

Abstract
In most cases aminoacyl-tRNA synthetases (aaRSs) are negatively charged, as are the tRNA substrates. It is apparent that there are driving forces that provide a long-range attraction between like charge aaRS and tRNA, and ensure formation of "close encounters." Based on numerical solutions to the nonlinear Poisson-Boltzmann equation, we evaluated the electrostatic potential generated by different aaRSs. The 3D-isopotential surfaces calculated for different aaRSs at 0.01 kT/e contour level reveal the presence of large positive patches—one patch for each tRNA molecule. This is true for classes I and II monomers, dimers, and heterotetramers. The potential maps keep their characteristic features over a wide range of contour levels. The results suggest that nonspecific electrostatic interactions are the driving forces of primary stickiness of aaRSs–tRNA complexes. The long-range attraction in aaRS–tRNA systems is explained by capture of negatively charged tRNA into "blue space area" of the positive potential generated by aaRSs. Localization of tRNA in this area is a prerequisite for overcoming the barrier of Brownian motion.
Keywords: Electrostatic interactions, aminoacyl-tRNA synthetase, tRNA, electrostatic potential, encounter complex, Brownian motion
The aaRSs ensure the fidelity of the translation of the genetic code, covalently attaching appropriate amino acids to the corresponding nucleic acid adaptor molecules—tRNA. Despite common catalytic functions, class I and class II synthetases (Eriani et al. 1990; Mirande 1991) vary greatly in subunit organization, size of polypeptide chains, and amino acid sequence. On the other hand, all known 3D structures of tRNA are L-shaped or closely resemble it. Thus, differently shaped protein molecules bind and recognize L-shape tRNA molecules. A great body of structural information has accumulated on crystal structures of aaRSs complexed with cognate tRNA. These data provide evidence of specific tRNA recognition with a few base-specific interactions concentrated mostly at the anticodon and acceptor areas. In certain complexes (ProRS, PheRS; Goldgur et al. 1997; Yaremchuk et al. 2000) amino acids specifically interact with nucleotide bases of the anticodon loop only. However, it has been known that the bulk of the contacts in the tRNA–aaRSs complexes occur between the protein and the sugar/phosphate backbone of tRNA; these contacts are nonspecific, localized at widely spaced regions of tRNA, and largely electrostatic. An increase in the ionic strength significantly weakens complex formation and is indicative of the importance of electrostatic interactions in aaRS–tRNA complexes (Bonnet and Ebel 1975). Based on fast kinetic experiments, fluorescence titrations, and ultracentrifugation analysis, Krauss et al. (1976) hypothesized that binding of tRNA by aaRSs proceeds in two steps. The initial bimolecular step is rapid, and has a broad specificity, whereas the second unimolecular step is related to conformational changes and more precise adjustment and/or recognition. Drawing an analogy to protein–protein and protein–DNA systems (Pontius 1993; Schreiber and Fersht 1996), it is believed that the bimolecular step in tRNA–aaRSs complexation proceeds rapidly by components of these systems first binding nonspecifically, governed by long-range electrostatic interactions. Calculations of the electrostatic potential contours around tRNA molecules (Chin et al. 1999) enable one to visualize and treat them as large, negatively charged particles (∼−75e), corrected for their L-shape.
Based on the nonlinear Poisson-Boltzmann equation (Nicholls and Honig 1991) we evaluated the contribution of nonspecific electrostatic interactions in formation of aaRS–tRNA complexes at long distances. Our results explain how, at distances away from the molecular surfaces, monomeric, dimeric, and heterotetrameric aaRSs provide an association energy that holds macromolecules near each other, exerting control over tRNA motion towards the binding site.
Results and Discussion
Monomeric aaRSs generate one positively charged patch
The structure of class Ia monomeric arginyl-tRNA synthetase (ArgRS; Delagoutte et al. 2000) complexed with tRNA suggests extensive same-subunit contacts with tRNA. Calculated isopotential surfaces (±0.01 kT/e) display partition onto two distinctly charged regions (Fig. 1A ▶): large positively charged patch and, embracing it, a negatively charged area. ArgRS from the yeast Saccharomyces cerevisiae and one of its cognate isoacceptors tRNAArg form the largest contact area of ∼3000 Å2 among the known structures of aaRS–tRNA complexes (Delagoutte et al. 2000). ArgRS is the first enzyme for which the D-loop, in addition to the anticodon loop and acceptor stem, is strongly involved in synthetase recognition or binding (Fig. 1B ▶). All three vertices of the L-shaped tRNA molecule are in contact with the surface of the enzyme, thus maximizing the interface between aaRS and tRNA.
Figure 1.
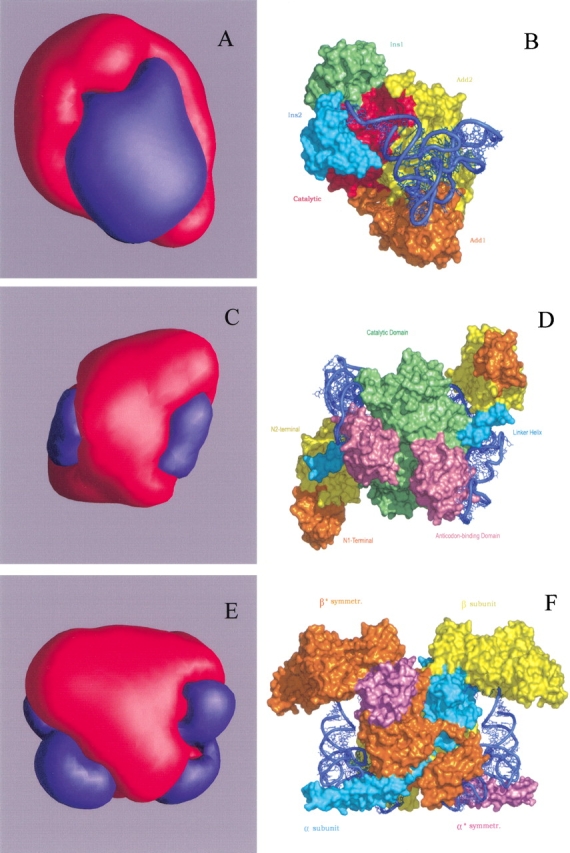
3D-isopotential surface representation of aaRSs and overall views of their complexes with cognate tRNA: class I monomeric ArgRS (A, B); class II dimeric ThrRS (C, D); class II heterotetrameric PheRS (E, F). Pairs of (A) and (B), etc., are displayed in different scale and have equivalent orientation. The parts (A), (C), (E) are scaled down by factors of 1.5, 2.5, and 3 compared to the molecular surface (B), (D), and (F), respectively. Structural domains of ArgRS and ThrRS and subunits of PheRS are marked with different colors. The tRNA is shown as blue ribbons. All isopotential surfaces are calculated and built at ±0.01 kT/e with GRASP (Nicholls et al. 1993). Patches of positive (+) and negative (−) potentials are shown in blue and red, respectively. Both molecular surfaces and ribbons are built with PyMol (DeLano 2002).
Dimeric aaRSs generate two positively charged patches
The class IIa Escherichia coli threonyl-tRNA synthetase (ThrRS) is a α2 dimeric enzyme with extensive cross-subunit tRNA binding (Fig. 1D ▶; Sankaranarayanan et al. 1999). The contact area of one tRNA molecule with two subunits is ∼2780 Å2. It is of interest that none of the cross-subunit interactions are base specific. At the isopotential level 0.01 kT/e, the homodimer displays only two positively charged patches (Fig. 1C ▶) corresponding to each tRNA molecule approaching the enzyme. However, each monomer considered by itself gives rise to the occurrence at the isopotential surface of additional small patches, both positively and negatively charged, not related to the tRNA binding sites. Thus, only integration of the potential fields generated by two individual monomers yields the unique configuration of ThrRS potential field.
Heterotetrameric PheRS also generates two positively charged patches
Heterotetrameric (αβ)2 PheRS from Thermus thermophilus is the most complex synthetase of aaRS family, containing 22 structural domains (Mosyak et al. 1995). One tRNAPhe molecule binds across all four subunits of the enzyme (Fig. 1F ▶; Goldgur et al. 1997). The acceptor stem of the tRNAPhe interacts with the active site located in the α-subunit and with the N-terminal domain of the β-subunit from the same heterodimer, while the anticodon loop of the tRNAPhe is specifically recognized by the C-terminus of the β*-subunit (where * indicates the second heterodimer). The N-terminal coiled-coil domain of the α*-subunit approaches tRNAPhe from the variable loop side. However, this plethora of domains gives rise to a strikingly homogeneous positively charged patches on the isopotential surface (Fig. 1E ▶). It is remarkable that PheRS, possessing four characteristic class II antiparallel folds, has only two tRNA binding sites (Goldgur et al. 1997). The unique structural organization is reflected also in the way the charged residues are distributed along the sequence to produce at 0.01 kT/e only two positively charged patches.
From comparison of all the figures by pairs (Fig. 1A,B, ▶etc.), one can conclude that the projections onto the molecular surface of the positive patches from the isopotential surface coincide with the tRNA binding sites for the above-mentioned aaRSs. In other words, the path of tRNA towards the molecular surface looks like motion along the radial lines that diverge from the tRNA binding sites.
We have studied various aaRSs both native and complexed with cognate tRNA molecules, among them seven class II and six class I enzymes. There are no exceptions for the observed empirical guideline: On the associated 3D-isopotential surfaces, each aaRS creates as many positive patches as there are tRNA molecules interacting with a given aaRS. The solutions of the nonlinear Poisson-Boltzmann equation and their visual representation indicate that nonspecific long-range electrostatic interactions are the dominant factor for general stickiness and formation of the encounter complex in all aaRS–tRNA systems. The isopotential surfaces at ∼0.01 kT/e are at a considerable distances from molecular surface of the enzymes and fall in the range between ∼40 Å (ArgRS) and ∼100 Å (PheRS). However, the binding energy that results from Coulomb interactions between aaRSs and tRNA placed into the "blue space area" of aaRS appears to be at least three times higher than the thermal energy kT. This fact makes it possible for tRNA to overcome the barrier of Brownian motion. The assembly of tRNA and aaRSs can happen only when these reactants are properly positioned, that is, tRNA is localized in the immediate vicinity of the positively charged patches generated by aaRS. The interactions mediating the attractive forces via nonspecific electrostatic interactions have no restrictions on the rotational freedom of tRNA. Such orientation-independent interactions at long distances can also rotate and realign while still guaranteeing an attraction between two molecules. In the range of 0.01–0.1 kT/e, each aaRS keeps congruent topology of the isopotential 3D contours. The combination of electrostatic potentials generated by characteristic structural domains give rise to unique configuration of the common positive potential that steers tRNA towards their positions in bound complexes. For certain aaRSs complexes, the projection of the positive patch onto the area of molecular surface where the tRNA binding site is located, occurs along the trajectories that does not coincide with radial lines. As an example, for IleRS (Silvian et al. 1999), tRNA from clearly defined positively charged patch on the isopotential surface, while in "blue space area" of the electrostatic potential field, moves to the molecular surface along the trajectory that is oblique-angled to the radial lines emanating from the tRNA binding site. A more complicated trajectory is dictated by topography of the enzyme surface and by the landscape of electrostatic energy. Computer simulations based on rigid-body minimization and molecular docking will offer a clearer view of the tRNA trajectory under the action of the Coulomb forces generated by nonspecific long-range electrostatic interactions.
Materials and methods
Structures
Coordinates of the aaRS–tRNA complexes were obtained from the Protein Data Bank (Bernstein et al. 1977; Berman et al. 2000; http://www.rcsb.org/pdb) and identified by their PDB codes: 1f7u for ArgRS–tRNAArg; 1qf6 for ThrRS–tRNAThr; 1eiy for PheRS–tRNAPhe.
Charge parameter sets
Parameter set (force field) amber96 that describes partial charges and atomic size in molecular simulation program AMBER (Weiner et al. 1986) was applied for calculation of the electrostatic potentials. A comparison with that of GRASP demonstrates that the patterns are in a good agreement.
Calculation of electrostatic potentials
All electrostatic potentials were calculated with the program Delphi (Nicholls and Honig 1991) by solving the nonlinear Poisson-Boltzmann equation (NLPB). All charges and dielectric boundary were mapped into a cubic grid (65 × 65 × 65 grid points/side). An interior relative permittivity constant of 4 was accepted. Dielectric permittivity for solvent was 80. The univalent salt concentration was taken as 0.10 M.
Electrostatic energy evaluation for aaRS–tRNA complexes
The hydrogen atoms were added to PDB structures and the energy of aaRS and tRNA was minimized (grad = 0.1 kcal/mole; amber96 force field, Biopolymer and Discover modules of InsightII). The tRNA molecules were placed into "blue space area" (area of positive potential) generated by aaRSs molecules. The rigid-body minimization was performed using X-PLOR (Brunger 1993). The aaRSs and tRNA were taken as rigid groups. Evaluation of the electrostatic energy contributions (E) of individual aaRS and tRNA to the binding energy of the complex, have been performed by using program Delphi on a lattice with one grid point per 1 Å. The (ΔE) is calculated from difference between the electrostatic energy of the docked complex aaRS–tRNA and electrostatic energy of the two individual unbound species (aaRS and tRNA):
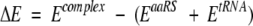 |
1 |
Visualization
The 3D-isopotential molecular surfaces around aaRSs are displayed with GRASP (Nicholls et al. 1993) program. The standard GRASP color codes are used for Figure 1 ▶: blue color is for positive potential and red is for negative. The aaRSs molecular surfaces and ribbons of tRNA are built and colored with the PyMol program (DeLano 2002).
Acknowledgments
We thank B. Honig and D. Fass for reading of the manuscript and critical comments. We are grateful to V. Neporent and D. Safro for technical assistance. This work was supported by a research grant from Edward and Anna Mitchell Research Fund. This work was partially supported by Yeda Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
3D, three-dimensional
PDB, Protein Data Bank of experimentally determined 3D structures of proteins
aaRS, aminoacyl-tRNA synthetase
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0301203.
References
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer, E.F., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The Protein Data Bank: A computer based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Bonnet, J. and Ebel, J.-P. 1975. Influence of various factors on the recognition specificity of tRNAs by yeast valyl-tRNA synthetase. Eur. J. Biochem. 58 193–201. [DOI] [PubMed] [Google Scholar]
- Brunger, A. 1993. X-PLOR version 3.1 manual. Yale University, New Haven, CT.
- Chin, K., Sharp, K., Honig, B., and Pyle, A.-M. 1999. Calculating the electrostatic properties of RNA provides new insights into molecular interactions and function. Nat. Struct. Biol. 6 1055–1061. [DOI] [PubMed] [Google Scholar]
- Delagoutte, B., Moras, D., and Cavarelli, J. 2000. tRNA aminoacylation by arginyl-tRNA synthetase: Induced conformations during substrates binding. EMBO J. 19 5599–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. The PyMol Molecular Graphics System. DeLano Scientific, San Carlos, CA. http://www.pymol.org.
- Eriani, G., Delarue, M., Poch, O., Gangloff, J., and Moras, D. 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347 203–206. [DOI] [PubMed] [Google Scholar]
- Goldgur, Y., Mosyak, L., Reshetnikova, L., Ankilova, V., Lavrik, O., Khodyreva, S., and Safro, M. 1997. The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure 5 59–68. [DOI] [PubMed] [Google Scholar]
- Krauss, G., Riesner, D., and Maass, G. 1976. The mechanism of discrimination between the cognate and non-cognate tRNA by phenylalanyl-tRNA synthetase from yeast. Eur. J. Biochem. 68 81–93. [DOI] [PubMed] [Google Scholar]
- Mirande, M. 1991. Aminoacyl-tRNA synthetase family from prokaryotes and eukaryotes: Structural domains and their implications. Prog. Nucleic Acid Res. Mol. Biol. 40 95–142. [DOI] [PubMed] [Google Scholar]
- Mosyak, L., Reshetnikova, L., Goldgur, Y., Delarue, M., and Safro, M. 1995. Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat. Struct. Biol. 2 537–547. [DOI] [PubMed] [Google Scholar]
- Nicholls, A. and Honig, B. 1991. A rapid finite difference algorithm, utilizing successive over-relaxation, to solve the Poisson-Boltzmann equation. J. Comp. Chem. 12 435–445. [Google Scholar]
- Nicholls, A., Bharadwaj, R., and Honig, B. 1993. GRASP: Graphical representation and analysis of surface properties. Biophys. J. 64 A166. [Google Scholar]
- Pontius, B.W. 1993. Close encounters: Why unstructured, polymeric domains can increase rates of specific macromolecules association. Trends Biosci. 18 181–186. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan, R., Dock-Bregeon, A.C., Romby, P., Caillet, J., Springer, M., Rees, B., Ehresmann, C., Ehresmann, B., and Moras, D. 1999. The structure of threonyl–tRNA synthetase–tRNAThr complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell 97 371–381. [DOI] [PubMed] [Google Scholar]
- Schreiber, G. and Fersht, A. 1996. Rapid electrostatically assisted association of proteins. Nat. Struct. Biol. 3 427–431. [DOI] [PubMed] [Google Scholar]
- Silvian, L.F., Wang, J., and Steitz, T.A. 1999. Insights into editing from an Ile-tRNA synthetase structure with tRNAIle and mupirocin. Science 285 1074–1077. [PubMed] [Google Scholar]
- Weiner, S.J., Kollman, P.A., Nguyen, D.T., and Case, D.A. 1986. An all atom force field for simulations of proteins and nucleic acids J. Comput. Chem. 7 230–252. [DOI] [PubMed] [Google Scholar]
- Yaremchuk, A., Cusack, S., and Tukalo, M. 2000. Crystal structure of a eukaryote/archaeon-like prolyl-tRNA synthetase and its complex with tRNAPro (CGG). EMBO J. 19 4745–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]