

Abstract
Pantothenate biosynthesis is essential for the virulence of Mycobacterium tuberculosis, and this pathway thus presents potential drug targets against tuberculosis. We determined the crystal structure of pantothenate synthetase (PS) from M. tuberculosis, and its complexes with AMPCPP, pantoate, and a reaction intermediate, pantoyl adenylate, with resolutions from 1.6 to 2 Å. PS catalyzes the ATP-dependent condensation of pantoate and β-alanine to form pantothenate. Its structure reveals a dimer, and each subunit has two domains with tight association between domains. The active-site cavity is on the N-terminal domain, partially covered by the C-terminal domain. One wall of the active site cavity is flexible, which allows the bulky AMPCPP to diffuse into the active site to nearly full occupancy when crystals are soaked in solutions containing AMPCPP. Crystal structures of the complexes with AMPCPP and pantoate indicate that the enzyme binds ATP and pantoate tightly in the active site, and brings the carboxyl oxygen of pantoate near the α-phosphorus atom of ATP for an in-line nucleophilic attack. When crystals were soaked with, or grown in the presence of, both ATP and pantoate, a reaction intermediate, pantoyl adenylate, is found in the active site. The flexible wall of the active site cavity becomes ordered when the intermediate is in the active site, thus protecting it from being hydrolyzed. Binding of β-alanine can occur only after pantoyl adenylate is formed inside the active site cavity. The tight binding of the intermediate pantoyl adenylate suggests that nonreactive analogs of pantoyl adenylate may be inhibitors of the PS enzyme with high affinity and specificity.
Keywords: M. tuberculosis, pantothenate synthetase, substrate complexes, reaction intermediate complex, dimer
Pantothenate (vitamin B5) is an essential precursor for the biosynthesis of coenzyme A (CoA) and acyl carrier proteins (ACP). Both CoA and ACP play critical roles in many cellular processes such as energy metabolism and fatty acid metabolism (Jackowski 1996). Micro-organisms and plants must synthesize pantothenate, while animals obtain this essential nutrient from their diet (Maas 1960). Therefore, the pantothenate biosynthetic pathway offers targets for developing drugs against microbial pathogens. Tuberculosis (TB) is becoming a global health threat due to the emergence of multidrug resistant strains of Mycobacterium tuberculosis (MTB) and the deadly synergy of TB with HIV (Grosset 1996). This points to the critical need to develop new drugs against novel targets of MTB. Because both CoA and ACP are essential in fatty acid biosynthesis that plays a key role in persistent growth and pathogenicity of MTB, pantothenate synthetase is an appropriate target for developing new drugs against TB. Recently, Jacobs and coauthors (Sambandamurthy et al. 2002) reported that an auxotrophic mutant of MTB defective in the de novo biosynthesis of pantothenate is highly attenuated both in immunocompromised mice and in immunocompetent mice. This indicates that a functional pantothenate biosynthetic pathway is essential for virulence of MTB.
The pantothenate biosynthetic pathway is best characterized in Escherichia coli. It involves four steps catalyzed by enzymes encoded by panB, panC, panD, and panE genes (Merkel and Nichols 1996). PanC encodes a pantothenate synthetase (PS), which catalyzes the last step of pantothenate biosynthesis, the ATP-dependent condensation of pantoate, and β-alanine to form pantothenate. E. coli PS was found to be a dimer from the crystal structure (von Delft et al. 2001), although earlier studies indicated that it could be a tetramer in solution (Miyatake et al. 1978). The panC gene product in several other organisms has also been identified and characterized, including that from Saccharomyces cerevisiae, higher plants Oryza sativa, and Lotus japonicus (Genschel et al. 1999), fungus Fusarium oxysporum (Perez-Espinosa et al. 2001), and M. tuberculosis (Zheng and Blanchard 2001). The PS enzymes from the higher plants and from MTB have also been found to be dimers in solution.
Zheng and Blanchard (2001) purified the MTB PS enzyme, and analyzed its kinetics. They found the kinetic mechanism to be “Bi Uni Uni Bi Ping Pong,” with ATP binding followed by pantoate binding, release of pyrophosphate, and then binding of β-alanine followed by the release of pantothenate and AMP. The enzyme-catalyzed reaction proceeds through two steps: the formation of an enzyme bound intermediate, pantoyl adenylate, from ATP and pantoate, followed by nucleophilic attack on the intermediate by β-alanine to form pantothenate and AMP. The existence of pantoyl adenylate has been confirmed by 31P NMR spectroscopy.
The E. coli PS enzyme structure was found to belong to the cytidylyltransferase superfamily (von Delft et al. 2001). It has two distinct domains: a large N-terminal domain having a Rossmann fold, and a smaller C-terminal domain of two layers with a helical layer on top of a three-stranded antiparallel β-sheet. Based on the structural comparison of the E. coli PS with other members of cytidylyltransferase superfamily having known structures, the authors deduced the ATP and pantoate binding sites of the E. coli PS, and proposed a hinged domain mechanism for opening and closing of the enzyme active site cavity.
In this study, we determined the crystal structure of the MTB PS enzyme, and its complexes with AMPCPP, a nonhydrolysable analog of ATP, with pantoate, and with a reaction intermediate, pantoyl adenylate. The MTB PS enzyme has the same fold as the E. coli enzyme, but the domains have a closed conformation in the apo enzyme and all the complexes, in contrast to the domain movement proposed by von Delft et al. (2001). Based on these structures, we propose an alternative mechanism for the MTB PS enzyme to open and close the active site cavity, in which a flexible region acts as a gate to the active site cavity. In addition, we propose potential inhibitors to the PS enzyme based on the structure of the reaction intermediate complex.
Results
Expression and purification of the MTB PS
The M. tuberculosis panC gene (Rv3602c) encoding the pantothenate synthetase was cloned into a pET30a expression vector for overexpression in E. coli, which produced a recombinant protein with an N-terminal tag of 44 amino acid residues. The N-terminal tag was cleaved by enterokinase digestion. Electrospray mass spectroscopy analysis of the protein gave a molecular weight of 31,611 ± 6 Daltons, indicating that nine residues from the C-terminus of the protein were also cleaved off by enterokinase digestion (calculated molecular weight of 31,610 Daltons). During PCR cloning, a few single point mutants were also generated from the PCR reactions. One of the mutants, E77G, in which the Glu at position 77 was replaced by a Gly (sequence number refers to that in the panC gene), was used for all the crystal structure studies in this article.
Pantothenate synthetase activity assays
To find out the effect of the C-terminal truncation, as well as that of the E77G mutation, on enzyme activity, we performed the pantothenate synthetase activity assays on the wild-type and E77G mutant proteins, both before and after enterokinase digestion. The results are listed in Table 1. The wild-type and E77G mutant have essentially identical activity; thus the mutation of Glu77 to a Gly does not have any detectable effect on the enzyme activity, under the experimental conditions. For both wild-type and E77G mutant enzymes, enterokinase digestion does not affect the enzyme activity. Therefore, the presence of the N-terminal tag of 44 residues or deletion of nine residues from the C-terminus does not affect the catalytic function of the enzyme.
Table 1.
Pantothenate synthetase activity assays (sec−1)
| Full lengtha | Truncatedb | |
| Wild type | 1.74 ± 0.12 | 1.66 ± 0.15 |
| E77G | 1.52 ± 0.02 | 1.58 ± 0.34 |
| Butyratec | ||
| 10 mM | 1.41 | |
| 50 mM | 1.11 | |
| 100 mM | 0.973 | |
| Glycerolc | ||
| 274 mM | 1.51 | |
| 685 mM | 1.51 | |
a Full-length protein is the protein before enterokinase digestion, with the N-terminal fusion tag.
b Truncated protein is the protein after enterokinase digestion.
c The effects of Na butyrate and glycerol on the activity are measured with the full-length E77G mutant protein. These are single measurements.
We also assayed the enzymatic activity in the presence of glycerol or butyrate. From electron density maps, we found that a few glycerol molecules bound in the protein, including one tightly bound at the pantoate binding site (see below). Therefore, activity assays were performed in the presence of glycerol. However, up to 5% of glycerol (685 mM) did not have any measurable effect on the enzyme activity (Table 1). Activity assays in the presence of sodium butyrate were also carried out to see whether butyrate can inhibit the enzyme activity by competing for the binding of β-alanine. The results indicate that butyrate inhibits the enzyme activity only very weakly (Table 1). In the presence of 100 mM butyrate, the enzyme still retained more than one half of its activity.
Overall structure
The crystal structure of the MTB PS enzyme indicates that it is a dimer (Fig. 1 ▶). This is consistent with our gel filtration studies (data not shown), as well as the finding of Zheng and Blanchard (2001), which indicated that the protein is a dimer in solution. Each subunit of the dimer has two well-defined domains. The N-terminal domain runs from residue 1 to 186, and forms a Rossmann fold. For simplicity, the β-strands and α-helices are numbered consecutively from N-terminus to C-terminus, except helix α3′ which is in a region that is ordered in one subunit (designated subunit A), but disordered in the other (subunit B). 310 helices are numbered according to the preceding β-strands. Strands β1 to β4, β6, and β7 form a highly twisted central parallel β-sheet that is flanked by α-helices. Strand β5 is at the dimer interface and forms a two-stranded intersubunit β-sheet with the corresponding β5 from the other subunit (Fig. 1B ▶).
Figure 1.
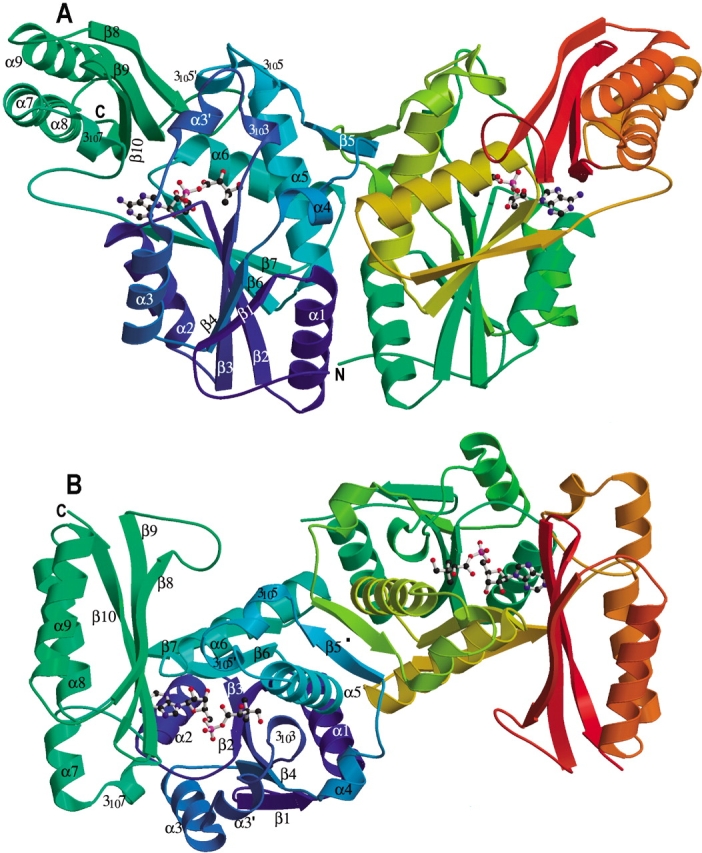
Ribbon diagram of the M. tuberculosis pantothenate synthetase dimer. (A) A side view of the dimer structure showing that it resembles the shape of a butterfly. (B) An orthogonal view of (A) from top, with the twofold NCS symmetry axis (labeled with a dot) approximately perpendicular to the paper plane. Secondary structure elements for the subunit A (left) are labeled. Those for subunit B are identical except that the short helix α3′ is not present. The figure was prepared from the coordinates of the intermediate complex (data set 5), with the program Molscript (Kraulis 1991) and Raster3D (Merritt and Murphy 1994). The molecule in the active site of each subunit, shown in ball-and-stick, is the reaction intermediate, pantoyl adenylate.
Strand β7 leads directly to the C-terminal domain, which starts with a hairpin loop followed by helix 3107. Helix α7 and α8 are nearly a straight continuous helix, except that there is a one-turn 310 helix between them to make a kink in the otherwise continuous α-helix. A tight turn connects helix α8 to α9, which is followed by three antiparallel β-strands that form a nearly flat layer. The two-layered C-terminal domain partially covers the active site cavity on the N-terminal domain.
Besides the nine residues that were digested off by enterokinase, there are 10 more residues from the C-terminus that do not have clear electron density. Based on its susceptibility to protease digestion and lack of clear electron density, it is reasonable to assume that these residues from the C-terminus of the protein are flexible, and do not have a defined structure in solution. It is clear from the structure that both the N- and C-termini are away from the active site cavity, and therefore, are unlikely to affect pantothenate synthesis. This is consistent with the results of activity assays that neither removal of the last nine residues by enterokinase digestion, nor the fusion of the enzyme to a 44-residue N-terminal tag affects the enzymatic activity.
Domain interactions
Residues on the β-strands of the C-terminal domain have extensive interactions with the N-terminal domain. There is a hydrophobic core composed of residues Leu123, Leu127, Tyr162, Val166, all in the N-terminal domain and residues Tyr249, Leu257, and Leu269 of the C-terminal domain (Fig. 2 ▶). Surrounding this hydrophobic core are three salt bridges, Glu159 to Arg267, Glu128 to Arg278, Glu126 to Arg253, and six additional hydrogen bonds linking the two domains. In addition, there are several water-mediated hydrogen bonding interactions. The relative positions of the two domains stay the same for the two subunits in the dimer and for all crystals including the one cocrystallized with β-alanine, which has a different space group (P212121) and solvent conditions.
Figure 2.

Sequence alignment between the E. coli and M. tuberculosis PS proteins. The sequences were aligned with CLUSTALW (Thompson et al. 1994) and the figure was generated using ALSCRIPT (Barton 1993). Identical residues are highlighted in black. Helices and β-strands of the two structures are marked with cylinders and arrows, respectively. The secondary structure elements and numbering for the E. coli PS are from von Delft et al. (2001). Residues involved in binding of AMPCPP, pantoate, or pantoyl adenylate are boxed in thin lines, while those involved in interactions between N- and C-terminal domains are boxed in thick lines in the M. tuberculosis sequence. A vertical arrow marks the start of the C-terminal domain. All residues involved in binding of substrates and the reaction intermediate are conserved between two sequences, indicating that they have identical reaction mechanism. However, residues involved in domain interactions are not all conserved. See text for more details.
Dimerization
The dimer of the MTB PS resembles a butterfly when viewed from one side (Fig. 1A ▶). The dimer interface is extensive, having a buried area of about 2150 Å2 measured with the program CNS (Brunger et al. 1998). A cluster of eight hydrophobic side chains at the center of the dimer interface form the core of the dimer interaction, Phe175, Leu 177, Leu144, and Val118. This cluster is surrounded by hydrogen bonds and salt bridges, as well as the specific β-strand interactions of the two-stranded intersubunit β-sheet that is composed of the strand β5 from each subunit. A patch of arginine residues lie at the bottom of the protein near the dimer interface, most of them partially disordered. These arginine residues form a large patch of positively charged surface at one side of the molecule. We can speculate that they interact with other molecules, or regulate enzyme activity.
The two subunits of the dimer are similar to each other, with an RMSD of 0.9 for all Cα-atoms except the disordered residues. Most of the deviation lies at residues 74 to 88, and residues 260 to 265, where the crystal packing environments are different. Residues 74 to 88 (around helix α3′) form one wall of the active site cavity. In subunit A these residues are ordered with residues 74 to 76 having high B factors, while in subunit B residues 75 to 80 do not have clear electron density, and the rest of the residues up to residue 88 are partially disordered with high B factors. These disordered residues became ordered in the complex with the reaction intermediate (Fig. 3 ▶), suggesting their function as a gate to the active site cavity (see more discussions below).
Figure 3.
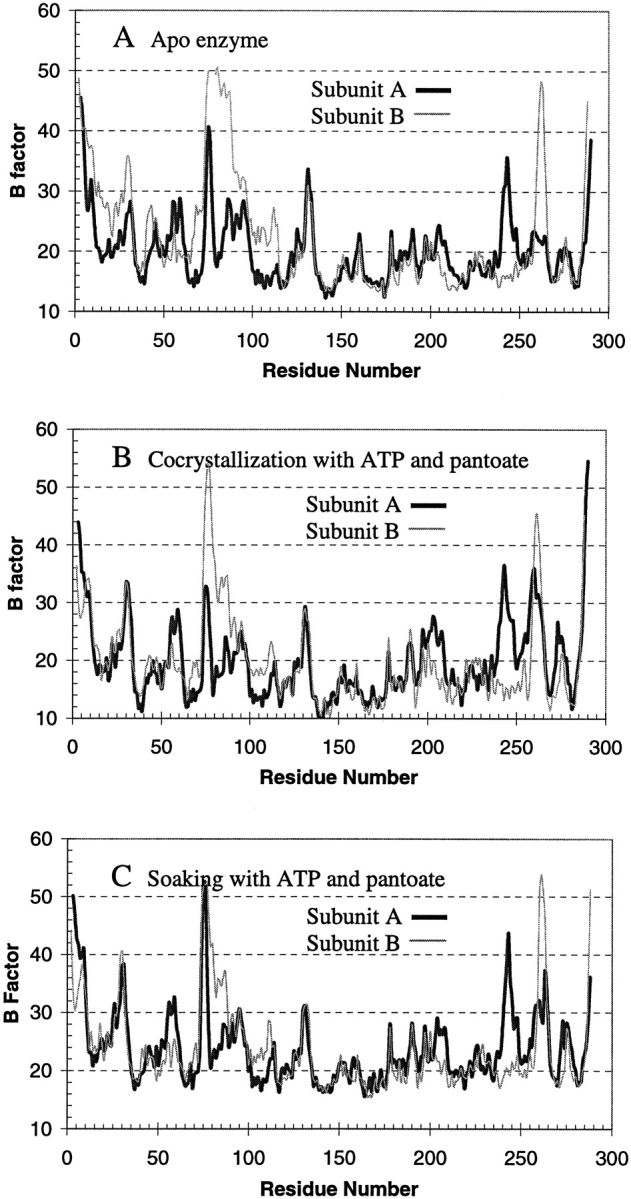
Average B factors of main chain atoms plotted against residue number. The dark thick curves are those of subunit A, and the light thin curves are those of subunit B. Subunit B of the apo enzyme (A, thin curve) has high B factors for residues from 75 to 88, with residues 76–78 totally disordered. However, these residues are ordered and have lower B factors in the crystal structure of the reaction intermediate complex from cocrystallization (B). Soaking of the apo enzyme crystals in solutions containing ATP and pantoate (C) also resulted in the disordered residues becoming ordered, and gave a profile of the B factors more similar to that of cocrystallized intermediate complex (B) than to that of the apo enzyme (A).
Active site cavity
Typical for nucleotide binding (Rossmann fold) proteins, the active site is located at the C-terminus of the central parallel β-sheet. The active site cavity is large and is in a cleft between strands β2 and β6 (Fig. 4A ▶). One wall is formed by β2-loop-α2 and the C-terminus of β7 and the following loop. The other wall is formed by β6-loop-α6 and 3105′-loop-α5. The flexible residues in the loop-3103-loop-α3′-loop region (termed flexible wall) fill a gap. Helix 3107 and the β-strands from the C-terminal domain partially cover the top of the active site cavity. The bottom of the active site cavity is mainly hydrophobic, while the top half of the cavity has several charged residues, including His44 and His47 at the N-terminus of α2 (the HIGH motif; Barker and Winter 1982; von Delft et al. 2001), Lys160 and Asp161 at the N-terminus of α6, and Arg198 of 3107 (KMSKS motif; von Delft et al. 2001). These charged residues are important in binding of ATP and in the catalytic reaction. Arg198 is near the surface at the entrance of the active site cavity. Together with three other arginine side chains, Arg132, Arg273, and Arg278, they form a patch of positive charges covering the active site cavity. These positively charged side chains may steer the negatively charged substrates to the active site cavity.
Figure 4.




Active site cavity and the binding of AMPCPP, pantoate, and pantoyl adenylate. (A) A stereo view of the active site cavity of subunit A of the complex with both AMPCPP and pantoate. The substrates (both with partial occupancy) are shown as ball-and-stick models. The active site cavity is surrounded by β2-loop-α2, β7-loop, β6-loop-α6, 3105′-loop- α5, and β3-loop-3103–α3′-loop, and covered by 3107 and the β-sheet of C-terminal domain. Residues around helix α3′ (shown in cyan) are disordered in subunit B, which has a fully occupied AMPCPP and a glycerol molecule in the active site. (B) A section of the initial difference electron density map (Fo − Fc) in the active site of subunit B superimposed on the refined model, calculated at 1.7 Å and contoured at the 2σ level. Side chains of Lys160, Ser196, and Arg198 have moved relative to those in the apo enzyme to interact with the phosphate groups, and thus also have positive initial difference electron density. The electron density figures are prepared with PYMOL (DeLano 2002). (C) Detailed binding interactions between AMPCPP (shown with carbon atoms in gold) and protein active site residues of subunit B. The Mg2+ ion is shown as a yellow sphere, and water molecules are shown as red spheres. Hydrogen bonds between AMPCPP and protein atoms, and some water-mediated hydrogen bonds are shown as dashed lines. A glycerol molecule found next to the α-phosphate of AMPCPP, at the pantoate binding site, is also shown. (D) A section of the initial difference electron density (Fo − Fc) around the bound pantoate molecule in the active site of subunit A of the pantoate–β-alanine complex (data set 7) shows that pantoate is very well ordered with full occupancy. The nearby residues did not move relative to those of the apo enzyme, and therefore did not have initial difference density. The electron density was calculated at 1.7 Å and contoured at 2σ. (E) The pantoate molecule (shown in gold for the carbon atoms) is tightly bound and fits snugly in its binding site. Two glutamine side chains form hydrogen bonds to the hydroxyl groups and one carboxyl oxygen of the pantoate. The two methyl groups and the hydrophobic side of pantoate interact with the side chains of Pro38, Met40, and Phe157. (F) A section of the initial difference electron density (Fo − Fc) around the bound pantoyl adenylate molecule in the active site of subunit B of the intermediate complex (data set 6) shows that intermediate is very well ordered with full occupancy. The electron density was calculated at 1.7 Å and contoured at 2σ. (G) The pantoyl adenylate molecule (shown with carbon atoms in gold) is tightly bound and fits snugly in the active site cavity. The adenosine and pantoyl groups are at identical positions as those in the AMPCPP complex and pantoate complex, respectively, and have identical interactions with the active site residues. However, the α-phosphate moved down to have a covalent bond to the pantoate, which allows the phosphate group to have a hydrogen bond to the amide nitrogen of Met40.
ATP binding interactions
When crystals were soaked in solutions containing AMPCPP, difference electron density that fits the AMPCPP molecule was found in the active site cavity (Fig. 4B ▶). The occupancy of the bound AMPCPP depends on the soaking time. Soaking for less than 8 h gave only very low occupancy of AMPCPP in the active site cavity of subunit B and no density for AMPCPP found in subunit A. However, overnight soaking resulted in good occupancy of AMPCPP in both active sites of the dimer (Table 2, data set 2). Soaking crystals in solutions containing both AMPCPP and pantoate gave a fully occupied AMPCPP, but with a glycerol in the pantoate binding site (see below) in subunit B, which has a flexible wall (residues 75–88). However, subunit A has partial occupancy of both AMPCPP and pantoate, as indicated by their electron densities and B factors. Increasing the concentration of pantoate in the soaking solution gave only a slightly higher occupancy of pantoate but a lower occupancy of AMPCPP (Table 2, data set 4 versus data set 3), indicating that AMPCPP and pantoate cannot coexist in the active site.
Table 2.
Atomic refinement of models for M. tuberculosis pantothenate synthetase and complexes
| Data set | 1 (Apo) | 2 (AMPCPP) | 3a (AMPCPP + pantoate) | 4a (AMPCPP + pantoate) | 5 (ATP + pantoate) | 6b (ATP + pantoate + butyrate) | 7 (pantoate + β-alanine) | 8c (β-alanine) |
| Resolution (Å) | 20–1.6 | 20–1.8 | 20–1.6 | 20–1.7 | 20–2.0 | 20–1.7 | 20–1.8 | 20–.21 |
| Rwork (Rfree)d | 19.2 (21.1) | 19.2 (22.3) | 18.8 (21.0) | 18.4 (21.5) | 18.2 (21.8) | 18.2 (20.4) | 19.3 (22.5) | 20.2 (23.7) |
| rmsd bonds (Å) | 0.010 | 0.010 | 0.011 | 0.013 | 0.014 | 0.014 | 0.013 | 0.012 |
| rmsd angles (°) | 1.52 | 1.46 | 1.56 | 1.59 | 1.58 | 1.59 | 1.60 | 1.53 |
| No. protein atoms | 4223 | 4228 | 4204 | 4184 | 4239 | 4221 | 4228 | 4121 |
| Ave B factors, protein atoms (Å2) | 2.6 | 28.8 | 25.0 | 25.4 | 20.9 | 25.4 | 32.6 | 33.7 |
| No. other molecules (Ave. B factors, Å2) | 4 glycerol (31.3) 4 sulfate (45.5) 5 ethanol (40.9) 469 water (34.2) |
2 AMPCPP (40.7, 30.4)c 2 sulfate (79.9) 2 Mgf (76.8, 52.5) 5 glycerol (40.3) 4 ethanol (46.6) 305 water (36.3) |
2 AMPCPP (41.4, 23.0)e 1 pantoate (35.2) 2 sulfate (58.0) 1 Mg (27.6) 2 glycerol (26.6) 4 ethanol (44.3) 408 water (35.6) |
2 AMPCPP (47.5, 22.4)e 1 pantoate (32.1) 1 sulfate (54.6) 2 Mg (62.9, 28.1) 3 glycerol (28.5) 5 ethanol (41.7) 344 water (35.8) |
2 pantoyl adenylate (18.3, 17.2)e 1 glycerol (18.3) 3 sulfate (63.0) 5 ethanol (33.7) 1 Mng (73.6) 299 water (29.1) |
2 pantoyl adenylate (28.3, 19.9)e 1 glycerol (23.9) 3 sulfate (59.1) 5 ethanol (41.3) 401 water (35.9) |
2 pantoate (28.3, 32.4)e 2 β-alanine (54.1, 50.8) 3 glycerol (44.2) 3 sulfate (66.0) 5 ethanol (50.0) 305 water (40.5) |
2 β-alanine (51.2, 58.6)e 2 sulfate (64.9) 192 water (36.5) |
a See Table 3 for differences in experimental conditions between data sets 3 and 4. Relative to data set 4, data set 3 has a lower B factor for the AMPCPP molecule bound in the active site of subunit A, but a higher B factor of the pantoate molecule (also bound in subunit A).
b Butyrate was not located in the electron density maps due to its low binding affinity. Soaking experiment (data set 6) have a higher B factor for the pantoyl adenylate in the active site of subunit A, while cocrystallization experiment (data set 5) gave two pantoyl adenylate molecules with about equal B factors.
c Cocrystallization with β-alanine gave two weakly bound β-alanine molecules in the active sites. However, they are found at the pantoate binding sites instead.
dR-factors were calculated using data in the resolution range for refinement without a σ cutoff. Rfree was calculated with a subset of data (8 percent) never used in the refinement. Rwork was calculated against the data used in the refinement.
e When two numbers are listed for the average B factors, the first number refers to the average B factor for the molecule found in the active site of subunit A. The B factors were refined with occupancy of 1 for the atoms.
f Magnesium ions are identified from their binding to the phosphate groups and geometry.
g Positive electron density was found between an aspartate side chain and a glutamate side chain at a crystal packing interface, with distances to one oxygen atom of each side chain of about 2.2 Å. Because Mn2+ is present in the crystallization solution, a Mn2+ was modeled into the electron density. The high B factor probably indicates low occupancy of the ion. That is probably why clear electron density was not found in other crystals grown in the presence of Mg2+, although they have identical crystal packing.
The active site is well designed for the binding of ATP. There is very little movement of the residues when AMPCPP is bound. Except for the residues on the flexible wall, there is no movement of more than 0.4 Å of the main chain atoms around the active site cavity. The AMPCPP molecule bound in the active site has a horse-shoe shape (Fig. 4C ▶). Its adenine group is flanked by Gly46 on helix α2 and Lys160 on the loop after β6. The N1 atom of adenine forms a hydrogen bond to the amide nitrogen of Val187. The N6 atom forms hydrogen bonds to the carbonyl oxygen of Met195 and Val187. The N3 atom faces the bottom of the active site cavity, which has two hydrophobic side chains, Val184 and Leu50. The ribose O2* atom has a hydrogen-bond to the Oδ2 atom of Asp161, and the O3* atom forms a hydrogen bond to the amide nitrogen of Gly158, and water-mediated hydrogen bonds to the carbonyl oxygen atoms of Phe156 and Pro38. The phosphate groups turn back towards the top of the active site cavity to the C-terminal domain, and are located near the N-terminal ends of helices α2 and 3107. Binding of the phosphate groups involves mainly charged side chains. The side chains of Lys160 and Arg198 are partially disordered in the apo enzyme structure, but become ordered and move to enable salt bridges to the β- and γ-phosphate groups of AMPCPP, respectively. The O3γ atom also forms hydrogen bonds to the amide nitrogen of Arg198 and the Oγ of Ser196. The β-phosphate group has hydrogen bonds to the side chains of His44 and Lys160. The α-phosphate does not have direct hydrogen bonds with the protein atoms, but water-mediated hydrogen bonds to amide nitrogen of Met40, carbonyl oxygen of Gly41 and Nɛ2 of His47 side chain. The Nɛ2 atom of His47 is ~3.4 Å from the carbon atom between the α- and β-phosphate of AMPCPP, and is therefore likely to function as a proton donor to the leaving pyrophosphate group during the enzyme-catalyzed reaction.
A solvent peak positioned as though it is a magnesium ion was found adjacent to the O1γ, O2β, and O2α atoms of the phosphate groups at distances of 2.06, 2.08, and 2.09 Å, respectively. It also has two water molecules at distances of 2.03 and 2.09 Å. These two water molecules form hydrogen bonds to the side chain oxygen atoms of Asp161 with normal hydrogen bond distances of 2.67 and 2.80 Å. A third water molecule was found to give octahedral coordination to the magnesium ion. However, this water is weakly occupied and has normal hydrogen bond distance of 2.66 Å to the magnesium.
Pantoate binding site
In the crystal that was soaked with pantoate and β-alanine, there is a fully occupied pantoate molecule in both of the active sites (Fig. 4D ▶). The binding site for pantoate is located in a pocket deep in the active site cavity. This is the same binding site proposed for the E. coli PS protein (von Delft et al. 2001), although the pantoate molecule has a different conformation and therefore different binding interactions. The pantoate molecule is tightly bound, with side chains of two glutamine residues, Gln72 and Gln164 forming hydrogen bonds to its hydroxyl groups and one carboxyl oxygen (Fig. 4E ▶). The two methyl groups face the apolar groups at the bottom of the active site cavity, Pro38 and Phe157 side chains. The side chain of Met40 packs against the apolar side of the pantoate molecule. This pantoate binding site in the apo enzyme is occupied by a glycerol molecule. The binding interactions of the glycerol molecule are similar to those of the pantoate molecule, with the two glutamine side chains having hydrogen bonds to the three hydroxyl groups and the apolar side of glycerol interacting with the apolar side chains. As stated above, crystals soaked in solutions containing both AMPCPP and pantoate gave only partially occupied pantoate in subunit A (Table 2).
Binding interactions of the reaction intermediate, pantoyl adenylate
Soaking the crystals with solutions containing both ATP and pantoate, or crystallization in the presence of both ATP and pantoate, resulted in a reaction intermediate, pantoyl adenylate, in the active sites. When the crystals were grown in the presence of ATP and pantoate, both active sites in the dimer have a fully occupied pantoyl adenylate molecule (Table 2, data set 5). However, crystals soaked with ATP and pantoate have one active site (subunit B) fully occupied by pantoyl adenylate but the other partially occupied, indicated by their difference in B factors (Table 2, data set 6). Subunit A of the dimer also has a lower occupancy of AMPCPP when crystals were soaked in solutions containing AMPCPP. This may be due to the crystal packing, which makes the flexible wall of the active site cavity (3103-loop-α3′-loop region) ordered, and the active site less accessible from the bulk solvent channel.
Pantoyl adenylate has extensive binding interactions with the active site residues (Fig. 4F,G ▶), with both the pantoate, and the adenylate moiety of the ATP interacting with the protein active site atoms. The pantoate moiety sits in the same position as the pantoate molecule and has identical interactions with the protein atoms, as does the adenosine moiety. However, the α-phosphate group moves closer to pantoate to form a covalent bond with the carboxyl group of pantoate. This allows the α-phosphate to form a direct hydrogen bond to the amide nitrogen of Met40. The pantoyl adenylate molecule is almost linear and fits snugly in the bottom of the active site cavity. The tight binding of pantoyl adenylate stabilizes this highly reactive intermediate.
Discussion
Structural comparison of MTB and E. coli PS
M. tuberculosis PS has a nearly identical structure to that of the E. coli PS. This is not surprising based on their sequence identity of 46% (Fig. 2 ▶). From the sequence alignment, there are 10 more residues at the N-terminus and about 20 more residues at the C-terminus of the MTB PS. The N-terminal residues form a coil before the first β-strand in the crystal structure. However, the C-terminal residues are disordered and were not found in our electron density maps. These C-terminal residues are away from the active site cavity, and as shown in Results, are unlikely to have catalytic function, but may be involved in interactions with other proteins.
Even though the residues at the dimer interface are poorly conserved in the PS family (von Delft et al. 2001), the dimer interfaces are quite similar in PS proteins from MTB and E. coli. When the N-terminal domains of subunit A are superimposed, the N-terminal domain of the other subunits align very well with only a slight rotation of less than 2°. However, the C-terminal domain has a rotation larger than 30° between the two proteins, when the N-terminal domains are aligned. The C-terminal domain of MTB PS covers the active site cavity with only a small opening (closed conformation), while that of E. coli PS is away from the active site cavity (open conformation).
Mechanism for opening and closing of the active site cavity
In contrast to the big movement between domains found in the crystal structure of the E. coli PS, the relative positions of the two domains of the MTB PS remain the same in all eight crystals we have studied; this is so for the two subunits in the dimer, and for all subunits in the complexes with substrates and reaction intermediate bound in the active site. This closed form observed in the apo MTB PS is unlikely to be induced by the binding of glycerol in the pantoate binding site, because there is also an ethanediol bound in the same site in the E. coli PS structure (von Delft et al. 2001), which is in an open form. In addition, domain movement does not seem to be necessary for substrates to enter the active site, because substrates can diffuse into the active sites and react when crystals are soaked in solutions containing substrates. Considering the tight interactions between the N- and C-terminal domains (see domain interactions above), it is not surprising that the domains have fixed positions relative to each other. It is worth noting that a few domain interactions, including two salt bridges (Arg267 to Glu159, Arg253 to Glu126), two additional hydrogen bonds (Tyr162 OH to Ile255 O and Tyr249 OH to Glu128 Oɛ2) and some hydrophobic interactions, are not present, if the E. coli PS were to form the same closed conformation (Fig. 2 ▶). One way to determine experimentally whether the domains are fixed in a closed form is to mutate the protein by inserting a long loop between domains and check for changed domain interactions.
It is possible that the E. coli PS uses the hinged movement of the C-terminal domain to open and close the active site, while the MTB PS uses the flexible region, residues 75 to 88, as a gate to the active site cavity. As stated above, the flexible region (gate residues) makes the active site accessible to the substrates even in the crystal lattice with the protein in a closed conformation. These residues are more ordered in subunit A because of crystal packing, and this subunit has a lower occupancy of AMPCPP and of the reaction intermediate when crystals are soaked in solutions containing substrates or the analog. However, crystallization in the presence of both ATP and pantoate resulted in both active sites having almost the same occupancy of pantoyl adenylate (Table 2). More importantly, the gate residues became more ordered when there was an intermediate in the active site, even in the apo enzyme crystals that were soaked with ATP and pantoate (Fig. 3 ▶). In contrast, these residues were disordered when the active site is occupied by AMPCPP and glycerol, or pantoate alone. Many other enzymes have been found to have a flexible loop that becomes ordered and closes the active site upon substrate binding (Kempner 1993; Gill and Eisenberg 2001).
Enzyme catalytic mechanism
As described in Results, AMPCPP and pantoate cannot bind simultaneously in the active site. This must be due to the steric and charge repulsions between the α-phosphate group and the carboxyl group of pantoate. Modeling both ATP and pantoate in one active site based on the fully occupied complexes will put one carboxyl oxygen atom of pantoate within 3.2 Å of the α-phosphorus atom of ATP. In subunit A, which has partial occupancy of both AMPCPP and pantoate, the carboxyl oxygen of pantoate is less than 3 Å from the α-phosphorus atom, and is in a good position for an in-line nucleophilic attack (Fig. 4A ▶).
Because residues of the flexible wall (gate residues) are disordered in subunit B, the active site cavity is open and allows AMPCPP to diffuse easily into the active site. The extensive binding interactions of AMPCPP to the protein atoms make its binding thermodynamically more favorable than the binding of pantoate to the protein. Therefore, AMPCPP has full occupancy in the active site cavity of subunit B of the complex with both AMPCPP and pantoate. On the other hand, the gate residues are less flexible in subunit A due to the crystal packing, thus making it kinetically less favorable for AMPCPP to bind in its active site cavity, and allowing pantoate to have partial occupancy in subunit A.
From these considerations, it is likely that the reaction starts by the binding of the bulky ATP molecule in the active site. The smaller sized pantoate then binds and initiates a nucleophilic attack on the α-phosphate. The magnesium ion and the positively charged side chains around the β- and γ-phosphate groups draw the negative charges toward the leaving pyrophosphate, thus facilitating the nucleophilic attack. His47 is likely to function as a general acid by donating a proton to the leaving pyrophosphate. Formation of a bond between pantoate and α-phosphate and breaking the bond between the α- and β-phosphate allows a direct hydrogen bond to form between the α-phosphate and the amide nitrogen of Met40. This favors the formation of the intermediate.
The structure of the reaction intermediate complex confirmed the kinetics results of Zheng and Blanchard (2001) that pantoyl adenylate is a kinetically competent intermediate. This molecule is highly unstable in solution due to the rapid lactonization (Wieland et al. 1963). However, inside the active site of the enzyme the molecule is tightly bound with hydrogen bonds to the active site residues, therefore preventing lactonization from occurring. With the reaction intermediate in the active site, the flexible wall becomes ordered. This closes the active site cavity, and thus prevents the reaction intermediate from being hydrolyzed.
Steady-state kinetics studies with β-alanine and its analogs (Zheng and Blanchard 2001) suggest that the enzyme has very high selectivity for β-alanine, indicating that there is a defined binding site for this substrate. However, crystallization in the presence of 20 mM β-alanine gave only weak occupancy of β-alanine at the binding site of pantoate instead. Soaking crystals with both pantoate and β-alanine gave weak binding of β-alanine in one of the active sites, but its amino group is 7.5 Å away from the carboxyl group of pantoate, indicating that it is not a true binding site either. It is possible that the binding of β-alanine can occur only after the reaction intermediate is formed. We attempted to soak crystals with a solution containing ATP, pantoate and butyrate, hoping to mimic the binding of β-alanine with butyrate. However, butyrate binds to the enzyme only very weakly (see activity assays) and cannot be located in the electron density map. From the crystal structure of the reaction intermediate complex, it is clear that β-alanine can make a direct nucleophilic attack on the intermediate only from above the carboxylate group (Fig. 4 ▶). Therefore, the binding site of β-alanine must be right above the carboxylate of the intermediate. A β-alanine molecule can be modeled based on the crystal structure of pantoyl adenylate complex. Its carboxyl group is near the side chains of His135 and Arg198, suggesting that these side chains may be involved in binding of β-alanine. A structure of a complex with a nonreactive analog of the reaction intermediate and β-alanine would be a better choice to elucidate this last step of the reaction mechanism.
Inhibitor design
One goal of studying pantothenate biosynthetic pathway in MTB is to design new drugs against these novel targets of this dreadful human pathogen. A mutant of MTB defective in de novo biosynthesis of pantothenate has been found to be highly attenuated in a mouse model (Sambandamurthy et al. 2002), although anti-TB drugs against enzymes in this pathway need to be carefully validated, because of the existence of the bacterial transporter of pantothenate. The crystal structure of the PS protein in complex with the reaction intermediate shows that the reaction intermediate, pantoyl adenylate, has strong interactions with the active site atoms (Fig. 4G ▶). This tight binding is necessary to stabilize this highly reactive compound. It can be expected that a nonreactive analog of pantoyl adenylate will be a good inhibitor. Intuitively, elimination of the scissile bond by substituting the oxygen atom between the carbonyl and phosphate with a methylene (CH2) group should give a compound with comparable binding affinity as pantoyl adenylate, because that oxygen does not contribute to binding of the intermediate.
A nonreactive analog of pantoyl adenylate is expected to be very specific to pantothenate synthetase as well. Because there is no pantothenate biosynthesis in humans, this analog will be unlikely to inhibit any human enzyme. Pantoate is synthesized from α-ketoisovalerate through two steps of enzymatic reaction catalyzed by enzymes encoded by panB and panE genes, neither of which is present in humans. By combining the adenylate group with pantoate, the analog should be unique, and might be expected to bind tightly only to pantothenate synthetase.
Materials and methods
Cloning, expression, and purification
The M. tuberculosis panC gene (Rv3602c) encoding the pantothenate synthetase was amplified from the genomic DNA of MTB strain H37Rv using FailSafe polymerase (Epicentre) with Premix J. The following primers were used for PCR cloning, CCTGCCATGGCGATTCCTGCGTTCCATCCC (5′ primer) and GCTCAAGCTTCAGTTTCTCCAATGTGATTCGAGGATT GCCCGG (3′ primer), which generate NcoI and HindIII restriction sites (underlined). Amplified DNA was ligated into a pCR4Blunt-TOPO vector (Invitrogen) to give an intermediary vector that was then digested with restriction enzymes NcoI and HindIII. The fragment containing panC gene was purified on the agarose gel, and ligated into a pET30a plasmid (Novagen) linearized with enzymes NcoI and HindIII. The resulting plasmid pET30-PanC adds a 6× His tag and an S tag to the N-terminus of the recombinant protein, and changes the second amino acid encoded in the panC gene from a threonine to an alanine. The plasmid was purified from Novablue cells (Novagen), and then transformed into BL21(DE3) competent cells (Novagen) for protein overexpression. DNA sequence of the panC gene was confirmed by sequencing (Davis Sequencing).
BL21(DE3) cells transformed with the pET30-PanC plasmid were grown in LB medium containing 50 μg/mL Kanamycin at 37°C to an OD600 of ~0.8. IPTG was then added to a final concentration of 0.4 mM to induce the protein expression. The cells were induced for 3 h and harvested by centrifugation. Cell pellets were resuspended in 20 mM HEPES, pH 7.8, 500 mM NaCl, and 0.5 mM PMSF. Cells were lysed by lysozyme treatment followed by sonication. Cell lysate was centrifuged at ~37,000 g for 30 min, and the supernatant was loaded onto a Ni2+-charged HiTrap chelating column (Pharmacia) preequilibrated with 20 mM HEPES, pH 7.8, 500 mM NaCl, and 10 mM imidazole. The column was washed thoroughly with the same buffer, and eluted with a linear gradient of imidazole from 10 to 300 mM in 20 mM HEPES, pH 7.8, and 500 mM NaCl. The fractions containing PS protein were pooled and concentrated to about 5 mg/mL, and the protein was further purified on a Superdex 75 column (Pharmacia) equilibrated with 20 mM HEPES, pH 7.8, and 150 mM NaCl for enzyme activity assays and for crystallization.
Digestion of the N-terminal fusion tag was carried out in a solution containing 50 mM Tris, pH 7.5, 50 mM NaCl, 2 mM CaCl2, and 0.15% (v/v) Tween-20 with enterokinase (New England Biolabs) overnight. Enterokinase was removed with trypsin inhibitor agarose (Sigma) after adjusting NaCl concentration to 500 mM. The solution was filtered, and loaded onto a Ni2+-charged HiTrap chelating column to separate the digested protein from the undigested protein and cleaved N-terminal tag. The protein was further purified on a Superdex 75 column. Full-length and enterokinase digested proteins were analyzed by SDS-PAGE and Electrospray mass spectroscopy.
Enzyme activity assays
Pantothenate synthetase activity was measured following similar procedures as in Wieland et al. (1963). The formation of AMP from the PS catalyzed reaction was coupled to the reactions of myokinase, pyruvate kinase, and lactate dehydrogenase, and the decrease in absorbance of NADH at 340 nm was measured at room temperature using a Beckman DU 650 spectrophotometer. NADH, ATP, β-alanine, potassium phosphoenolpyruvate, myokinase (rabbit muscle), pyruvate kinase (rabbit muscle), and lactate dehydrogenase (rabbit muscle) were purchased from Sigma. Sodium pantoate was prepared by mixing NaOH with d-pantoyl lactone (Aldrich) to adjust the final pH of the solution to between 7 and 8. The reaction was run in 1 mL of solution containing 100 mM HEPES, pH 7.8, 10 mM MgCl2, 10 mM ATP, 5 mM β-alanine, 5 mM pantoate, 1 mM potassium phosphoenolpyruvate, 0.2 mM NADH, 18 units each of pyruvate kinase, lactate dehydrogenase, and myokinase, and 5–10 μL of purified pantothenate synthetase. The reaction components except PS were mixed in a cuvette and the background reaction was measured on a spectrophotometer. PS was then added and quickly mixed, and the reaction progress was monitored by measuring the absorbance at 340 nm. The initial reaction rates were calculated using an extinction coefficient of NADH of 6220 M−1cm−1. Similar procedures were followed for the inhibition studies with butyrate and glycerol. Certain amount of sodium butyrate or glycerol was mixed with other components prior to the addition of PS.
Crystallization and data collection
For protein crystallization, the purified protein was exchanged into a dilute buffer of 5 mM HEPES, pH 7.5, 10 mM NaCl by three cycles of dilution and concentration, and then concentrated to above 10 mg/mL. Crystallization experiments were carried out using the hanging-drop vapor-diffusion method. In each drop, 3–5 μL of protein solution was mixed with an equal volume of well solution. Clusters of small crystals were obtained from the enterokinase-digested protein with Wizard I crystal screen kit solution 15 (Emerold BioStructures). Crystals were improved by modifying the crystallization condition, and the best crystals were obtained from drops set up with well solutions containing 10%–15% PEG 3000, 5% glycerol, 2% ethanol, 20 mM MgCl2, 150 mM Li2SO4, and 100 mM imidazole, pH 8.0 at 20°C. These crystals are thick plates with dimensions of about 0.5 × 0.3 × 0.1 mm. For cocrystallization with the reaction intermediate, a similar crystallization solution was used, with 10 mM each of ATP and pantoate added, and with 20 mM MnCl2 substituted for MgCl2. The crystals are small (about 0.1 mm in the longest dimension) but diffract to 2.0 Å. Cocrystallization with β-alanine was done with a well solution of 15% PEG 3000, 2% ethanol, 20 mM MgCl2, 100 mM imidazole, pH 8.0 and 20 mM β-alanine.
To get crystals in complex with substrates, crystals grown in the absence of substrates or analogs were soaked in solutions containing substrates or an ATP analog, AMPCPP. Soaking experiments were carried out by adding solutions containing substrates or AMPCPP directly to the drop, or to a drop of pseudomother liquor with transferred crystals, and incubating overnight or longer. Crystals were soaked in solutions containing AMPCPP alone, both AMPCPP and pantoate, both pantoate and β-alanine, ATP together with pantoate and sodium butyrate, and pantothenate alone.
For data collection, crystals were soaked for 2–5 min in a cryogenic solution similar to the well solutions, or substrate soaking solutions, but with 30% glycerol added, and then flash-frozen in a cryostream of N2 gas at 100 K. Diffraction data were collected at 100 K on a Rigaku FRD generator with an R-AXIS IV++ detector. Data reduction and scaling were done with the programs DENZO and SCALEPACK (Otwinowski and Minor 1996). Data processing statistics are listed in Table 3. The apo enzyme crystallized in space group P21. Crystallization in the presence of both ATP and pantoate also gave crystals of space group P21 with similar cell dimensions. However, crystals obtained in the presence of β-alanine are in space group P212121, presumably because of the difference in the crystallization conditions. Data collected on crystals soaked with pantothenate did not show any density for pantothenate in the initial difference density map, and therefore are not reported. All data sets collected gave two molecules per asymmetric unit.
Table 3.
X-ray diffraction data for M. tuberculosis pantothenate synthetase and complexesa
| Data setb | 1 (Apo) | 2 (AMPCPP) | 3c (AMPCPP + pantoate) | 4c (AMPCPP + pantoate) | 5 (ATP + pantoate) | 6 (ATP + pantoate + butyrate) | 7 (pantoate + β-alanine) | 8 (β-alanine) |
| Resolution (Å) | 80–1.6 | 50–1.8 | 50–1.6 | 50–1.7 | 50–2.0 | 50–1.7 | 50–1.8 | 50–2.1 |
| Rmergee (last bin)d | 0.049 (0.312) | 0.059 (0.471) | 0.066 (0.463) | 0.049 (0.381) | 0.125 (0.426) | 0.077 (0.489) | 0.086 (0.374) | 0.081 (0.597) |
| Completenessd % | 89.1 (51.1) | 99.4 (99.8) | 93.7 (58.8) | 95.2 (86.5) | 94.3 (91.4) | 98.8 (94.1) | 97.7 (97.7) | 99.9 (99.9) |
| I/σd | 36.4 (4.4) | 21.1 (2.6) | 25.2 (2.6) | 22.9 (2.8) | 10.3 (2.8) | 23.7 (2.98) | 14.9 92.4) | 25.3 (3.9) |
| Redundancyd | 6.6 (4.6) | 3.4 (3.2) | 6.4 (3.5) | 3.5 (2.9) | 4.0 (3.8) | 7.5 (5.7) | 4.0 (2.5) | 7.7 (7.6) |
| Space Group | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P212121 |
| Cell | ||||||||
| a | 48.27 | 48.31 | 48.27 | 48.13 | 48.26 | 48.18 | 48.05 | 69.75 |
| b | 70.93 | 70.91 | 70.82 | 70.57 | 70.86 | 70.80 | 70.69 | 98.65 |
| c | 81.83 | 81.88 | 81.80 | 81.77 | 81.82 | 81.75 | 81.59 | 82.10 |
| β | 98.69 | 99.31 | 99.28 | 99.23 | 99.34 | 99.43 | 99.10 | |
a All data collected at cryogenic temperature of 100 K.
b Data sets 2, 3, 4, 6, and 7 are collected on crystals grown in identical conditions to that of data set 1, but soaked in various solutions. Data sets 5 and 8 are from cocrystallization crystals.
c For data set 3, the crystal was soaked in 10 mM each of AMPCPP and pantoate overnight; from data set 4, the crystal was soaked in 10 mM of AMPCPP and 20 mM of pantoate for 24 h.
d The numbers in parentheses are for the last bin of data, which is from 1.66 to 1.6 Å, 1.76 to 1.7 Å, 1.86 to 1.8 Å, 2.07 to 2.0 Å, or 2.18 to 2.1 Å
eRmerge = Σ|(Ihkl − 〈Ihkl〉|/ΣIhkl, where 〈Ihkl〉 is the average of Ihkl over all symmetry equivalents.
Molecular replacement, model building, and refinement
The crystal structure of the apo enzyme was determined by the molecular replacement method using the program AMORE (Navaza 1994). Subunit A of the E. coli PS structure (von Delft et al. 2001, PDB ID code 1IHO) was used as a model for molecular replacement, and the two domains were separated as two independent search models. A set of data was first collected to 2.2 Å (not shown) and was used for molecular replacement. The rotation function was run with data from 8 to 2.8 Å, while translation function was run with data from 8 to 2.2 Å. The positions of the two N-terminal domains and one C-terminal domain were found from the rotation and translation searches, and the other C-terminal domain was generated by a noncrystallographic-symmetry (NCS) operation on the position of the known C-terminal domain with the NCS symmetry operators derived from the two N-terminal domains.
The R-factor was 47.3% after one round of rigid body refinement with program CNS using data from 20 to 2.5 Å resolution. The map calculated from the rigid body-refined model showed clear continuous density. A set of data to 1.6 Å was collected at this point (data set 1 in Table 3), and phases were refined and extended to 1.6 Å using the program DM (CCP4 1994; Cowtan 1994) with solvent flattening, histogram matching, multiresolution modification and NCS averaging. The refined phases were given to the automatic refinement procedure ARP/wARP (Perrakis et al. 1999) for automatic model building. This built more than 90% of residues in the final model. The model was then manually rebuilt using the program O (Jones et al. 1991), and refined using CNS. The structure of the apo enzyme was used to calculate phases for the data sets of various complexes by rigid body refinement into the new data sets, except that for β-alanine complex data (data set 8 in Table 3), which is in a different space group, molecular replacement with the program AMORE was used. The final refinement statistics for all the refined coordinate sets are reported in Table 2.
Over 91% of the residues fall in the most favorable region of the Ramachandran plot for all the refined coordinate sets. One residue, Leu127, however, falls in the disallowed region with φ, ψ angles of about 46° and −110°, respectively. This residue has well-defined electron density and is well ordered in the crystal structure. It consistently shows up at this same position of Ramachandran plot for all the refined coordinate sets. The corresponding residue, Leu118 of the E. coli PS structure also falls at the similar position of the Ramachandran plot.
Data deposition
The atomic coordinates have been deposited to the Protein Data Bank with identification codes: 1MOP, 1N2B, 1N2E, 1N2G, 1N2H, 1N2I, 1N2J, and 1N2O.
Acknowledgments
We thank NIH and HHMI for support. The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
MTB, Mycobacterium tuberculosis
TB, tuberculosis
ACP, acyl carrier proteins
CoA, coenzyme A
PS, pantothenate synthetase
AMPCPP, α,β-methyleneadenosine 5′-triphosphate
IPTG, isopropyl β-D-1-thiogalactopyranoside
NCS, noncrystallographic-symmetry
PMSF, phenylmethanesulfonyl fluoride
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0241803.
References
- Barker, D.G. and Winter, G. 1982. Conserved cysteine and histidine residues in the structures of the tyrosyl and methionyl-tRNA synthetases. FEBS Lett. 145 191–193. [DOI] [PubMed] [Google Scholar]
- Barton, G.J. 1993. ALSCRIPT: A tool to format multiple sequence alignments. Protein Eng. 6 37–40. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, N., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project Number 4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D50 760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan, K. 1994. DM: An automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newslett. Protein Crystallogr. 31 34–38. [Google Scholar]
- DeLano, W.L. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA.
- Genschel, U., Powell, C.A., Abell, C., and Smith, A.G. 1999. The final step of pantothenate biosynthesis in higher plants: Cloning and characterization of pantothenate synthetase from Lotus japonicus and Oryza sativum (rice). Biochem. J. 341 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, H.S. and Eisenberg, D. 2001. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 40 1903–1912. [DOI] [PubMed] [Google Scholar]
- Grosset, J. 1996. Current problems with tuberculosis treatment. Res. Microbiol. 147 10–16. [DOI] [PubMed] [Google Scholar]
- Jackowski, S. 1996. Biosynthesis of pantothenic acid and coenzyme A. Escherichia coli and Salmonella typhimurium: Cellular and molecular biology, 2nd ed., vol. 1, pp. 687–694. American Society for Microbiology, Washington, DC.
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Kempner, E.S. 1993. Movable lobes and flexible loops in proteins. FEBS Lett. 326 4–10. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Maas, W.K. 1960. The biosynthesis of pantothenic acid. Proc. Int. Congr. Biochem. 11 161–168. [Google Scholar]
- Merkel, W.K. and Nichols, B.P. 1996. Characterization and sequence of the Escherichia coli panBCD gene cluster. FEMS Microbiol. Lett. 143 247–252. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D50 869–873. [DOI] [PubMed] [Google Scholar]
- Miyatake, K., Nakano, Y., and Kitaoka, S. 1978. Enzymological properties of pantothenate synthetase from Escherichia coli B. J. Nutr. Sci. Vitaminol. 24 243–253. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1996. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis, A., Morris, R.M., and Lamzin, V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6 458–463. [DOI] [PubMed] [Google Scholar]
- Perez-Espinosa, A., Roldan-Arjona, T., and Ruiz-Rubio, M. 2001. Pantothenate synthetase from Fusarium oxysporum f. sp. lycopersici is induced by alpha-tomatine. Mol. Genet. Genomics 265 922–929. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy, V.K., Wang, X., Chen, B., Russell, R.G., Derick, S., Collins, F.M., Morris, S.L., and Jacobs, W.R. 2002. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 8 1171–1174. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Delft, F., Lewendon, A., Dhanaraj, V., Blundell, T.L., Abell, C., and Smith, A.G. 2001. The crystal structure of E. coli pantothenate synthetase confirms it as a member of the cytidylyltransferase superfamily. Structure 9 439–450. [DOI] [PubMed] [Google Scholar]
- Wieland, T., Lowe, W., Kreiling, A., and Pfleiderer, G. 1963. On pantothenic acid synthetase from E. coli V. Pantoyladenylate as the acylating component in the enzymatic synthesis of pantothenic acid. Biochem. Z. 339 1–7. [PubMed] [Google Scholar]
- Zheng, R. and Blanchard, J.S. 2001. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry 40 12904–12912. [DOI] [PubMed] [Google Scholar]