

Abstract
The relative strengths of interactions involving polypeptide chains can be estimated with reasonable accuracy with statistical potentials, free-energy functions derived from the frequency of occurrence of structural arrangements of residues or atoms in collections of protein structures. Recent published work has shown that the energetics of side-chain/backbone interactions can be modeled by the φ/Ψ propensities of the 20 amino acids. In this report, the more commonly used φ/Ψ probabilities are demonstrated to fail in evaluating the free energies of protein conformations because of an overriding preference for all helical structures. Comparison of the hypothetical reactions implied by these two different statistics—propensities versus probabilities—leads to the conclusion that the Boltzmann hypothesis may only be applicable for the calculation of statistical potentials after the starting conformation has been specified. This conclusion supports a simple conjecture: The surprising success of the Boltzmann hypothesis in explaining the energetics of protein structures is a direct consequence of a real equilibrium, one extending over evolutionary time that has maintained the stability of each protein within a narrow range of values.
Keywords: φ/Ψ angles, Ramachandran, propensities, statistical potentials, knowledge-based potentials, threading, side-chain interactions, backbone interactions
For the past several decades, protein chemists have been turning to high-resolution protein structures to obtain a more quantitative understanding of the energetics of interactions involving polypeptide chains. Through a detailed analysis of the frequencies with which different types of amino acid residues (or atoms) are found in specific structural arrangements, scoring functions known as statistical potentials, knowledge-based potentials, or empirical potentials have been developed that are surprisingly accurate at predicting the occurrence of these structural arrangements in proteins not included in the original set (Pohl 1971; Sippl 1995; Vajda et al. 1997). In some cases, statistical potentials are clearly superior to equivalent energy terms used in molecular mechanics and molecular dynamics simulations (Kuszewski et al. 1996; Moult 1997).
To convert the frequencies of occurrence of a structural interaction into an estimate of its free energy, the Boltzmann hypothesis is invoked. In effect, the specific interaction in the database of known protein structures being analyzed is conjectured to behave in a manner equivalent to a simple system at thermodynamic equilibrium, so amino acids populate each structural feature with a probability that can be calculated by the familiar Boltzmann weighting factor of statistical thermodynamics:
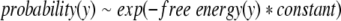 |
1 |
In general terms, this assumption is similar to that used by physicists to calculate potentials of mean force for simpler systems, such as liquids or solids (Chaikin and Lubensky 1995).
What is remarkable in the case of proteins is the surprising number of different structural interactions that seem to display Boltzmann-like behavior when treated in isolation, ignoring all other interactions. In other words, if the apparent free energy is calculated by applying Equation 1 to the frequency of a structural feature in one set of proteins, this free-energy term often predicts the probability of occurrence of the same feature in proteins not included in the calculation set. For the purposes of the discussion below, a measurable level of success in predicting structural features with such statistical potentials is described by saying that the underlying interaction "conforms" to the Boltzmann hypothesis. In effect, the structural features behave as if the hypothesis of an underlying Boltzmann distribution is true. Because there is no rigorous way of establishing that the estimated free energies agree with actual free-energy terms, a requirement for a real physical equilibrium, conformance to the Boltzmann hypothesis must be understood as a qualitative statement.
Examples found in the protein science literature of structural interactions that conform to the Boltzmann hypothesis include hydrogen bonds (Sippl et al. 1996), hydrophobicity (Rose et al. 1985; Miller et al. 1987; Casari and Sippl 1992), proline-isomerization (MacArthur and Thornton 1991), internal cavities (Rashin et al. 1997), various types of side-chain/side-chain interactions (Miyazawa and Jernigan, 1985; Vajda et al. 1997; Kannan and Vishveshwara 2000), and even interactions at the level of specific atom types (Samudrala and Moult 1998; Lu and Skolnick 2001). Why so many types of physical interactions can be semiquantitatively described as simple equilibrium ensembles independent of (or averaged over) all other interactions remains something of a mystery (Finkelstein et al. 1995).
Recent work from this laboratory has reported that the φ/Ψ propensities of the 20 naturally occurring amino acids also conform to the Boltzmann hypothesis (Shortle 2002). The propensities for amino acid x are calculated for a region y of the Ramachandran map with the following equation:
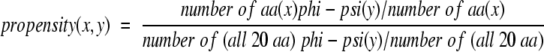 |
2 |
Simple summing of the values of −log p(x,y) for each amino acid assigned a specific set of φ/Ψ values provides a surprisingly good scoring function for conformations. When used to identify the correct conformation out of 300,000 alternative conformations, this sum yields a fairly high success rate for fragments of 20- to 40-residue length taken from proteins of known structure.
Like all applications of the Boltzmann hypothesis to the calculation of statistical potentials, there is an implied hypothetical reaction whose free-energy change is being estimated. In the case of φ/Ψ propensities, it was argued that the value of the propensity approximates an equilibrium constant for a hypothetical exchange reaction in which an "average" amino acid side-chain is removed from a polypeptide chain with fixed φ/Ψ angles and replaced with a specific side-chain (Shortle 2002). When the propensity is greater than one, this is a favorable reaction accompanied by a reduction in free energy. When the propensity is less than one, the free energy for the exchange is positive. The surprising accuracy of φ/Ψ propensities in scoring peptide conformations indicates at some approximate level, protein structures behave as if this exchange reaction, in a background with all other interactions averaged, is at equilibrium and obeys Equation 1.
An extensive review of the literature indicates that this example of Boltzmann-like behavior has not been fully appreciated, even though several investigators have conducted statistical analyses of amino acid preferences for different ranges of φ/Ψ angles. Although a number of reports document variations in amino propensities (Chou and Fasman 1974; Matsuo and Nishikawa 1993; Munoz and Serrano 1994; Swindells et al. 1995; Bahar et al. 1997), the energetic significance of these seemingly small differences was not clearly demonstrated. More commonly, published reports have calculated amino acid–specific φ/Ψ probabilities (Robson and Pain 1974; Rooman et al. 1991; Kang et al. 1993; Abagyan and Totrov 1994; Evans et al. 1995; Kuszewski et al. 1996; Feldman and Hogue 2002), not propensities, in which the probability is defined as
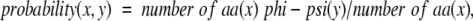 |
3 |
As can be seen from Equation 2, these φ/Ψ probabilities can be thought of as propensities that have not been normalized relative to an "average" amino acid.
In this report, φ/Ψ propensities and φ/Ψ probabilities are compared with respect to their ability to predict the correct conformation of fragments taken from known protein structures. It quickly becomes apparent that φ/Ψ probabilities always favor the α-helix over all other regions of the Ramachandran map, whereas propensities do not show this limitation. The failure of φ/Ψ probabilities to conform to the Boltzmann hypothesis, along with the success of propensities, can be used to support a simple explanation for the surprising success of the Boltzmann hypothesis when applied to the energetics of protein structure.
Results and Discussion
To calculate φ/Ψ propensities and probabilities, the Ramachandran map was subdivided into sets of bins. As described previously, the β-, the α-, and the L-handed helical regions were progressively subdivided (Shortle 2002). In all cases, the most densely populated regions were more finely subdivided than were the less populated regions, ultimately leading to a total of 137 discreet bins. As described in Materials and Methods, a set of 2017 nonredundant high-resolution protein structures were divided into a test set of 40 structures and a library set used in calculation of propensities/probabilities (1977), with all 2017 structures used as templates for conformational sampling by threading.
The amino acid sequences of proteins in the test set were broken down into arbitrary segments of varying lengths, always beginning at residue 3 and then shifting the first residue by 10, so that fragments began at position 3, 13, 23, etc. The score of every conformation in the template set (∼460,000 to 500,000) was computed by summing the −log of the propensity or probability. The best 450 scoring fragments were saved, and the ranking of the correct wild-type conformation of each sequence segment was determined in this list.
The results of this fragment identification assay are shown in Figure 1 ▶. For φ/Ψ propensities, the correct wild-type conformation is recognized as having the best score (Fig. 1A ▶) or scoring in the top 0.1% (Fig. 1B ▶) with increasing frequency (1) when the Ramachandran map is more finely subdivided or (2) when longer fragments are used. The results using φ/Ψ probabilities are just the reverse (Fig. 1C ▶). No wild-type conformation was correctly recognized; therefore, scoring in the top 0.1% of conformations was used as the criterion of "successful" identification. Although 1% to 2% of sequence segments of length 10 receive a very good score, except for the probability from a four-bin Ramachandran map, the longer fragments score less well. And with finer subdivision, probabilities become less, not more, accurate at finding the correct conformation.
Figure 1.

Graphs of the fraction of sequence fragments, of 10-, 20-, or 30-residue length, that scored at either the top (A) or in the top 0.1% (B, C) out of >460,000 conformations sampled. (A, B) Propensities were used. (C) Probabilities were used. The symbol designates the number of bins into which the φ/Ψ map was subdivided.
The reason for this failure of φ/Ψ probabilities has a simple explanation. For all amino acids, including proline and glycine, the α-helical region of the Ramachandran plot is the most densely populated, when measured as counts per unit area, and therefore the most probable. As the plot is more finely subdivided, this difference in density becomes the overriding issue. In addition, because helices always have the best score, the odds that a random sequence segment will be all helical declines with increasing segment length. Inspection of the structure of test protein segments that were identified by 15-bin probabilities confirmed that they were always 100% helical.
A deeper explanation for the large differences between φ/Ψ propensities and φ/Ψ probabilities requires comparison of the hypothetical reaction being modeled by each. In the language of frequentist statistics, a probability is the ratio of object counts in two sample subspaces (Kachigan 1986). In other words, two sets of objects from the full sample space must be selected. For specific φ/Ψ probabilities defined by Equation 3, the denominator represents a selection for a specific amino acid type, whereas the numerator further selects for a specific set of φ/Ψ values for that amino acid. These two selection events, carried out over a protein sequence, define the implicit reaction:
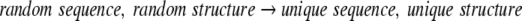 |
4 |
Beginning with a totally unspecified or average state, an amino acid residue is first selected from the sequence of the protein, then the φ/Ψ angles of the residue are assigned.
Although the reference state is described as having a "random structure," this is not the only interpretation. Because the φ/Ψ angles of each residue are not considered in the process of counting all instances of that amino acid type in the set of protein structures, this information is lost. Consequently, any arbitrary interpretation could be given to the reference state. Nevertheless, the general notion of "randomness" is the simplest way to communicate this lack of specific information about the initial conformation.
A propensity, however, is not a probability, because it can assume values greater than one. Rather it is the ratio of two probabilities, so first one must address the two reactions implicit in its definition. The numerator of Equation 2 is equal to a φ/Ψ probability, so it implies the reaction of Equation 4. The denominator defines a nonspecific φ/Ψ probability:
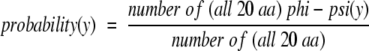 |
5 |
The denominator of Equation 5 corresponds to the full sample space of protein sequences, so the subspace selection is equivalent to no selection. On the other hand, the numerator selects for a specific set of φ/Ψ values. Therefore, the implicit reaction is
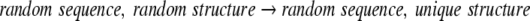 |
6 |
The negative logarithm of the φ/Ψ propensity yields the following equation:
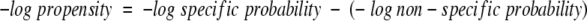 |
7 |
Therefore, when the propensity is converted to a free-energy estimate, it yields the difference between these two reactions, which is
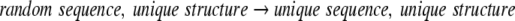 |
8 |
in agreement with the statement above that the propensity approximates the equilibrium constant for an exchange of an average side chain with a unique one at constant φ/Ψ angles.
If the success of a statistical potential in fragment identification is interpreted to mean the corresponding interaction conforms to the Boltzmann hypothesis, then three conclusions follow:
The Boltzmann hypothesis applies to random sequence/unique structure → unique structure, unique sequence (Equation 8)
The Boltzmann hypothesis does not apply to random sequence/random structure → unique sequence, unique structure (Equation 4)
Therefore, the Boltzmann hypothesis cannot apply to random sequence/random structure → random sequence / unique structure (Equation 6) because this reaction is simply the difference between the second and the first. In other words, Equation 4 has been divided into two steps, Equations 6 and 8.
The last conclusion is equivalent to saying that the evidence does not support a hypothetical equilibrium between any sequence/any structure and unique folds with any sequence. Protein folds are not generated in an equilibrium process dominated by the principles of physical chemistry. To a molecular biologist, this makes perfect sense, because abundant anecdotal evidence supports the belief that most folds have evolved from preexisting folds; they are the product of a long evolutionary history. There is probably little spontaneous de novo formation of new folds from an ensemble of nonnative conformations. So we see that application of the Boltzmann hypothesis appears to require specification of a unique conformation; when it is applied in situations that effectively predict the frequency of that conformation independent of a unique sequence, it will fail to give a sensible answer. This conclusion indicates that Boltzmann-like behavior may arise only in real proteins after they have appeared in a biological context.
A simple conjecture follows directly. The Boltzmann hypothesis applies to the energetics of protein structures because each protein fold has been subjected to a dynamic equilibrium involving its sequence interacting with its structure over evolutionary time. Most proteins are marginally stable, having unfolding free energies of 5 to 15 kcal/mole. Over millions of years, the sequence of every protein changes, due both to selection for functional modifications and to random genetic drift. If a protein is to remain in this narrow range of biologically useful stability, there will be intermittent selection for compensatory stabilizing mutations to increase stability after mutational losses and occasionally selection for loss of stability after mutations that make the protein too stable.
This simple conjecture does not address the most perplexing part of the puzzle: why each type of interaction analyzed in isolation should conform to the Boltzmann hypothesis. One possibility is that all classes of interactions (e.g., hydrogen bonds, hydrophobic interactions, tight packing, side-chain/backbone interactions) are relatively weak and roughly equal in strength, so that each one must be near its optimum before the window of stability is reached by a folded protein. In a fully cooperative system, the location of a favorable (or unfavorable) interaction is not important. Because only its free-energy contribution to net stability counts, any change in sequence that raises the free energy through one type of interaction can be corrected by a change in sequence elsewhere that lowers the free energy, through the same or different types of interactions. If a near-optimum must be maintained for each type of interaction, protein structures may actually participate in a dynamic equilibrium over evolutionary time in order that the stability of the biologically functional structure be held constant.
Materials and methods
A list of protein crystal structures (resolution ≤2.2 Å) with <40% sequence identity were obtained from the PISCES Web site of Dr. Ronald Dunbrack, Fox Chase Cancer Center, Philadelphia. Of the 2485 structures listed, 2067 were available and readable for use. A test set of 40 proteins were selected at random from this list to give 10 all-α proteins (1lycA, 1clc, 1gkmA, 1ew6A, 1k1fA, 2cy3, 1jnrA, 1i07a, 1evyA, 1hh5A), 10 all-β proteins (1hh2p, 1dhkB, 1edqA, 3tss, 1rmg, 2sns,1bebA, 1kv7A, 1gkpA, 1igqA) 10 α/β proteins (1k1eA, 1qj4A, 1l4uA, 1i1qB, 1g60A, 1fl2A, 1dosA, 1qj5A, 1eonA, 1esc), and 10 α+β proteins (1ggxA, 1fsvA, 1jy0A, 119l, 1jnrB, 1tif, 1ezm, 1a73A, 1tig, 1jyrA). When broken into arbitrary segments beginning at residue 3 and offset by 10, these 40 proteins formed 996 fragments of length 10, 956 fragments of length 20, and 916 fragments of length 30. The 40 proteins in the test set were excluded from the set of 1967 proteins used to calculate φ/Ψ propensities and probabilities, providing statistics on a total of 504,819 amino acid residues. Details of subdivision of the Ramachandran map into 4, 6, 9, and 15 bins have been published (Shortle 2002). The φ/Ψ values of the 26-, 74-, and 137-bin subdivisions will be published elsewhere (Fang and Shortle 2003).
Acknowledgments
I thank Charlie Strauss for several stimulating discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0306903.
References
- Abagyan, R. and Totrov, M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1002. [DOI] [PubMed] [Google Scholar]
- Bahar, I., Kaplan, M. and Jernigan, R.L. 1997. Short-range conformational energies, secondary structure propensities, and recognition of correct sequence-structure matches. Proteins 29292–308. [DOI] [PubMed] [Google Scholar]
- Casari, G. and Sippl, M.J. 1992. Structure-derived hydrophobic potential: A hydrophobic potential derived from x-ray structures of globular proteins is able to identify native folds. J Mol. Biol. 224 725–732. [DOI] [PubMed] [Google Scholar]
- Chaikin, P.M. and Lubensky, T.C. 1995. Principles of condensed matter physics. Cambridge University Press, Cambridge, UK.
- Chou, P.Y. and Fasman, G.D. 1974. Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry 13 211–222. [DOI] [PubMed] [Google Scholar]
- Evans, J.S., Mathiowetz, A.M., Chan, S.I., and Goddard III, W.A. 1995. De novo prediction of polypeptide conformations using dihedral probability grid Monte Carlo methodology. Protein Sci 4 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Q. and Shortle, D. 2003. Prediction of protein structure by emphasizing local side-chain/backbone interactions in ensembles of turn fragment. Proteins (in press). [DOI] [PubMed]
- Feldman, H.J. and Hogue, C.W. 2002. Probabilistic sampling of protein conformations: New hope for brute force? Proteins 46 8–23. [PubMed] [Google Scholar]
- Finkelstein, A.V., Badretdinov, A.Y., and Gutin, A.M. 1995. Why do protein architectures have Boltzmann-like statistics? Proteins 23 142–150. [DOI] [PubMed] [Google Scholar]
- Kachigan, S. 1986 Statistical analysis. Radius Press, New York, NY.
- Kang, H.S., Kurochkina, N.A. and Lee, B. 1993. Estimation and use of protein backbone angle probabilities. J. Mol. Biol. 229 448–460. [DOI] [PubMed] [Google Scholar]
- Kannan, N. and Vishveshwara, S. 2000. Aromatic clusters: A determinant of thermal stability of thermophilic proteins. Protein Eng. 13 753–761. [DOI] [PubMed] [Google Scholar]
- Kuszewski, J., Gronenborn, A.M., and Clore, G.M. 1996. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 5 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H. and Skolnick, J. 2001. A distance-dependent atomic knowledge-based potential for improved protein structure selection. Proteins 44 223–232. [DOI] [PubMed] [Google Scholar]
- MacArthur, M.W. and Thornton, J.M. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218 397–412. [DOI] [PubMed] [Google Scholar]
- Matsuo, Y. and Nishikawa, K. 1993. Protein structural similarities predicted by a sequence-structure compatibility method. Protein Sci. 3 2055–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S., Janin, J., Lesk, A.M., and Chothia, C. 1987. Interior and surface of monomeric proteins. J. Mol. Biol. 196 641–656. [DOI] [PubMed] [Google Scholar]
- Miyazawa, S. and Jernigan, R.L. 1985. Estimation of effective inter-residue contact energies from crystal structures: Quasi-chemical approximation. Macromolecules 18 534–552. [Google Scholar]
- Moult, J. 1997. Comparison of database potentials and molecular mechanics force fields. Curr. Opin. Struct. Biol. 7 194–199. [DOI] [PubMed] [Google Scholar]
- Munoz, V. and Serrano, L. 1994. Intrinsic secondary structural propensities of the amino acids, using statistical φ-Ψ matrices: Comparison with experimental scales. Proteins 20 301–311. [DOI] [PubMed] [Google Scholar]
- Pohl, F.M. 1971. Empirical protein energy maps. Nat. New Biol. 234 277–279. [DOI] [PubMed] [Google Scholar]
- Rashin, A.A., Rashin, B.H., Rashin, A., and Abagyan, R. 1997. Evaluating the energetics of empty cavities and internal mutations in proteins. Protein Sci. 6 2143–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson, B. and Pain, R.H. 1974. Analysis of the code relating sequence to conformation in globular proteins: Development of a stereochemical alphabet on the basis of intra-residue information. Biochem. J. 141 869–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooman M.J., Kocher, J.-P.A., and Wodak, S.J. 1991. Prediction of protein backbone conformation based on seven structure assignments, I: Influence of local interactions. J. Mol. Biol. 221 961–979. [DOI] [PubMed] [Google Scholar]
- Rose, G.D., Geselowitz, A.R., Lesser, G.J., Lee, R.H., and Zehfus, M. H. 1985. Hydrophobicity of amino acid residues in globular proteins. Science 229 834–838. [DOI] [PubMed] [Google Scholar]
- Samudrala, R. and Moult, J. 1998. An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J. Mol. Biol. 275 895–916. [DOI] [PubMed] [Google Scholar]
- Shortle, D. 2002. Composites of local structural propensities: Evidence for local encoding of long range structure. Protein Sci. 11 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippl, M.J. 1995. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 5229–235. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J., Ortner, M., Jaritz, M., Lackner, P., and Flockner, H. 1996. Helmholtz free energies of atom pair interactions in proteins. Fold Design 1 289–298. [DOI] [PubMed] [Google Scholar]
- Swindells, M.B., MacArthur, M.W. and Thornton, J.M. 1995. Intrinsic φ, Ψ propensities of amino acids, derived from the coil regions of known structures. Na. Struct. Biol. 2 596–603. [DOI] [PubMed] [Google Scholar]
- Vajda, S., Sippl, M. and Novotny, J. 1997. Empirical potentials and functions for protein folding and binding. Curr. Opin. Struct. Biol. 7 222–228. [DOI] [PubMed] [Google Scholar]