

Abstract
The well-known preference of the peptide bond for the trans conformation has been attributed to steric effects. Here, we show that a proline residue with an N-formyl group (Hi−1−C′i−1=Oi−1), in which Hi−1 presents less steric hindrance than does Oi−1, likewise prefers a trans conformation. Thus, the preference of the peptide bond for the trans conformation cannot be explained by steric effects alone. Rather, an n → π* interaction between the oxygen of the peptide bond (Oi−1), and the subsequent carbonyl carbon in the polypeptide chain (C′i) also contributes to this preference. The Oi−1 and C′i distance and Oi−1···C′i=Oi angle are especially favorable for such an n → π* interaction in a polyproline II helix. We propose that this electronic effect provides substantial stabilization to this and other elements of protein structure.
Keywords: Bürgi-Dunitz trajectory, collagen, hyperconjugation, peptide bond, polyproline II helix, proline isomerization, stereoelectronic effect, steric effect
In folded proteins, only 0.03% of Xaai−1–nonProi peptide bonds are in the cis conformation (Stewart et al. 1990; Weiss et al. 1998; Jabs et al. 1999). This prevalence is 5.2% for Xaai−1–Proi peptide bonds. The greater stability of trans peptide bonds is assumed to arise solely from a steric effect (Schulz and Schirmer 1979; Creighton 1993; Kyte 1995; Fischer 2000). Simply put, the Cα substituents of each amino acid are forced into clashing proximity in the cis isomer, whereas this steric strain is relieved in the more pervasive trans isomer. The isomers of a prolyl peptide bond are more nearly isoenergetic, presumably because the Cδ protons of the pyrrolidine ring provide nearly as much steric encumbrance as do the Cα substituents.
Are steric effects alone responsible for the observed trans:cis ratio of prolyl peptide bonds? To answer this question, we synthesized N-formyl-l-proline methyl ester (FmProOMe; 1; Scheme 1 ▶). The amide bond in amide 1 is isologous to a prolyl peptide bond, except that the steric effects in amide 1 are reversed, favoring the cis rather than the trans isomer. This reversal occurs because the van der Waals radius of oxygen is greater than that of hydrogen and the C′i−1=Oi−1 bond is longer than the C′i−1−Hi−1 bond (Fig. 1A ▶). The synthesis of amide 1 as a methyl ester rather than a secondary amide avoids intramolecular hydrogen bonding to form a γ-turn, as has been observed in N-acetylproline (Madison and Schellman 1970; DeTar and Luthra 1977) and N-acetylproline N-methylamide (Matsuzaki and Iitaka 1971; Higashijima et al. 1977; Liang et al. 1992; Benzi et al. 2002).
Scheme 1.

Figure 1.
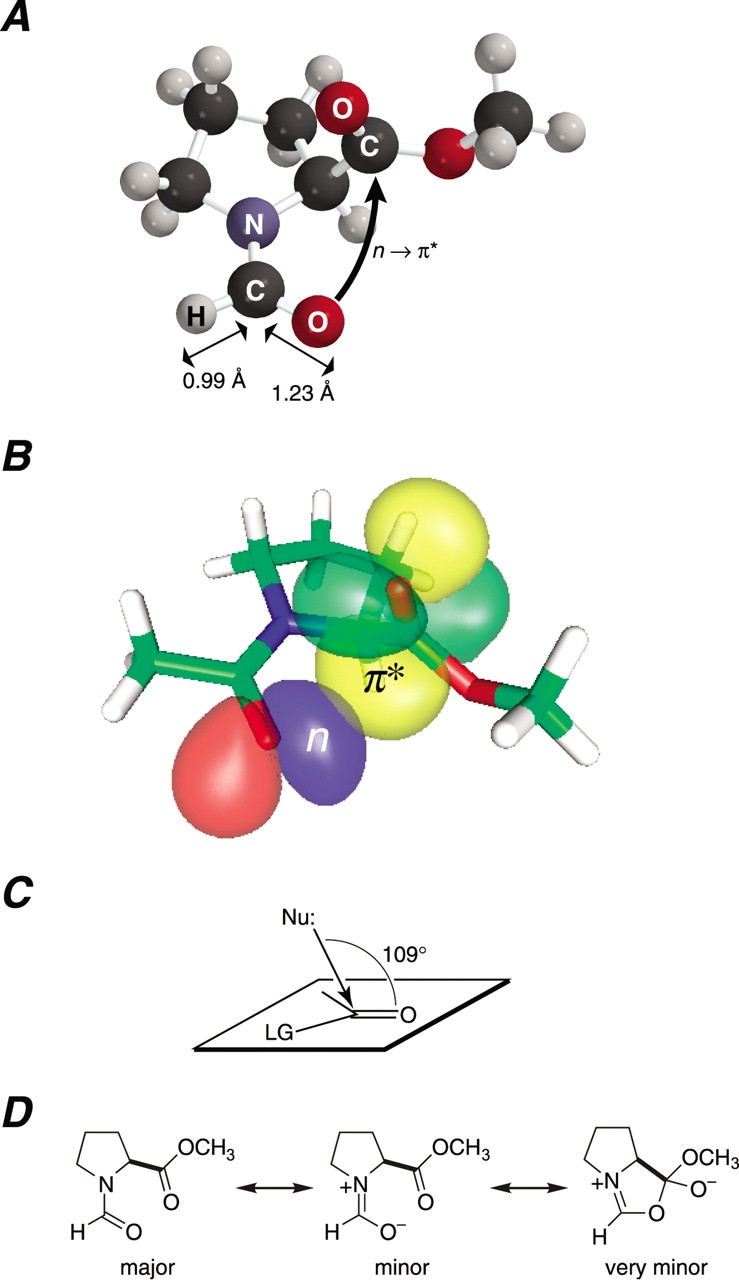
n → π* Interaction between Oi−1 and C′i. (A) Structure of the trans isomer of N-formyl-l-proline methyl ester (1) in its Cγ-exo conformation. The C′i−1–H and C′i−1 =Oi−1 bond lengths are from the structure of crystalline dimethyl formamide (Borrmann et al. 2000). (B) Depiction of the n and π* natural bond orbitals of the trans isomer of N-acetyl-l-proline methyl ester (2) in its Cγ-exo conformation. The Oi−1· · · C′i distance is δBD = 2.87 Å and the Oi−1· · · C′i=Oi angle is τBD = 99.35° (DeRider et al. 2002). (C) Bürgi–Dunitz trajectory for the attack of a nucleophile on a carbonyl group to displace a leaving group (LG; Bürgi et al. 1973, 1974a,b; Bürgi and Dunitz 1983; Eliel and Wilen 1994). (D) Major and minor resonance forms of amide 1 (Pauling 1960) and a very minor structure that arises from the hyperconjugative delocalization of an n → π* interaction.
In addition to diminishing steric effects, we also sought to enhance those effects by increasing the steric bulk on either side of the amide bond of amide 1. Specifically, replacing the formyl proton with a methyl group yields N-acetyl-l-proline methyl ester (AcProOMe; 2; Scheme 1 ▶). Replacing the two Cδ protons with methyl groups yields N-formyl-5,5-dimethyl-l-proline methyl ester (Fm[dmPro]OMe; 3). We synthesized amides 2 and 3, and compared their values of Ktrans/cis = [trans]/[cis] to that for amide 1. The resulting data reveal the manifestation of a previously unappreciated electronic effect on protein structure.
Results and Discussion
Existence of an electronic effect
Steric repulsion is perhaps the most well-known factor in molecular conformational stability. Based on steric considerations alone, the value of Ktrans/cis for amide 1 should be <1. Yet, we find that the value of Ktrans/cis, as determined by 1H-NMR spectroscopy, is actually >1 in water, dioxane, and chloroform (Table 1, Fig. 2 ▶). This result provides the first experimental evidence that steric effects alone are not the sole determinant of the preference of a peptide bond for the trans conformation.
Table 1.
Values of Ktrans/cis for amides 1–3
| Solvent | |||
| Amide | D2O | Dioxane-d8 | CDCl3 |
| FmProOMe (1) | 1.8 | 1.4 | 1.4 |
| AcProOMe (2) | 5.3 | 4.2 | 4.0 |
| Fm(dmPro)OMe (3) | 25 | 9 | 13 |
Values of Ktrans/cis (±20%) were determined by integration of 1H-NMR spectra obtained at 25°C. Resonances of the trans and cis isomers were assigned by using NOESY 1D spectroscopy with observed NOEs between the formyl (C′i−1–H) or acetyl (Cαi−1–H) and α (Cαi–H) or δ (Cδi–H) protons.
Figure 2.

1H NMR spectrum of N-formyl-l-proline methyl ester (1) in dioxane-d8 at 25°C. Values of Ktrans/cis were determined from integration of the indicated resonances.
What is the explanation for the value of Ktrans/cis for amide 1 being >1? In a peptide bond, the oxygen (Oi−1) bears a partial negative charge, and the carbon (C′i) bears a partial positive charge (Pauling 1960; Momany et al. 1975; Zimmerman and Scheraga 1976). The favorable Coulombic interaction between Oi−1 and C′i could, of course, increase the value of Ktrans/cis (Zimmerman and Scheraga 1976). The true picture is, however, more complex.
No less important than steric effects in dictating molecular conformation are the stabilizing effects of hyperconjugative delocalization (Cramer 1998; Weinhold 2001). A familiar manifestation of hyperconjugation in a biomolecule is the “anomeric effect,” which arises from the delocalization of a nonbonding pair of electrons (n) from the ring oxygen of pyranose sugars to the σ* orbital of the adjacent C–O bond (Deslongchamps 1983; Petillo and Lerner 1993). This n → σ* interaction stabilizes the α anomer.
We have analyzed in detail the conformational energetics of amides like 1 with density functional theory calculations using the natural bond orbital (NBO) paradigm (DeRider et al. 2002). These previous calculations reveal that the interaction between Oi−1 and C′i has a large dependence on the Oi−1· · · C′i=Oi angle as well as the Oi−1· · · C′i distance, and is better described by quantum mechanics than by simple electrostatics. More precisely, the increase in the value of Ktrans/cis arises from extensive hyperconjugative delocalization of a nonbonding pair of electrons (n) from the amide oxygen to the π* orbital of the ester carbon (Fig. 1B ▶).
The n → π* interaction in amide 1 is reminiscent of the Bürgi–Dunitz trajectory of organic chemistry, which describes the most favorable approach of a nucleophile (e.g., Oi−1) to the carbon of a carbonyl group (e.g., C′i =Oi) during an acyl transfer reaction (Fig. 1C ▶; Bürgi et al. 1973, 1974a,b; Bürgi and Dunitz 1983). Accordingly, the hyperconjugative delocalization that arises from an n → π* interaction between Oi−1 and C′i can be depicted as a very minor resonance form of amide 1 (Fig. 1D ▶). A similar structure is a likely intermediate during the formation of a 2-oxazolin-5-one, or “azlactone,” which is a cleavage product that can arise during the chemical synthesis of peptides (Dakin and West 1928a,b,c,Kemp 1979).
Strength of n → π* interaction
The strength of the n → π* interaction in amides 1 and 2 can be estimated from Ktrans/cis values for isologous amides that lack a C′i=Oi group. The isopropyl and methyl groups of N-isopropyl-N-methylformamide present steric hindrance to Oi−1 and Hi−1 that are similar to those in amide 1 (Scheme 2 ▶). In contrast to amide 1, the trans isomer of N-isopropyl-N-methylformamide (4)
Scheme 2.

lacks any stabilization from an n → π* interaction, and its value of Ktrans/cis is threefold lower than that of amide 1 (Laplanche and Rogers 1963; Stewart and Siddall III 1970). A comparison of N-isopropyl-N-methylacetamide and amide 2 yields a similar result (Scheme 3 ▶). From these data,
Scheme 3.

we estimate that the n → π* interaction contributes approximately 0.7 kcal/mole (= RTln3) at 25°C to the stability of the trans isomer of amides 1 and 2. This value is in gratifying agreement with the previous density functional theory calculations (DeRider et al. 2002), which indicate that n → π* delocalization provides 0.42 kcal/mole of stabilization energy when the pyrrolidine ring of amide 2 is in the Cγ-endo (major: 66%) pucker and 1.29 kcal/mole in the Cγ-exo (minor: 34%; Fig. 1B ▶) pucker. These calculations likewise assign a value of 0.7 kcal/mole (= 0.42 kcal/mole × 66% + 1.29 kcal/mole × 34%) to the strength of the n → π* interaction. It is also noteworthy that the preference for a trans isomer of amide 2 has an almost entirely enthalpic origin (Eberhardt et al. 1993), which is consistent with the enthalpic contribution expected from an n → π* interaction as well as steric effects. Finally, we recognize that an amide carbon is somewhat less electron-deficient than is an ester carbon. As a consequence, the conformational stability provided by an n → π* interaction will be lower, but still manifested, in an amide. That stability could be similar in magnitude to that from a cation–π interaction (Shi et al. 2002a), another nonclassical noncovalent interaction (Dougherty 1996).
Replacing the formyl or Cδ protons of amide 1 with bulkier methyl groups increases steric effects in a manner that should favor the trans isomer (Scheme 1 ▶). The value of Ktrans/cis does indeed increase in the order 1 < 2 < 3 (Table 1). We conclude that steric effects are manifested in amides 1–3. Likewise, the identity of the residue preceding a proline residue is a primary determinant of the prevalence of a trans prolyl peptide bond (Grathwohl and Wüthrich 1981; Yao et al. 1994), as increasing steric hindrance tends to increase the relative stability of the trans isomer in peptides. For example, Glyi−1–Proi and Alai−1–Proi peptide bonds have Ktrans/cis values in water of 6.2 and 12, respectively (Reimer et al. 1998). Finally, 5,5-dimethylproline is known to provide a conformational lock for the cis conformation in peptides (An et al. 1999). Clearly, steric effects make a dominant contribution to the preference for the trans isomer of peptide bonds.
Implications of n → π* interaction
Proline has a unique influence on protein structure (MacArthur and Thornton 1991; Reiersen and Rees 2001). This influence arises largely from the constraint imposed by the pyrrolidine ring on its φ torsion angle, which tends to range from −75° in the Cγ-endo pucker to −60° in the Cγexo pucker (Vitagliano et al. 2001). This constraint preorganizes proline for a favorable n → π* interaction (DeRider et al. 2002). Moreover, the subtle modulation of this interaction, as occurs in diastereomers of 4-hydroxyproline and 4-fluoroproline, has been shown to affect the value of Ktrans/cis (Bretscher et al. 2001; DeRider et al. 2002). An n → π* interaction is also likely to increase the value of Ktrans/cis for Xaai−1–nonProi peptide bonds, albeit to a lesser extent.
The influence of an n → π* interaction extends beyond being a determinant of the value of Ktrans/cis. A meaningful n → π* interaction can be defined as one in which the Oi−1· · · C′i distance is δBD ≤ 3.2 Å and the Oi−1· · · C′i=Oi angle is 99° ≤ τBD ≤ 119°, which is ±10° of the Bürgi–Dunitz trajectory (Fig. 1C ▶). Such an optimal n → π* interaction can exist in a polyproline II helix (PII), right-handed α-helix (αR), and left-handed α-helix (αL; Fig. 3A ▶). Each of these secondary structures could gain stability from this electronic effect.
Figure 3.
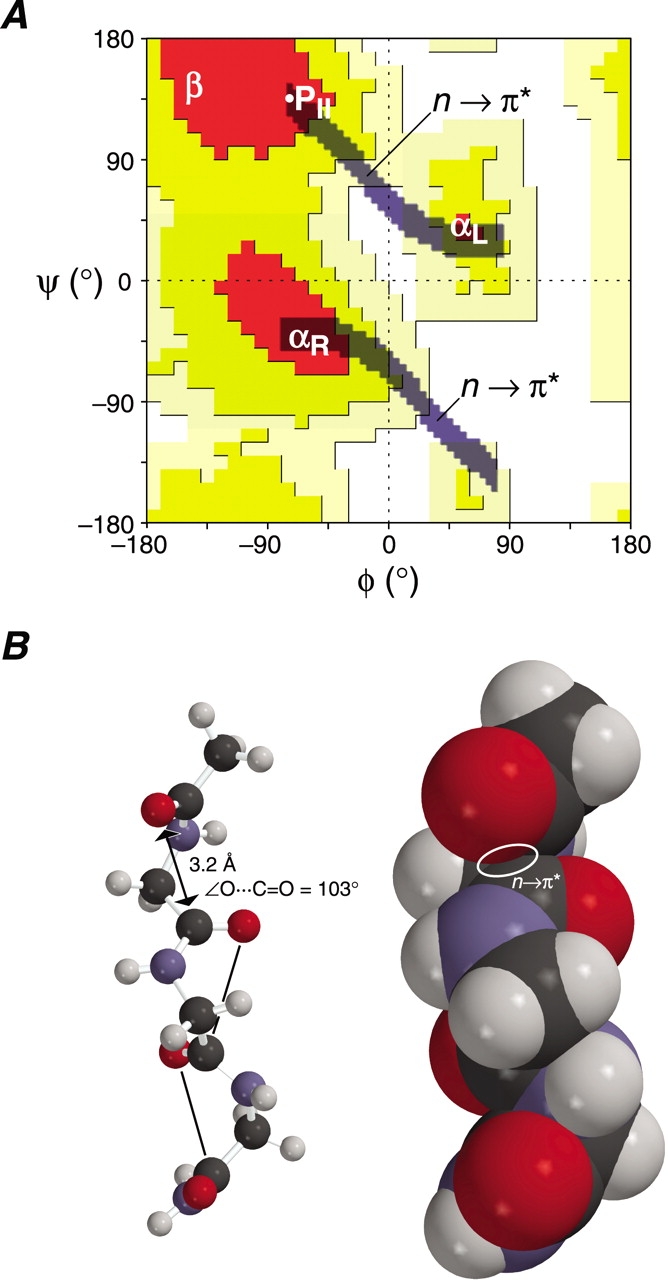
Implications of the n → π* interaction between Oi−1 and C′i. (A) Ramachandran plot (Ramachandran and Sasisekharan 1968; Richardson 1981) showing the two “n → π*” regions of the trans isomer of AcGlyNH2. In these regions, the Oi−1· · · C′i distance is δBD ≤ 3.2 Å and the Oi−1· · · C′i=Oi angle is 99° ≤ τBD ≤ 119°. The white dot indicates the φ and ψ angles for an ideal polyproline II helix (B). (B) Energy-minimized structure of AcGly3NH2 in the conformation of a polyproline II helix with φ = −75° and ψ = +145°. The structure is depicted as a ball-and-stick (left) or space-filling (right) model. The Oi−1· · · C′i distance (δBD = 3.2 Å) and Oi−1· · · C′i=Oi angle (τBD = 103°) is indicated in the ball-and-stick model.
An n → π* interaction is likely to be of particular importance to the conformational stability of a PII helix. An ideal PII helix has main-chain dihedral angles (φ = −75°, ψ = +145°) that produce a favorable n → π* interaction, with δBD = 3.2 Å and τBD = 103° (Fig. 3B ▶). Unlike an α-helix, a PII helix lacks intrastrand hydrogen bonds. Yet, the PII helix is prevalent in polyproline (Isemura et al. 1968; Tiffany and Krimm 1968), has been observed in Lys7 and Ala7 peptides (Rucker and Creamer 2002; Shi et al. 2002b), and is a common motif in folded proteins (Siligardi and Drake 1995; Kleywegt and Jones 1996). An n → π* interaction between Oi−1 and C′i does not rely on solvation. Likewise, specific hydration is not critical for the conformational stability of a PII helix, which can form in methanol, trifluoroethanol, propionic acid, and benzyl alcohol (Knof and Engel 1974; Antonyraj et al. 1998; Nakamura et al. 2001). Analogous solvent-independent stereoelectronic effects enhance the conformational stability of the collagen triple helix (Engel and Prockop 1998; Holmgren et al. 1998; Bretscher et al. 2001; Jenkins and Raines 2002), in which each residue has φ and ψ torsion angles that are similar to those in a PII helix (Bella et al. 1994).
In the analysis above, we have treated the C′i(Oi)–Ni+1 amide as merely a ketone. Of course, the immonium resonance form C′i(Oi− ) = Ni+1+, which has been estimated to contribute as much as 40% to the structure of an amide (Pauling 1960), must also play a role in determining the most favorable position for Oi−1 (Baldwin 1976). Ideally, the Oi−1· · · C′i=Ni+1+ angle, like the Oi−1· · · C′i=Oi angle, would be near 109°. Remarkably, the Oi−1· · · C′i=Ni+1+ angle in a PII helix is identical to the Oi−1· · · C′i=Oi angle (Fig. 3B ▶), as both are τBD = 103°. Thus, a PII helix is stabilized by hyperconjugative delocalization of n from Oi−1 to the π* orbitals of both C′i=Oi and C′i=Ni+1+.
A favorable Coulombic interaction between Oi−1 and C′i has been proposed to enhance the stability of the right-handed twist of β-strands (Maccallum et al. 1995). The Oi−1· · · C′i distance (δBD = 3.45 Å) and Oi−1· · · C′i=Oi angle (τBD = 125°) in a right-twisted β-strand (φ = −90°, ψ = +125°) are, however, inappropriate for a strong n → π* interaction (Fig. 3A ▶). We therefore believe that an interaction between Oi−1 and C′i is unable to provide substantial stability to the right-handed twist of β-strands.
Finally, we note that the contribution of an n → π* interaction to conformational stability can be cooperative. Both the negative charge on Oi and the C′i=Oi bond length increase as a result of an n → π* interaction between Oi−1 and C′i (DeRider et al. 2002). These effects can in turn serve to increase the stabilization provided by an n → π* interaction between Oi and C′i+1.
Materials and methods
General
Chemicals and solvents were from Aldrich. Reactions were monitored by thin-layer chromatography using TLC plates (AL SIL G/UV) from Whatman, with visualization by illumination with ultraviolet light or staining with I2. NMR spectra were obtained with Bruker AC-250, Bruker AC-300, and Varian UNITY-500 spectrometers. Mass spectra were obtained with electrospray ionization (ESI) techniques at the University of Wisconsin Biotechnology Center.
Synthesis of N-formyl-l-proline methyl ester (1)
l-Proline (2.0 g, 17.4 mmole) was dissolved in formic acid (25 mL), and the resulting solution was cooled to 0°C. The cooled solution was added to a mixture of acetic anhydride (20 mL, 218 mmole) in formic acid (25 mL), which was also at 0°C. The resulting solution was allowed to warm to room temperature overnight. The solvent was then removed under reduced pressure. The residue was purified by flash chromatography (30 g silica gel, 5% v/v methanol in chloroform). Fractions containing N-formyl-l-proline were pooled, and the solvent was removed under reduced pressure to yield N-formyl-l-proline (1.73 g, 63%) as a clear oil. Rf = 0.6 (10% v/v methanol in chloroform). 1H-NMR (CDCl3, 300 MHz) δ 11.7 (s, 1H), 8.34 (s, 0.6H), 8.27 (s, 0.4H), 4.45–4.52 (m, 1H), 3.63–3.77 (m, 1H), 3.55 (apparent triplet, J = 7 Hz, 1H) 2.22–2.38 (m, 1H), 1.89–2.19 (m, 3H). 13C-NMR (CDCl3, 75 MHz) δ 173.7, 173.6, 163.1, 162.2, 58.9, 56.6, 46.8, 44.1, 29.4, 29.0, 23.6, 22.5. MS (ESI) m/z 166.0 (M + Na+).
N-Formyl-l-proline was converted to amide 1 by esterification with diazomethane. Diazomethane was generated in situ with an Aldrich minidiazald reactor. (CAUTION! Accumulated diazomethane is highly explosive. See Aldrich Technical Information Bulletin No. AL-180.) Diazald (1.5 g, 7.0 mmole) was mostly dissolved in ether (10 mL). Residual solids were removed by decanting the yellow solution into a dropping funnel with clear-seal glass joints. At a rate of ∼1 drop every 2–3 sec, the diazald solution was added to the diazald reactor, which contained KOH (1.00 g, 17.9 mmole) dissolved in water/ethanol (40:60 v/v, 2.5 mL) heated to 70°C. The ethereal diazomethane immediately distilled over to the second chamber and condensed against a coldfinger at −78°C. The yellow diazomethane droplets then dripped into a solution of N-formyl-l-proline (0.50 g, 3.2 mmole) in ether/acetonitrile (50:50 v/v, 25 mL) cooled to 0°C. The reaction continued until the product solution turned light yellow. The reaction was then quenched with glacial acetic acid. The product was isolated by removal of the volatile components under reduced pressure. The residue was purified by flash chromatography (30 g silica gel, ethyl acetate/hexanes [50:50–100:0 v/v]). Fractions containing amide 1 were pooled, and the solvent was removed under reduced pressure to yield amide 1 as a pale yellow oil (0.53 g, 97%). Rf = 0.45 (ethyl acetate/hexane [80:20 v/v], I2 staining). 1H-NMR (D2O, 500 MHz) δ 8.28 (s, 0.64H), 8.23 (s, 0.36H), 4.76 (dd, J = 8, 3 Hz, 0.36H) 4.52 (m, 0.64H), 3.70–3.85 (m, 4.3H). 3.55 (m, 0.7H), 2.22–2.46 (m, 1.3H), 1.98–2.12 (m, 2.6H). 13C-NMR (dioxane-d8, 125 MHz) δ 172.9, 172.4, 161.3, 160.5, 58.7, 56.5, 52.2, 51.8, 46.1, 43.8, 29.8, 29.6, 24.3, 23.1. MS (ESI) m/z 180.0 (M + Na+). IR Data: (neat) vmax 1742.8 (s), 1672.1 (s).
Synthesis of acetyl-l-proline methyl ester (2)
Amide 2 was synthesized as described previously (Panasik Jr. et al. 1994).
Synthesis of N-formyl-5,5-dimethyl-l-proline methyl ester (3)
2-(3-Methyl-3-nitro-butyl)-[1,3]dioxolane was prepared as described previously (Bonnett et al. 1959). 2-(3-Methyl-3-nitro-butyl)-[1,3]dioxolane (68 g, 0.36 mole) was dissolved in methanol (0.25 L). Raney nickel was added to this solution, and the flask was filled with H2(g) from a balloon. (CAUTION! Raney nickel is highly pyrophoric and will ignite methanol vapors if dry.) Periodically, the reaction was assayed for completion by 13C-NMR spectroscopy. Completion times ranged from overnight to 10 days, depending on the scale and catalyst loading. Upon completion, the catalyst was carefully filtered away. The solvent was removed under reduced pressure to yield 3-[1,3]dioxolan-2-yl-1,1-dimethyl-propylamine as a pale yellow oil (56.5 g, 99%). 1H-NMR (CDCl3, 300 MHz) δ 4.84 (t, J = 5 Hz, 1H), 3.82–3.98 (m, 4H), 1.65–1.75 (m, 2H), 1.42–1.51 (m, 2H), 1.10 (s, 6H). 13C-NMR (CDCl3, 75.5 MHz) 104.0, 64.1, 48.3, 38.1, 29.67, 28.5. MS (ESI) m/z 160.2 (M + H+).
3-[1,3]Dioxolan-2-yl-1,1-dimethyl-propylamine (15.3g, 96 mmole) was dissolved in hot water (40 mL) and the pH lowered to ∼3 with 2 N HCl. The resulting solution was heated at reflux for 30 min, and then made basic by the addition of 6 N KOH and extracted (4×) with chloroform. The organic layer was dried over MgSO4(s), and the solvent was removed under reduced pressure to yield a black oil. The black oil was purified by distillation under reduced pressure to yield 2,2-dimethyl-3,4-dihydro-2H-pyrrole as a clear pungent oil (7.5 g, 80%). 1H-NMR (CDCl3, 300 MHz) δ 7.33 (broad s, 1H), 2.52 (t, J = 7 Hz, 2H), 1.56 (t, J = 7 Hz, 2H), 1.2 (s, 6H). 13C-NMR (CDCl3, 75.4 MHz) δ 163.1, 72.9, 36.7, 34.3, 28.5.
2,2-Dimethyl-3,4-dihydro-2H-pyrrole (12.4 g, 128 mmole) was dissolved in water (70 mL) at 0°C. KCN (16.3 g, 251 mmole) was added to the solution, and the pH was lowered from ∼13.4 to ∼6 over 2 h by the addition of 2 N HCl (128 mL, 256 mmole). The pH continued to rise, but was maintained near 6 by the addition of 2 N HCl. After 3 h at 0°C, the solution was made basic by the addition of 2 N NaOH, and extracted (4×) with chloroform. The combined organic extract was dried with MgSO4(s), and the solvent removed under reduced pressure. The residue was purified by flash chromatography (0.4 kg silica gel, ethyl acetate/hexanes [65:35 v/v]). Fractions containing 5,5-dimethyl-pyrrolidine-2-carbonitrile were pooled, and the solvent was removed under reduced pressure to yield 5,5-dimethyl-pyrrolidine-2-carbonitrile as a pale yellow oil (7.47 g, 46%). 1H-NMR (CDCl3, 300 MHz) δ 4.07–4.12 (apparent dd, J = 8, 5 Hz, 1H), 2.12–2.36 (m, 2H), 1.79–1.90 (m, 2H), 1.61–1.72 (m, 1H), 1.30 (s, 3H), 1.17 (s, 3H). 13C-NMR (CDCl3, 75.4 MHz) δ 122.0, 59.5, 46.5, 38.3, 30.9, 29.0, 28.8. 13C-DEPT-135 δ 46.5, 29.0, 28.8 positive (CH or CH3), 38.3, 30.9 negative (CH2). MS (ESI) m/z 125.2 (M + H+).
5,5-Dimethyl-pyrrolidine-2-carbonitrile was hydrolyzed to form 5,5-dimethylproline as described previously (Bonnett et al. 1959). Racemic 5,5-dimethylproline was resolved with d-tartrate as described previously (An et al. 1999) to yield 5,5-dimethyl-l-proline in greater than 97% ee. 5,5-Dimethyl-l-proline was converted to N-formyl-5,5-dimethyl-l-proline methyl ester by the procedure used to convert l-proline to N-formyl-l-proline methyl ester (vide supra). 1H NMR (D2O, 500 MHz) δ 8.25 (s, 0.96H), 8.10 (s, 0.04H), 4.48–4.58 (m, 1H), 3.76 (s, 3H), 2.2–2.45 (m, 1H), 1.85–2.15 (m, 3H), 1.47 (s, 3H), 1.42 (s, 3H). 1H-NMR (dioxane-d8, 500 MHz) δ 8.22 (s, 0.89H), 8.09 (s, 0.11H), 4.37–4.45 (m, 1H), 3.71 (s, 0.45H), 3.65 (s, 2.55H), 2.12–2.23 (m, 1H), 1.84–1.93 (m, 1.8H), 1.75–1.83 (m, 1.2H), 1.46–1.49 (2 singlets, 3H total), 1.35–1.38 (2 singlets, 3H total). 13C-NMR (dioxane-d8, 125 MHz) δ 173.4, 172.5, 161.2, 159.8, 61.6, 61.0, 52.2, 51.8, 40.6, 40.0, 28.9, 28.8, 27.3, 26.6, 24.9. 1H-NMR (CDCl3, 250 MHz) δ 8.30 (s, 0.92H), 8.22 (s, 0.8H), 4.39–4.59 (m, 1H), 3.78 (s, 0.33H), 3.74 (s, 2.67H), 2.15–2.35 (m, 0.96H), 1.78–2.08 (m, 3.04H), 1.54–1.6 (pair of singlets, 3H total), 1.41–1.44 (pair of singlets, 3H total). 13C-NMR (CDCl3, 62.9 MHz) δ 171.8, 161.2, 160.0, 60.5, 58.1, 52.0, 39.3, 29.1, 28.7, 26.3. 13C NMR DEPT-135 δ 160.0, 58.1, 52.0, 29.1, 28.7 are positive (CH or CH3). δ 39.3, 26.3 are negative (CH2). MS (ESI) m/z 208.1 (M + Na+), 160.1.
Determination of Ktrans/cis values
cis and trans resonances of amides 1–3 were identified by using NOESY1D spectroscopy as described in the supporting information. Values of Ktrans/cis were determined by integration of 1H NMR spectra obtained at 25°C.
Depiction of natural bond orbitals
The n and π* NBOs of the trans isomer of AcProOMe (2) in its Cγ-exo conformation were depicted in Figure 1B ▶ with the program gOpenMol, version 2.2 (Laaksonen 1992; Bergman et al. 1997) by using NBO 4.0 output from the B3LYP/6-311+G(2d,p) level of theory (DeRider et al. 2002).
Calculation of O i−1· · · Ci′ distances and Oi′· · · Ci′=Oi angles
The φ (C′i−1–Ni–Cαi–C′i) and ψ (Ni–Cαi–C′i–Ni+1) torsion angles in the trans (ω = 180°) isomer of AcGlyNH2 were varied in 5°-increments before energy minimization with the program MacSpartanPro, version 1.0.4 (Wavefunction). The Oi−1· · · C′i distance (δBD) and Oi−1· · · C′i=Oi angle (τBD) were recorded in each energy-minimized structure. Those structures with Oi−1· · · C′i distances δBD ≤ 3.2 Å and Oi−1· · · C′i=Oi angles 99° ≤ τBD ≤ 119° were depicted in Figure 3A ▶ on a Ramachandran plot (Ramachandran and Sasisekharan 1968; Richardson 1981).
Electronic supplemental material
NOESY 1D, 1H-, and 13C-NMR spectra and IR spectra are available for amides 1–3.
Acknowledgments
This article is dedicated to the memory of Peter A. Kollman. This work was supported by grant AR447276 (NIH). We are grateful to S.H. Gellman, J.A. Hodges, C.L. Jenkins, G.R. Krow, B.G. Miller, G.N. Phillips, F. Weinhold, and W.M. Westler for contributive discussions. M.P.H. was supported by Chemistry–Biology Interface Training Grant GM08506 (NIH). NMR spectra were obtained at the Magnetic Resonance Facility in the Department of Chemistry, which was supported by grants CHE-8813550 (NSF), CHE-9629688 (NSF), and S10 RR04981–01 (NIH). IR spectra were obtained at the Biophysics Instrumentation Facility, which was supported by grants BIR-9512577 (NSF) and S10 RR13790 (NIH).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: See www.proteinscience.org.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0241903.
References
- An, S.S.A., Lester, C.C., Peng, J.-L., Li, Y.-J., Rothwarf, D.M., Welker, E., Thannhauser, T.W., Zhang, L.S., Tam, J.P., and Scheraga, H.A. 1999. Retention of the cis proline conformation in tripeptide fragments of bovine pancreatic ribonuclease A containing a non-natural proline analogue, 5,5-dimethylproline. J. Am. Chem. Soc. 121 11558–11566. [Google Scholar]
- Antonyraj, K.J., Karunakaran, T., and Raj, P.A. 1998. Bactericidal activity and poly-L-proline II conformation of the tandem repeat sequence of human salivary mucin glycoprotein (MG2). Arch. Biochem. Biophys. 356 197–206. [DOI] [PubMed] [Google Scholar]
- Baldwin, J.E. 1976. Approach vector analysis: A stereochemical approach to reactivity. Chem. Commun. 738–741.
- Bella, J., Eaton, M., Brodsky, B., and Berman, H.M. 1994. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science 266 75–81. [DOI] [PubMed] [Google Scholar]
- Benzi, C., Improta, R., Scalmani, G., and Barone, V. 2002. Quantum mechanical study of the conformational behavior of proline and 4R-hydroxyproline dipeptide analogues in vacuum and in aqueous solution. J. Comput. Chem. 23 341–350. [DOI] [PubMed] [Google Scholar]
- Bergman, D.L., Laaksonen, L., and Laaksonen, A. 1997. Visualization of solvation structure in liquid mixtures. J. Mol. Graph. Model. 15 301–306. [DOI] [PubMed] [Google Scholar]
- Bonnett, R., Clark, V.M., Giddey, A., and Todd, A. 1959. Experiments towards the synthesis of corrins. I. Preparation and reactions of some Δ 1-pyrrolines. A novel proline synthesis. J. Chem. Soc. 2087–2093.
- Borrmann, H., Persson, I., Sandström, M., and Stålhandske, C.M.V. 2000. The crystal and liquid structures of N,N-dimethylthioformamide and N,N-dimethylformamide showing a stronger hydrogen bonding effect for C–H· · · S than for C–H· · · O. J. Chem. Soc. Perkin Trans. 2 393–402. [Google Scholar]
- Bretscher, L.E., Jenkins, C.L., Taylor, K.M., and Raines, R.T. 2001. Conformational stability of collagen relies on a stereoelectronic effect. J. Am. Chem. Soc. 123 777–778. [DOI] [PubMed] [Google Scholar]
- Bürgi, H.B. and Dunitz, J.D. 1983. From crystal statics to chemical dynamics. Acc. Chem. Res. 16 153–161. [Google Scholar]
- Bürgi, H.B., Dunitz, J.D., and Shefter, E. 1973. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 95 5065–5067. [Google Scholar]
- Bürgi, H.B., Dunitz, J.D., Lehn, J.M., and Wipff, G. 1974a. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 30 1563–1572. [Google Scholar]
- Bürgi, H.B., Lehn, J.M., and Wipff, G. 1974b. An ab initio study of nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 96 1965–1966. [Google Scholar]
- Cramer, C.J. 1998. Hyperconjugation. In Encyclopedia of computational chemistry (ed. P.V.R. Schleyer), pp. 1294–1298. John Wiley & Sons, New York.
- Creighton, T.E. 1993. Proteins: Structure and molecular properties, 2nd ed., p. 5. W. H. Freeman, New York.
- Dakin, H.D. and West, R. 1928a. A general reaction of amino acids. J. Biol. Chem. 78 91–105. [Google Scholar]
- ———. 1928b. A general reaction of amino acids. II. J. Biol. Chem. 78 745–756. [Google Scholar]
- ———. 1928c. Some aromatic derivatives of substituted acetylaminoacetones. J. Biol. Chem. 78 757–764. [Google Scholar]
- DeRider, M.L., Wilkens, S.J., Waddell, M.J., Bretscher, L.E., Weinhold, F., Raines, R.T., and Markley, J.L. 2002. Collagen stability: Insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations. J. Am. Chem. Soc. 124 2497–2505. [DOI] [PubMed] [Google Scholar]
- Deslongchamps, P. 1983. Stereoelectronic effects in organic chemistry, pp. 7–20. Pergamon Press, New York.
- DeTar, D.F. and Luthra, N.P. 1977. Conformations of proline. J. Am. Chem. Soc. 99 1232–1244. [DOI] [PubMed] [Google Scholar]
- Dougherty, D.A. 1996. Cation–π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 271 163–168. [DOI] [PubMed] [Google Scholar]
- Eberhardt, E.S., Loh, S.N., and Raines, R.T. 1993. Thermodynamic origins of prolyl peptide bond isomers. Tetrahedron Lett. 34 3055–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliel, E.L. and Wilen, S.H. 1994. Stereochemistry of organic compounds, p. 877. Wiley-Interscience, New York.
- Engel, J. and Prockop, D.J. 1998. Does bound water contribute to the stability of collagen? Matrix Biol. 17 679–680. [DOI] [PubMed] [Google Scholar]
- Fischer, G. 2000. Chemical aspects of peptide bond isomerisation. Chem. Soc. Rev. 29 119–127. [Google Scholar]
- Grathwohl, C. and Wüthrich, K. 1981. NMR studies of the rates of proline cis-trans isomerization in oligopeptides. Biopolymers 20 2623–2633. [Google Scholar]
- Higashijima, T., Tasumi, M., and Miyazawa, T. 1977. 1H Nuclear magnetic resonance studies of N-acetyl-L-proline N-methylamide. Molecular conformations, hydrogen bondings, and thermodynamic quantities in various solvents. Biopolymers 16 1259–1270. [DOI] [PubMed] [Google Scholar]
- Holmgren, S.K., Taylor, K.M., Bretscher, L.E., and Raines, R.T. 1998. Code for collagen’s stability deciphered. Nature 392 666–667. [DOI] [PubMed] [Google Scholar]
- Isemura, T., Okabayashi, H., and Sakakibara, S. 1968. Steric structure of L-proline oligopeptides. I. Infrared absorption spectra of the oligopeptides and poly-L-proline. Biopolymers 6 307–321. [DOI] [PubMed] [Google Scholar]
- Jabs, A., Weiss, M.S., and Hilgenfeld, R. 1999. Non-proline cis peptide bonds in proteins. J. Mol. Biol. 286 291–304. [DOI] [PubMed] [Google Scholar]
- Jenkins, C.L. and Raines, R.T. 2002. Insights on the conformational stability of collagen. Nat. Prod. Rep. 19 49–59. [DOI] [PubMed] [Google Scholar]
- Kemp, D.S. 1979. Racemization in peptide synthesis. In The Peptides (eds. E. Gross and J. Meienhofer), vol. 1. Academic Press, New York.
- Kleywegt, G.J. and Jones, T.A. 1996. Phi/Psi-chology: Ramachandran revisited. Structure 4 1395–1400. [DOI] [PubMed] [Google Scholar]
- Knof, S. and Engel, J. 1974. Conformational stability, partial specific volumes and spectroscopic properties of poly-L-proline, poly-L-hydroxyproline and some of its O-acyl-derivatives in various solvent systems. Israel J. Chem. 12 165–177. [Google Scholar]
- Kyte, J. 1995. Structure in protein chemistry, p. 197. Garland Publishing, New York.
- Laaksonen, L. 1992. A graphics program for the analysis and display of molecular dynamics trajectories. J. Mol. Graph. 10 33–34. [DOI] [PubMed] [Google Scholar]
- Laplanche, L. and Rogers, M.T. 1963. Configurations in unsymmetrically N,N-disubstituted amides. J. Am. Chem. Soc. 85 3728–3730. [Google Scholar]
- Liang, G.B., Rito, C.J., and Gellman, S.H. 1992. Variations in the turn-forming characteristics of N-acyl proline units. Biopolymers 32 293–301. [DOI] [PubMed] [Google Scholar]
- MacArthur, M.W. and Thornton, J.M. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218 397–412. [DOI] [PubMed] [Google Scholar]
- Maccallum, P.H., Poet, R., and Milner-White, E.J. 1995. Coulombic attractions between partially charged main-chain atoms stabilise the right-handed twist found in most β-strands. J. Mol. Biol. 248 374–384. [DOI] [PubMed] [Google Scholar]
- Madison, V. and Schellman, J. 1970. Location of proline derivatives in conformational space. I. Conformational calculations; optical activity and NMR experiments. Biopolymers 9 511–567. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, T. and Iitaka, Y. 1971. The crystal structure of acetyl-L-proline-N-methylamide. Acta Crystallogr. Sect. B 27 507–516. [Google Scholar]
- Momany, F.A., McGuire, R.F., Burgess, A.W., and Scheraga, H.A. 1975. Energy parameters in polypeptides. VII. Geometric parameters, partial atomic charges, nonbonded interactions, hydrogen bond interactions, and intrinsic. J. Phys. Chem. 79 2361–2381. [Google Scholar]
- Nakamura, T., Wakahara, M., Oka, M., Hayashi, T., Hattori, M., and Hirano, Y. 2001. Conformational analysis of model polypeptides having repitive Ala-Pro-Pro-Pro sequence. In Peptide science 2000 (ed. T. Shioiri), pp. 321–324. Japanese Peptide Society, Tokyo.
- Panasik Jr., N., Eberhardt, E.S., Edison, A.S., Powell, D.R., and Raines, R.T. 1994. Inductive effects on the structure of proline residues. Int. J. Pept. Protein Res. 44 262–269. [DOI] [PubMed] [Google Scholar]
- Pauling, L. 1960. The nature of the chemical bond, 3rd ed., pp. 281–282. Cornell University Press, Ithaca, NY.
- Petillo, P.A. and Lerner, L.E. 1993. Origin and quantitative modeling of anomeric effect. In The anomeric effect and associated stereoelectronic effects (ed. G.R.J. Thatcher), pp. 156–175. American Chemical Society, Washington, DC.
- Ramachandran, G.N. and Sasisekharan, V. 1968. Conformation of polypeptides and proteins. Adv. Protein Chem. 23 283–437. [DOI] [PubMed] [Google Scholar]
- Reiersen, H. and Rees, A.R. 2001. The hunchback and its neighbours: Proline as an environmental modulator. Trends Biochem. Sci. 26 679–684. [DOI] [PubMed] [Google Scholar]
- Reimer, U., Scherer, G., Drewello, M., Kruber, S., Schutkowski, M., and Fischer, G. 1998. Side-chain effects on peptidyl-prolyl cis/trans isomerization. J. Mol. Biol. 279 449–460. [DOI] [PubMed] [Google Scholar]
- Richardson, J. 1981. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 34 167–339. [DOI] [PubMed] [Google Scholar]
- Rucker, A.L. and Creamer, T.P. 2002. Polyproline II helical structure in protein unfolded states: Lysine peptides revisited. Protein Sci. 11 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz, G.E. and Schirmer, R.H. 1979. Principles of protein structure, pp. 25–26. Springer-Verlag, New York.
- Shi, Z., Olson, C.A., and Kallenbach, N.R. 2002a. Cation–π interaction in model α -helical peptides. J. Am. Chem. Soc. 124 3284–3291. [DOI] [PubMed] [Google Scholar]
- Shi, Z., Olson, C.A., Rose, G.D., Baldwin, R.L., and Kallenbach, N.R. 2002b. Polyproline II structure in a sequence of seven alanine residues. Proc. Natl. Acad. Sci. 99 9190–9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siligardi, G. and Drake, A.F. 1995. The importance of extended conformtions and, in particular, the PII conformation for the molecular recognition of peptides. Biopolymers (Peptide Sci) 37 281–292. [DOI] [PubMed] [Google Scholar]
- Stewart, D.E., Sarkar, A., and Wampler, J.E. 1990. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 214 253–260. [DOI] [PubMed] [Google Scholar]
- Stewart, W.E. and Siddall III, T.H. 1970. Nuclear magnetic resonance studies of amides. Chem. Rev. 70 517–551. [Google Scholar]
- Tiffany, M.L. and Krimm, S. 1968. Circular dichroism of poly-L-proline in an unordered conformation. Biopolymers 6 1767–1770. [DOI] [PubMed] [Google Scholar]
- Vitagliano, L., Berisio, R., Mazzarella, L., and Zagari, A. 2001. Structural bases of collagen stabilization induced by proline hydroxylation. Biopolymers 58 459–464. [DOI] [PubMed] [Google Scholar]
- Weinhold, F. 2001. A new twist on molecular shape. Nature 411 539–541. [DOI] [PubMed] [Google Scholar]
- Weiss, M.S., Jabs, A., and Hilgenfeld, R. 1998. Peptide bonds revisited. Nat. Struct. Biol. 5 676. [DOI] [PubMed] [Google Scholar]
- Yao, J., Feher, V.A., Espejo, B.F., Reymond, M.T., Wright, P.E., and Dyson, H.J. 1994. Stabilisation of a type VI turn in a family of linear peptides in water solution. J. Mol. Biol. 243 746–753. [DOI] [PubMed] [Google Scholar]
- Zimmerman, S.S. and Scheraga, H.A. 1976. Stability of cis, trans, and nonplanar peptide groups. Macromolecules 9 408–416. [DOI] [PubMed] [Google Scholar]