

Abstract
FSD-1 (full sequence design 1) is a protein folded in a ββα motif, designed on the basis of the second zinc finger domain of Zif268 by a substitution of its metal coordination site with a hydrophobic core. In this work, we analyzed the possibility of introducing the DNA recognition motif of the template zinc finger (S13RSDH17) into FSD-1 sequence in order to obtain a small DNA-binding module devoid of cross-link(s) or metal cofactors. The hybrid protein was unfolded, as judged by CD and NMR criteria. To reveal the role of each of the five amino acids, which form the N-capping motif of the α-helix, we analyzed conformational and stability properties of eight FSD-1 mutants. We used a shielded methyl group of Leu 18 and a CD signal at 215 nm as a convenient measure of the folded state. Glu 17→His substitution at the N3 in S13NEKE17 variant decreased the folded structure content from 90% to 25% (equivalent to 1.8 kcal • mole−1 destabilization) by disruption of N-capping interactions, and had the most significant effect among single mutants studied here. The Ncap Asn 14 substitution with Arg considerably decreased stability, reducing structure content from 90% to 40% (1.4 kcal • mole−1 destabilization) by disruption of a helix-capping hydrogen bond and destabilization of a helix macrodipole. The N1 Glu 15→Ser mutation also produced a considerable effect (1.0 kcal • mole−1 destabilization), again emphasizing the significance of electrostatic interactions in α-helix stabilization.
Keywords: Zinc finger, Zif268, FSD-1, helix capping, NMR, CD
Zinc fingers are independently folded small domains representing the most abundant protein domain in the human genome (Venter et al. 2001). Cys2His2 transcription factors belong to the classical zinc finger family. They are most often arranged tandemly, and individual domains serve to recognize neighboring 3 to 4 bp along the major groove of double-stranded DNA. In recent years, there has been major progress in designing the classical zinc fingers to be able to recognize a desired DNA sequence (Pabo et al. 2001). Classical Cys2His2 zinc fingers are of particular biophysical interest, as they have served to establish stereochemical constraints of metal binding and thermodynamics of protein folding, or to design novel metal-binding sites (Berg and Godwin 1997).
The ββα fold of Cys2His2 zinc fingers is composed of ∼30 amino acid residues, and it is stabilized by zinc ion. The role of zinc is pivotal for the fold stability, as in its absence, the domain is unfolded and essentially devoid of DNA-binding properties (Berg and Godwin 1997). Using an advanced computational screening of a very large rotamer library, Dahiyat and Mayo (1997) showed that it is possible to obtain a stable fold of the classical zinc finger without stabilization by a metal ion or a covalent cross-link. In the designed peptide, called FSD-1, only 6 amino acids are identical and 11 are similar, as compared with the sequence of the second zinc finger domain of murine transcription factor Zif268 (Zif268.2), which was used as a template. Among the most essential differences between Zif268.2 and FSD-1 is a difference in the region that is involved in DNA recognition by Zif268.2 (residues 13–17). This segment forms the first turn of the α-helix, and in Zif268.2, its sequence is S13RSDH17, whereas in FSD-1 it is R13NEKE17. We explored the possibility of obtaining a small DNA-binding module devoid of metal stabilization by introducing a S13RSDH17 sequence into the FSD-1 framework. This hybrid peptide, however, appeared to be significantly unfolded, as judged from CD and NMR spectra. To reveal the source of instability, we constructed eight mutants in the N-capping motif of the α-helix and measured their conformational properties.
Results and Discussion
We studied R13NEKE17 (FSD-1), S13RSDH17 and eight additional mutants that differed in the five-residue sequence (positions 13 to 17) involved in DNA recognition (Elrod-Erickson et al. 1998). All of the mutants (shown in Table 1) will be abbreviated by single-letter amino acid codes of this sequence, and FSD-1 (RNEKE) serves in the present study as the wild-type protein. The structure of FSD-1 has been determined by NMR (Dahiyat and Mayo 1997) and consists of a short β-hairpin motif followed by an α-helix. The stable fold of FSD-1 has been designed replacing the Cys2His2 zinc-binding site of Zif268.2 with a small hydrophobic core. FSD-1 and Zif286.2 show completely unrelated modes of structure stabilization. Both structures, however, superimpose well and, despite different sequences, the position of the α-helix N-capping motif is preserved.
Table 1.
Structure content of FSD-1 mutants measured by CD and NMR techniques
| Percent folded by CD | |||
| FSD-1 variant | pH 5.5 | pH 7.5 | Percent folded by NMR |
| RNEKE | 100 | 94 | 100 |
| SNEKE | 90 | 85 | 83 |
| SNEDE | 73 | 69 | 75 |
| SNSKE | 59 | 61 | 51 |
| SNSDE | 53 | 55 | 49 |
| SREKE | 40 | 45 | nd |
| SNEKH | 25 | 47 | nd |
| SRSDE | 25 | 30 | nd |
| SNSDH | 6 | 19 | nd |
| SRSDH | 0 | 12 | 0 |
CD spectra were recorded at 4°C and pH 5.5 and pH 7.5, whereas NMR spectra were collected at 5°C and pH ∼5; (nd) Not determined.
The amino-terminal part of the α-helix of the representative conformer of FSD-1 (the most similar to an average structure) is presented in Figure 1 ▶. The structure of the α-helix is stabilized by a hydrophobic interaction between Phe 12 and Leu 18, and it can be classified as a N"→N4 capping motif (Aurora and Rose 1998). This motif is further stabilized by a hydrogen bond formed between a side chain of the Ncap residue, Asn 14 and the backbone amide of Glu 17. To identify the most crucial residues responsible for the decrease of SRSDH stability in relation to RNEKE, several mutations in the first turn of the α-helix were introduced.
Figure 1.

The N-capping motif of the representative conformation of FSD-1 (PDB code 1fsd). Amide protons of the residues in the first turn of α-helix are shown in ball-and-stick representation. Hydrogen bond between Ncap Asn 14 and N3 Glu 17 is shown as a broken line. (*) Position of a shielded methyl group of Leu 18.
Quantification of structure content
Thermal denaturation of RNEKE and other FSD-1 variants is weakly cooperative (Fig. 2 ▶), and the free energies of unfolding for all of the variants could not be determined reliably. However, we were able to determine the structure content of the peptides using CD and 1H NMR techniques. Far UV CD spectra of all of the variants measured at pH 5.5 and 4°C are as shown in Figure 3 ▶. Progressive decrease of spectra intensity in the 210–230 nm range reflects the decrease of a folded state population resulting from mutations introduced in the N-capping motif of the α-helix. To quantify effects of the mutations, we used mean residue ellipticity measured at 215 nm for a given variant and compared it with the respective value for RNEKE (Table 1).
Figure 2.

Thermal unfolding of FSD-1 variants monitored by CD. The temperature dependence of 215 nm ellipticity was measured at 5 • 10−6 M peptide in 10 mM sodium acetate (pH 5.5), in 10-mm cuvette.
Figure 3.

Circular dichroism spectra of FSD-1 variants. The spectra were measured at 5 • 10−5 M peptide in 10 mM sodium acetate (pH 5.5), in a 1-mm cuvette, at 4°C.
Because proton chemical shifts are very sensitive indicators of a protein’s structural integrity, NMR spectroscopy is a reliable method for detection of tertiary interactions in folded proteins. Due to low stabilities of FSD-1 variants, NMR spectra were measured at 5°C. A series of 1H NMR spectra of the most stable mutants is shown in Figure 4 ▶. Dispersion of signals in the aliphatic region reflects the population of the folded state. The chemical shift of the Leu 18 CH3 group in FSD-1 is moved upfield, due to the hydrophobic interaction with Phe 12 that bridges the N-capping motif of the α-helix (Fig. 2 ▶). Thus, the degree of Leu 18 CH3 group shielding was a convenient measure of the folded state and served to calculate structure content of the mutants relative to RNEKE (see Materials and Methods; Table 1). Comparison of Hα and HN chemical shifts of the three most stable FSD-1 variants (RNEKE, SNEKE, and SNEDE) revealed small differences and excluded the possibility of significant structural changes upon mutations in the N-capping motif (see Electronic Supplemental Material).
Figure 4.

Aliphatic region of 1H NMR spectra of the most stable FSD-1 variants. The four resolved methyl groups are as follows: (a) Ala 5; (b) Thr 4; (c) Thr 11; (d) Leu 18. All spectra were measured at 5°C (pH ∼5).
Positions of the four resolved methyl groups in 1H NMR spectra of the most stable FSD-1 variants are shown in Figure 4 ▶. Two features reflect clearly an extent of unfolding of FSD-1 variants; the methyl group of Leu 18 tends to be less shielded, and the difference between Thr 4 and Thr 11 methyl groups decreases. Structure contents calculated from CD and 1H NMR spectra are in good agreement (Table 1); however, chemical shifts could only be measured if the folded state was populated in more than ∼50%. 1H NMR spectrum of SNEKH (Fig. 4 ▶) reflects a lack of Leu 18 CH3 shielding and nearly degenerate chemical shifts of Thr 4 and Thr 11 methyl groups. This indicates that SNEKH is unfolded significantly and according to CD spectra population of structured state, is barely 25% (Table 1). This is also manifested in sharper signals of 1H NMR spectrum of SNEKH relative to the more stable mutants (Fig. 4 ▶).
SNEKE: Arg 13→Ser mutation in FSD-1
Substitution of Arg 13, a residue that directly precedes the α-helix (the N‘ position), with Ser, leads to relatively small, 10% decrease of the FSD-1 stability (Table 1). The resulting FSD-1 variant, SNEKE, was a subject of further modifications and introduction of single substitutions into the first turn of the α-helix. Because there are charged side chains located at positions 14–17, in further studies we decided to keep Ser at position 13 to exclude a possibility of long-range electrostatic effect. Below, we rationalize effects of the substitutions of residues in the Ncap, N1, N2, and N3 positions.
SREKE: The Ncap mutation (Asn 14→Arg)
The Ncap residue occupies one of the most important positions that stabilize the α-helix. Previous analysis showed that this site is very selective for 6 amino acid residues (Ser, Thr, Asp, Asn, Pro, Gly), whereas 11 are avoided (Val, Ile, Phe, Ala, Lys, Leu, Tyr, Arg, Glu, Met, Gln; Kumar and Bansal 1998). Small and polar residues, which can form hydrogen bond with amide protons from the first turn of α-helix, are preferred at the Ncap position (Doig et al. 1997; Kumar and Bansal 1998). Mutation of Asn 14 to Arg considerably decreased stability of SNEKE and comparison of CD spectra (Fig. 3 ▶) shows that it is accompanied by a reduction of structure content from 90% to 40% (Table 1), equivalent to destabilization by 1.4 kcal • mole−1. Furthermore, analysis of 1H NMR spectrum of SREKE failed to detect a folded structure (data not shown). Substitution of Asn 14 with Arg might cause two unfavorable effects, disruption of a helix-capping hydrogen bond, which involves a side chain carbonyl oxygen of the Ncap and a backbone amide of Glu 17 at the N3 site, and destabilization of helix macrodipole by positively charged Arg.
SNSKE: The N1 mutation (Glu 15→Ser)
Glutamate is one of the most favorable and commonly found residues at the N1 position (Kumar and Bansal 1998; Cochran et al. 2001). Side chain of Glu 15, which occupies the N1 site, is solvent exposed and carries a negative charge, which allows favorable interaction with a helix macrodipole. Furthermore, there are two positively charged residues Lys 16 and Arg 19 adjacent to Glu 15. Mutation of Glu 15→Ser should not introduce significant strain, as Ser is also a relatively common N1 residue (Penel et al. 1999). We observed that this mutation resulted in a decrease of structure content from 90% to 59%, equivalent to destabilization by 1.0 kcal • mole−1 (Table 1). This is a considerable effect and emphasizes the significance of electrostatic interactions stabilizing α-helical structure.
SNEDE: The N2 mutation (Lys 16→Asp)
The N2 position of α-helix is usually solvent exposed and prefers a polar side chain (Penel et al. 1999). Because Lys is positively charged and cannot accept hydrogen bond, it has a moderate propensity for the N2 site (Kumar and Bansal 1998; Cochran and Doig 2001). In contrast, Asp is a very common residue at this position, and it may potentially interact with main chain N2 and N3 amides (Kumar and Bansal 1998; Penel et al. 1999). Our measurements, however, showed that the Lys 16→Asp mutation led to destabilization of FSD-1 and decreased population of the folded state by 17% (destabilization of 0.7 kcal • mole−1; Table 1). Because N2 is surrounded by three acidic residues (Glu 15, Glu 17, and particularly closely located Asp 20), the presence of Asp 16 might create destabilizing electrostatic strain. Furthermore, Lys at N2 can form side chain to side chain interactions with N6, which is occupied by Asp 20 (Penel et al. 1999).
SNEKH: The N3 mutation (Glu 17→His)
Glu residue has a very high propensity to occur at the N3 position (Kumar and Bansal 1998; Penel et al. 1999). Its side chain is often found to be involved in the N-capping hydrogen bond with a backbone amide of the Ncap residue (Penel et al. 1999). Glu 17→His substitution in SNEKE has the most significant effect among single mutants studied here. It leads to decrease of structure content from 90% to 25% (1.8 kcal • mole−1 of destabilization) and it was not possible to detect any folded structure in SNEKH using 1H NMR spectra (Fig. 4 ▶). The effect of the mutation cannot be fully explained as a consequence of the presence of a destabilizing positive charge of His at the N3 position. The CD spectrum measured at a higher pH, which assured deprotonation of His side chain, showed that structure content is increased to only 47% (Table 1), which is equal to 1.0 kcal • mole−1 destabilization relative to SNEKE. We expect that another effect takes place upon Glu 17→His mutation. Solution structure of FSD-1 shows that in 2 of 41 conformers, the side chain of relatively disordered Glu 17 forms a hydrogen bond with the amide proton of Asn 14. Weak temperature gradient of Asn 14 amide proton strongly suggests that it is hydrogen bonded (see below). The arrangement of two hydrogen bonds involving a side chain of Ncap and a backbone of N3 and, reciprocally, a side chain of N3 and a backbone of Ncap supported by hydrophobic interaction between N" and N4 was found to be present in other proteins and is called a "big box" (Seale et al. 1994). Loss of this highly probable hydrogen bond upon Glu 17→His substitution may result in significant destabilization of the protein. Interestingly, the effect of this mutation is more destructive than loss of the N-capping hydrogen bond upon Asn 14 substitution with Arg.
Increasing the stability of SRSDH
Analysis of both CD and NMR spectra showed that SRSDH is almost completely unfolded. Chemical shifts of this variant are very close to random-coil values (data not shown). As shown above, two mutations: Asn 14→Arg (at Ncap) and Glu 17→His (at N3), have the strongest effect, decreasing the population of the folded state of FSD-1. To verify this, we introduced reciprocal substitutions, Arg 14→Asn and His 17→Glu, into SRSDH variant. The single mutation of the Ncap residue, Arg 14→Asn, produced a small, 6% increase of the folded state population of SNSDH relative to SRSDH. A slightly larger effect was observed upon single substitution of the N3 residue and structure content of His 17→Glu variant, SRSDE, increased to 25%. This mutation further emphasizes an important role of the N3 position in α-helix stabilization, as it corresponds to both significant destabilization of SNEKE and stabilization of SRSDH. Simultaneous introduction of two mutations, Arg 14→Asn and His 17→Glu, provided a SNSDE variant that is 53% folded, and this could be detected by 1H NMR analysis (Fig. 4 ▶). Our data show that positions of Ncap and N3 are essential for stabilization of the α-helix, and lack of favorable interactions in the N-capping motif leads to a fully unfolded variant of FSD-1.
Effect of pH on FSD-1 stability
Analysis of CD spectra of FSD-1 variants revealed that, in most cases, the difference in the folded state population between pH 5.5 and pH 7.5 did not exceed 5%. However, the peptides containing His residue at the N3 position showed meaningful stabilization of the structure upon increase of pH (Table 1). Deprotonation of His 17 at a higher pH leads to elimination of a positive charge located in the amino-terminal fragment of the α-helix. The largest effect was observed for SNEKH, and an increase in pH raised structure content by 22% (0.8 kcal • mole−1 stabilization). The effect observed emphasizes that electrostatic interactions with helix macrodipole cannot be neglected in designing α-helices.
Amide proton temperature coefficients of SNEKE
Our previous study showed that temperature coefficients could be used to judge hydrogen bonds formed by amide protons in proteins (Cierpicki and Otlewski 2001; Cierpicki et al. 2002). Values of δσHN/δT more positive than −4.6 ppb/K indicate the presence of a hydrogen bond with high probability. It was also shown that, unlike proteins, short protein fragments and peptides exhibit significant correlation between temperature coefficients of amide protons (δσHN/δT) and their conformational shifts (δσHN; Andersen et al. 1997). In such cases, values of temperature gradients are affected by a decrease in the population of the structured state upon warming.
Analysis of the thermal denaturation curve of SNEKE (Fig. 2 ▶) shows lack of a cooperative unfolding process typical for globular proteins. Thus, an increase in temperature should result in a decrease of a population of a folded state, affecting chemical shifts of amide protons. In turn, the correlation between δσHN/δT and δσHN should be expected.
The relationship between δσHN/δT and δσHN for SNEKE is shown in Figure 5 ▶. The linear dependence (correlation coefficient R = 0.90) is observed upon excluding four amides, which belong to residues 13–15 and carboxy-terminal Arg 28. The three consecutive residues, Arg 13, Asn 14, and Glu 15 are located within the initial fragment of the α-helix. The linear correlation is in agreement with the poor unfolding cooperativity of SNEKE. However, lack of the correlation observed for the three amides preceding the α-helix suggests that this short fragment may unfold in a noncooperative manner relative to the ββα fold of SNEKE. This compares well with several other cases, in which much better correlation was observed for secondary structure elements than for a full-length peptide (Andersen et al. 1997).
Figure 5.

Correlation between amide proton temperature coefficients (δσHN/δT) and conformational shifts of amide protons (δσHN) of SNEKE (R = 0.90). (Filled circles) Amides forming hydrogen bonds; (open circles) amides not involved in hydrogen bonds; (squares) four amides excluded from correlation.
We observe reasonable correlation between values of temperature coefficients and the presence of hydrogen bonds in SNEKE (Fig. 5 ▶). The most pronounced exception is the amide proton of Asn 14, which has the weakest temperature coefficient (δσHN/δT = 0.2 ppb/K), but according to the NMR structure, it fails to form a hydrogen bond. Our observation regarding the effect of Glu 17→His substitution led us to conclude that this amide might be involved in a hydrogen bond with the side chain of Glu 17. This example emphasizes that the values of δσHN/δT may also be useful to predict hydrogen bonds in proteins of relative low stability.
In this work, we carried out mutational analysis of an amino-terminal fragment of α-helix and its impact on structure content of FSD-1. Recent study of a coiled-coil dimerization domain within the human kinesin neck region also emphasized the significance of the N-capping motif in overall structure stability (Tripet and Hodges 2002). Our study revealed that the N-capping motif of Zif286.2 is far from being optimal. Contrary, the presence of a "big box" motif in FSD-1 provides an optimal network of interactions such as hydrogen bonds and electrostatics. The most crucial residues, Ncap and N3, are involved in hydrogen bonds, and their substitution has the strongest effect. The two other positions, N1 and N2, are solvent exposed and should be optimized to enable favorable interactions with both helix macrodipole and neighboring residues. The analysis presented here shows that the optimal design of the α-helix N-capping motif is crucial for obtaining structured proteins, and even trivial mutation may cause a significant effect.
Materials and methods
Bacterial strain Escherichia coli BL21(DE3)pLysS was purchased from Stratagene. pAED4 expression vector was a kind gift of Professor P.S. Kim (MIT, Cambridge, Massachusetts, USA). Restriction enzymes were supplied by New England Biolabs. Other DNA-modifying enzymes and the Sequenase v.2.0 DNA sequencing kit were from Amersham Pharmacia Biotech. Oligonucleotides were synthesized by MWG-Biotech. Acetonitrile was supplied by Roth; TFA and cyanogen bromide were obtained from Fluka. Other chemicals used were from Sigma.
Peptide expression and purification
The coding sequence of the FSD-1 variant, SRSDH, was annealed from two pairs of partially overlapping oligonucleotides and converted to fully double stranded using Sequenase v. 2.0. The resulting cassettes were digested with HindIII + BclI and BclI + BamHI and ligated into HindIII + BamHI-digested pAED4 expression vector. FSD-1 and its other variants were obtained through site-directed mutagenesis using synthetic oligonucleotides and a single-stranded template of SRSDH containing vector, and confirmed by DNA sequencing.
The peptides were expressed in E. coli BL21(DE3)pLysS from pAED4 vector as fusion with a hydrophobic leader sequence. Fusion proteins were purified from inclusion bodies, cleaved with cyanogen bromide, and the free peptides were double purified with high-performance liquid chromatography on C18 column (Vydac) with acetonitrile gradient in 0.1% TFA. The peptide identity was verified using MALDI mass spectrometry.
CD measurements
CD spectra of the peptides were recorded in the 190–250-nm wavelength range in 10 mM sodium acetate (pH 5.5), or 10 mM sodium phosphate (pH 7.5) at 4°C on a Jasco J-715 spectropolarimeter. Measurements were performed at a protein concentration of 5 • 10−5 M using a 1-mm cuvette. Spectra were averaged from 10 separate scans. Thermal stability of the peptides was measured in 10 mM sodium acetate (pH 5.5) at a protein concentration of 5 • 10−6 M using a 10-mm cuvette. Thermal denaturation was monitored following the ellipticity at 215 nm, using a band slit of 2 nm and a 16-sec response time. Automatic Peltier accessory, PFD 350S, allowed continuous monitoring of the thermal transition at a constant rate of 45deg • hour−1. Temperature of the sample was monitored directly using a probe immersed in the cuvette and controlled with PFD-350S/350L Peltier type FDCD attachment.
Structure content for a given FSD-1 variant was calculated from its CD spectrum, according to the following formula:
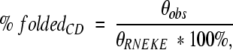 |
in which θobs and θRNEKE are the mean residue ellipticity at 215 nm for a given variant and RNEKE, respectively.
1H NMR spectroscopy
All protein samples were dissolved in 50 mM phosphate buffer and 90% H2O, 10% D2O mixture. Final concentrations of FSD-1 variants were ∼1 mM and the pH ranged between 5.2 and 5.4. NMR spectra were acquired on a Bruker DRX500 spectrometer. One-dimensional 1H spectra were measured at various temperatures ranging from 5 to 30°C and were referenced to DSS signal. Chemical-shift assignment of RNEKE, SNEKE, SNEDE, and SRSDH was carried out from the combination of two-dimensional TOCSY (Braunschweiler and Ernst 1983) and NOESY (Kumar et al. 1980) experiments. All NMR data were processed using Bruker Xwinnmr software and analysis of two-dimensional spectra was performed using the Sparky program (Goddard and Kneller 2002).
Structure content relative to FSD-1 (RNEKE) was calculated from chemical shifts of the upfield shifted methyl group of Leu 18 (σRNEKE = 0.673 ppm). Chemical shift of Leu 18 CH3 of the least stable variant, SRSDH, was determined from two-dimensaional TOCSY (σSRSDH = 0.86 ppm). Structure content was calculated according to the following formula:
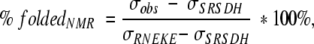 |
in which σobs is the observed Leu 18 CH3 group chemical shift.
Electronic supplemental material
Comparison of conformational shifts of amide protons and Hα protons of FSD-1, SNEKE, and SNEDE is available.
Acknowledgments
This work was supported by grant 6 PO4B 014 17 from the Polish Committee for Scientific Research. We thank the Foundation for Polish Science for support. J.O. is an International Scholar of the Howard Hughes Medical Institute. We thank Dr. Michal Dadlez for mass spectrometry analysis.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
FSD-1, full sequence design 1
NMR, nuclear magnetic resonance
Zif268.2, the second zinc finger domain of murine transcription factor Zif268
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0301703.
Supplemental material: See www.proteinscience.org.
References
- Andersen, N.H., Neidigh, J.W., Harris, S.M., Lee, G.M., Liu, Z., and Tong, H. 1997. Extracting information from the temperature gradients of polypeptide HN chemical shifts. 1. The importance of conformational averaging. J. Am. Chem. Soc. 119 8547–8561. [Google Scholar]
- Aurora, R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J.M. and Godwin, H.A. 1997. Lessons from zinc-binding peptides. Annu. Rev. Biophys. Biomol. Struct. 26 357–371. [DOI] [PubMed] [Google Scholar]
- Braunschweiler, L. and Ernst, R.R. 1983. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 53 521–528. [Google Scholar]
- Cierpicki, T. and Otlewski, J. 2001. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21 249–261. [DOI] [PubMed] [Google Scholar]
- Cierpicki, T., Zhukov, I., Byrd, R.A., and Otlewski, J. 2002. Hydrogen bonds in human ubiquitin reflected in temperature coefficients of amide protons. J. Magn. Reson. 157 178–180. [DOI] [PubMed] [Google Scholar]
- Cochran, D.A. and Doig, A.J. 2001. Effect of the N2 residue on the stability of the α-helix for all 20 amino acids. Protein Sci. 10 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran, D.A., Penel, S., and Doig, A.J. 2001. Effect of the N1 residue on the stability of the α-helix for all 20 amino acids. Protein Sci. 10 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiyat, B.I. and Mayo, S.L. 1997. De novo protein design: Fully automated sequence selection. Science 278 82–87. [DOI] [PubMed] [Google Scholar]
- Doig, A.J., MacArthur, M.W., Stapley, B.J., and Thornton, J.M. 1997. Structures of N-termini of helices in proteins. Protein Sci. 6 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod-Erickson, M., Benson, T.E., and Pabo, C.O. 1998. High-resolution structures of variant Zif268-DNA complexes: Implications for understanding zinc finger-DNA recognition. Structure 6 451–464. [DOI] [PubMed] [Google Scholar]
- Goddard, T.D. and Kneller, D.G. SPARKY 3. 2002. University of California, San Francisco, CA.
- Kumar, A., Ernst, R.R., and Wuthrich, K. 1980. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 95 1–6. [DOI] [PubMed] [Google Scholar]
- Kumar, S. and Bansal, M. 1998. Dissecting α-helices: Position-specific analysis of α-helices in globular proteins. Proteins 31 460–476. [DOI] [PubMed] [Google Scholar]
- Pabo, C.O., Peisach, E., and Grant, R.A. 2001. Design and selection of novel Cys2His2 zinc finger proteins. Annu. Rev. Biochem. 70 313–340. [DOI] [PubMed] [Google Scholar]
- Penel, S., Hughes, E., and Doig, A.J. 1999. Side-chain structures in the first turn of the α-helix. J. Mol. Biol. 287 127–143. [DOI] [PubMed] [Google Scholar]
- Seale, J.W., Srinivasan, R., and Rose, G.D. 1994. Sequence determinants of the capping box, a stabilizing motif at the N-termini of α-helices. Protein Sci. 3 1741–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet, B. and Hodges, R.S. 2002. Helix capping interactions stabilize the N-terminus of the kinesis neck coiled-coil. J. Struct. Biol. 137 220–235. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291 1304–1351. [DOI] [PubMed] [Google Scholar]