997. α-Hemolysin, γ-hemolysin and leukocidin from Staphylococcal aureus: Distant in sequence but similar in structure. Protein Sci. 6 2631–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouaux, J.E., Braha, O., Hobaugh, M.R., Song, L., Cheley, S., Shustak, C., and Bayley, H. 1994. Subunit stoichiometry of staphylococcal α-hemolysin in crystals and on membranes: A heptameric transmembrane pore. Proc. Natl. Acad. Sci. 91 12828–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M. and Sternberg, M.J. 1996. The disulphide β-cross: From cystine geometry and clustering to classification of small disulphide-rich protein folds. J. Mol. Biol. 264 603–623. [DOI] [PubMed] [Google Scholar]
- Heuck, A.P., Tweten, R.K., and Johnson, A.E. 2001. β-barrel pore-forming toxins: Intriguing dimorphic proteins. Biochemistry 40 9065–9073. [DOI] [PubMed] [Google Scholar]
- Hildebrand, A., Pohl, M., and Bhakdi, S. 1991. Staphylococcus aureus α-toxin. Dual mechanism of binding to target cells. J. Biol. Chem. 266 17195–17200. [PubMed] [Google Scholar]
- Hotze, E.M., Wilson-Kubalek, E.M., Rossjohn, J., Parker, M.W., Johnson, A.E., and Tweten, R.K. 2001. Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane β-sheet from a prepore intermediate. J. Biol. Chem. 276 8261–8268. [DOI] [PubMed] [Google Scholar]
- Krasilnikov, O.V., Merzlyak, P.G., Yuldasheva, L.N., Rodrigues, C.G., Bhakdi, S., and Valeva, A. 2000. Electrophysiological evidence for heptameric stoichiometry of ion channels formed by Staphylococcus aureus α-toxin in planar lipid bilayers. Mol. Microbiol. 37 1372–1378. [DOI] [PubMed] [Google Scholar]
- Malghani, M.S., Fang, Y., Cheley, S., Bayley, H., and Yang, J. 1999. Heptameric structures of two α-hemolysin mutants imaged with in situ atomic force microscopy. Micros. Res. Technol. 44 353–356. [DOI] [PubMed] [Google Scholar]
- Matsumura, M. and Matthews, B.W. 1989. Control of enzyme activity by an engineered disulfide bond. Science 243 792–794. [DOI] [PubMed] [Google Scholar]
- Menestrina, G., Serra, M.D., and Prevost, G. 2001. Mode of action of β-barrel pore-forming toxins of the staphyloccal α-hemolysin family. Toxicon 39 1661–1672. [DOI] [PubMed] [Google Scholar]
- Miles, G., Movileanu, L., and Bayley, H. 2002. Subunit composition of a bicomponent toxin: Staphylococcal leukocidin forms an octameric transmembrane pore. Protein Sci. 11 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya, M. and Gouaux, E. 2003. β-Barrel membrane protein folding and structure viewed through the lens of α-hemolysin. Biochem. Biophys. Acta 1609 19–27. [DOI] [PubMed] [Google Scholar]
- Olson, R., Nariya, H., Yokota, K., Kamio, Y., and Gouaux, E. 1999. Crystal structure of Staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat. Struct. Biol. 6 134–140. [DOI] [PubMed] [Google Scholar]
- Palmer, M., Weller, U., Messner, M., and Bhakdi, S. 1993. Altered pore-forming properties of proteolytically nicked staphylococcal α-toxin. J. Biol. Chem. 268 11963–11967. [PubMed] [Google Scholar]
- Pédelacq, J.-D., Maveyraud, L., Prévost, G., Baba-Moussa, L., González, A., Courcelle, E., Shepard, W., Monteil, H., Samama, J.-P., and Mourey, L. 1999. The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins. Structure 7 277–287. [DOI] [PubMed] [Google Scholar]
- Pédelacq, J.D., Prevost, G., Monteil, H., Mourey, L., and Samana, J.P. 2000. Crystal structure of the F component of the Panton-Valentine leucocidin. Int J. Med. Microbiol. 290 395–401. [DOI] [PubMed] [Google Scholar]
- Peitsch, M.C. 1996. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans. 24 274–279. [DOI] [PubMed] [Google Scholar]
- Peitsch, M.C. and Tschopp, J. 1995. Comparative molecular modelling of the Fas-ligand and other members of the TNF family. Mol. Immunol. 32 761–772. [DOI] [PubMed] [Google Scholar]
- Peters, K. and Richards, F.M. 1977. Chemical cross-linking: Reagents and problems in studies of membrane structure. Annu. Rev. Biochem. 46 523–551. [DOI] [PubMed] [Google Scholar]
- Prevost, G., Mourey, L., Colin, D.A., and Menestrina, G. 2001. Staphylococcal pore-forming toxins. Curr. Top. Microbiol. Immunol. 257 53–83. [DOI] [PubMed] [Google Scholar]
- Prinz, W.A., Aslund, F., Holmgren, A., and Beckwith, J. 1997. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in Escherichia coli cytoplasm. J. Biol. Chem. 272 15661–15667. [DOI] [PubMed] [Google Scholar]
- Rossjohn, J., Raja, S.M., Nelson, K.L., Feil, S.C., van der Goot, F.G., Parker, M.W., and Buckley, J.T. 1998. Movement of a loop in domain 3 of aerolysin is required for channel formation. Biochemistry 37 741–746. [DOI] [PubMed] [Google Scholar]
- Song, L., Hobaugh, M.R., Shustak, C., Cheley, S., Bayley, H., and Gouaux, J.E. 1996. Structure of Staphyloccal α-hemolysin, a heptameric transmembrane pore. Science 274 1859–1866. [DOI] [PubMed] [Google Scholar]
- Southall, N.T. 1997. Phosphatidyl choline micelle catalysis of staphylococcal α-hemolysin. MS. thesis, University of Chicago, Chicago, IL.
- Sugawara-Tomita, N., Tomita, T., and Kamio, Y. 2002. Stochastic assembly of two-component Staphylococcal γ-hemolysin into heteroheptameric transmembrane pores with alternate subunit arrangements in ratios of 3:4 and 4:3. J. Bacteriol. 184 4747–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita, T. and Kamio, Y. 1997. Molecular biology of pore-forming cytolysins from Staphylococcus aureus, α- and γ-hemolysins and leukocidin. Biosci. Biotechnol. Biochem. 61 565–572. [DOI] [PubMed] [Google Scholar]
- Valeva, A., Palmer, M., and Bhakdi, S. 1997a. Staphylococcal α-toxin: Formation of the heptameric pore is partially cooperative and proceeds through multiple intermediate stages. Biochemistry 36 13298–13304. [DOI] [PubMed] [Google Scholar]
- Valeva, A., Pongs, J., Bhakdi, S., and Palmer, M. 1997b. Staphylococcal α-toxin: The role of the N-terminus in formation of the heptameric pore—A fluorescence study. Biochim. Biophys. Acta 1325 281–286. [DOI] [PubMed] [Google Scholar]
- Valeva, A., Schnabel, R., Walev, I., Boukhallouk, F., Bhakdi, S., and Palmer, M. 2001. Membrane insertion of the heptameric staphyloccal α-toxin pore. A domino-like structural transition that is allosterically modulated by the target cell membrane. J. Biol. Chem. 276 14835–14841. [DOI] [PubMed] [Google Scholar]
- Vécsey-Semjén, B., Lesieur, C., Möllby, R., and van der Goot, F.G. 1997. Conformational changes due to membrane binding and channel formation by Staphylococcal α-toxin. J. Biol. Chem. 272 5709–5717. [DOI] [PubMed] [Google Scholar]
- Walker, B. and Bayley, H. 1995a. Key residues for membrane binding, oligomerization, and pore forming activity of Staphylococcal α-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 270 23065–23071. [DOI] [PubMed] [Google Scholar]
- ———. 1995b. Restoration of pore-forming activity in staphylococcal α-hemolysin by targeted covalent modification. Protein Eng. 8 491–495. [DOI] [PubMed] [Google Scholar]
- Walker, B., Krishnasastry, M., Zorn, L., and Bayley, H. 1992. Assembly of the oligomeric membrane pore formed by Staphylococcal α-hemolysin examined by truncation mutagenesis. J. Biol. Chem. 267 21782–21786. [PubMed] [Google Scholar]
- Walker, B., Braha, O., Cheley, S., and Bayley, H. 1995. An intermediate in the assembly of a pore-forming protein trapped with a genetically-engineered switch. Chem. Biol. 2 99–105. [DOI] [PubMed] [Google Scholar]