

Abstract
Pyridoxine 5′-phosphate oxidase catalyzes the terminal step in the synthesis of pyridoxal 5′-phosphate. The cDNA for the human enzyme has been cloned and expressed in Escherichia coli. The purified human enzyme is a homodimer that exhibits a low catalytic rate constant of ∼0.2 sec−1 and Km values in the low micromolar range for both pyridoxine 5′phosphate and pyridoxamine 5′-phosphate. Pyridoxal 5′-phosphate is an effective product inhibitor. The three-dimensional fold of the human enzyme is very similar to those of the E. coli and yeast enzymes. The human and E. coli enzymes share 39% sequence identity, but the binding sites for the tightly bound FMN and substrate are highly conserved. As observed with the E. coli enzyme, the human enzyme binds one molecule of pyridoxal 5′-phosphate tightly on each subunit.
Keywords: Pyridoxine 5′-phosphate oxidase, pyridoxal 5′-phosphate, pyridoxamine 5′-phosphate, X-ray crystallography, flavin mononucleotide
Mammalian PLP-containing enzymes are involved in the synthesis of neurotransmitters, modulation of steroid-receptor interaction, and regulation of immune function (McCormick 1996, 1997). In mammalian cells, PLP is formed from the vitamins pyridoxine, pyridoxamine, and pyridoxal, supplied from ingested nutrients, or by recycling the cofactor from degraded enzymes in a salvage pathway (Hill et al. 1996). In both of these metabolic scenarios, the terminal step in forming PLP is catalyzed by pyridoxine 5′-phosphate oxidase (PNPOx), which uses both pyridoxine 5′-phosphate (PNP) and PMP as substrates (McCormick and Chen 1999). A recent study in sheep brain has shown that PNPOx is expressed in many neural tissues, and it appears to be the same enzyme as expressed in other nonneural cells (Bahn et al. 2002).
PNPOx has been purified from sheep, rat and pig brain, rabbit liver, and Escherichia coli (Bowers-Komro and McCormick 1985; Choi et al. 1987; Zhao and Winkler 1995; Di Salvo et al. 1998), with the most extensive studies being performed on the rabbit liver, sheep brain, and E. coli enzymes. PNPOx catalyzes the transfer of a pair of electrons from C4′ of PNP (or PMP) to a tightly bound molecule of FMN, forming FMNH2. These electrons are then transferred in a second half-reaction to molecular oxygen, regenerating FMN and forming H2O2 (Di Salvo et al. 2002). Both the yeast and E. coli enzymes have been crystallized and their structures determined, but only the structure of the E. coli enzyme has been discussed (Safo et al. 2000, 2001; Di Salvo et al. 2002).
The purpose of this paper is to describe the expression, purification, characterization, and crystal structure of human PNPOx and to discuss the first structure of a mammalian PNPOx.
Results
Purification of human PNPOx
We obtained 40 mg of pure enzyme from 6 L of cells. The enzyme is stable for several weeks if stored as an ammonium sulfate pellet at 4°C, or at −80°C in 50 mM potassium phosphate (pH 7.5). Enzyme purity was tested under both reducing (2-mercaptoethanol added to the sample buffer) and nonreducing conditions by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Under conditions with 2-mercaptoethanol, at least two closely spaced bands are observed at ∼30 kD. Under nonreducing conditions, the intensity of the bands near 30 kD was decreased, but there are at least two other closely spaced bands at ∼60 kD. The observation that additional bands appear near 60 kD under nonreducing conditions indicates that at least some monomers are joined by a disulfide(s) bond to form dimers. The multiple closely spaced bands at both 30 kD and 60 kD indicate that some proteolysis has occurred resulting in the multiple bands. These bands were seen even though protease inhibitors were added before cell disruption and all purification steps were performed at zero to 4°C and completed in less than 2 d.
To determine if protease digestion had occurred at the N terminus, the bands from the SDS-PAGE gel were excised and the amino acid sequence was determined. The faster-moving band gave an amino acid sequence starting at residue 7 (Fig. 1▶), but the slower-moving band, and presumably larger protein, showed no residues upon Edman degradation.
Figure 1.
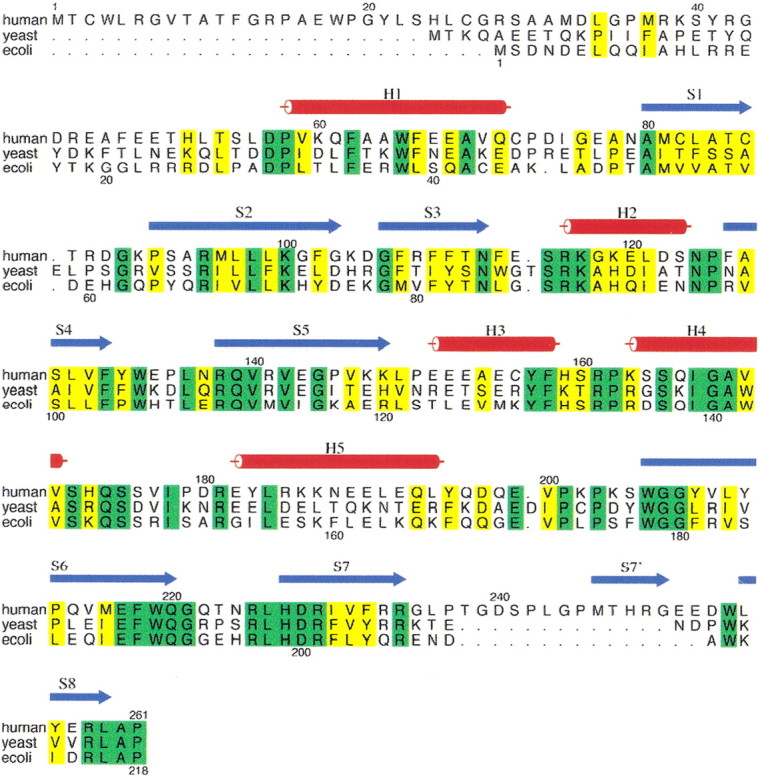
Multiple sequence alignment of human, yeast, and Escherichia coli PNPOx proteins. Proteins used in the sequence alignment are as follows (database ID numbers are shown in parentheses): E. coli, Escherichia coli (P28225); yeast, Saccharomyces cerevisiae (P38075); and human, Homo sapiens (AK001397). The numbering above and below the alignment corresponds to the human and E. coli sequences, respectively. The strands and helices of the human structure are shown in blue and red, respectively. The additional strand between S7 and S8 of the human PNPOx structure is labeled S7′. Identical residues in the three sequences are shaded in green, and conserved residues are in yellow. The figure was prepared using the program ALSCRIPT (Barton 1993).
The enzyme is yellow, exhibiting absorption maxima at 276 nm, 385 nm, and 448 nm (Fig. 2▶, curve 1). The A278 nm: A448 nm ratio of the pure enzyme is 5.65, which is similar to the 6.1 observed for this ratio with E. coli PNPOx (Di Salvo et al. 1998). The apo enzyme is colorless and shows no absorption above 300 nm. The molar extinction coefficient for the holo enzyme at 278 nm was determined from the amino acid composition and the absorption spectrum of the denatured and native human PNPOx. A value of 76,760 cm−1 M−1 was determined for the holo enzyme, which gives an absorbance of 2.4 for a 1-mg/mL solution, and 53,360 cm−1 M−1 for apo enzyme (no bound FMN), which gives an absorbance at 278 nm of 1.67 for a 1-mg/mL solution. The high molar absorbance coefficient is the result of the seven Trp residues and nine Tyr residues per subunit. If the enzyme is clipped at residue 6, then it is missing Trp 4 and Cys 3 and would exhibit a molar absorbance coefficient of 71,200 cm−1 M−1. Again, these values are similar to those of E. coli PNPOx (Di Salvo et al. 1998).
Figure 2.

Spectra of purified human PNPOx. Curve 1, spectrum of milligram per milliliter solution of holoPNPOx; curve 2, spectrum of the same concentration of apoPNPOx after the addition of PLP and passage down a P6 DG molecular sieve column to remove excess PLP.
Sulfhydryl groups
One of the major differences in the amino acid sequence between E. coli and human PNPOx is that the human enzyme contains six Cys residues, whereas the E. coli enzyme contains only a single Cys. We determined the number of —SH groups in the native enzyme, enzyme denatured with SDS, and the enzyme denatured with SDS in the presence of 1 mM dithiothreitol by titration with 5,5′-dithio-bis-2-nitrobenzoic acid (DTNB). The enzyme denatured in dithiothreitol showed a total of 5.4 —SH groups, whereas the enzyme denatured in the absence of dithiothreitol contained 4.3 —SH groups, further indicating that there are some disulfide bonds in this protein. One of the Cys residues is residue 3 in the amino acid sequence, and because some proteolysis had occurred at the N terminus, one Cys residue in the dimer could be missing, accounting for less than the full six —SH groups expected. There was no significant difference in catalytic activity of the enzyme stored in the presence or the absence of dithiothreitol. The native enzyme treated with DTNB for 2 min also showed no change in catalytic activity, indicating that there are no critical catalytic Cys residues.
Kinetic constants
Because the enzyme has a low turnover number, assays were conducted in a 10-cm pathlength cuvette at 37°C. Km and kcat values were determined for both PNP and PMP and are recorded in Table 1▶. For comparison, the values for E. coli PNPOx with PNP and PMP are shown in parentheses. The human enzyme is sluggish, having a turnover number of only 0.19 sec−1 for PNP and 0.20 sec−1 for PMP. These are very close to the values determined for the E. coli enzyme. The affinities for PNP and PMP are both in the low micromolar range, which is different from E. coli PNPOx, where the Km for PMP is 50-fold higher than PNP (Table 1▶). Also, PLP is a competitive inhibitor for both enzymes and exhibits similar Ki values (Table 1▶).
Table 1.
Kinetic constants for human PNPOx
| Substrate | Km or Ki (μM) | kcat (sec−1) |
|---|---|---|
| PNP | 1.8 (2.0)a | 0.19 (0.3)a |
| PMP | 1.0 (105)b | 0.20 (1.7)b |
| PLP | 3.2 (8.0)b | — |
Activity determined in 50 mM Tris buffer (pH 7.6).
a Values previously determined for Escherichia coli PNPOx at pH 7.6 (Yang and Schirch 2000).
b Values determined in Tris at pH 8.5 (Yang and Schirch 2000).
Tight binding of PLP
Our studies with E. coli PNPOx showed that both the holo and apo enzyme bind one molecule of PLP tightly on each subunit as shown by the lack of dissociation of PLP during repeated molecular sieve chromatography (Yang and Schirch 2000). We tested the human PNPOx to determine if it also binds PLP tightly as defined by the PLP not dissociating during size exclusion chromatography. The purified enzyme shows less than 0.1 molecule of PLP bound per subunit. When this purified enzyme is incubated with a twofold excess of PLP and chromatographed on a 1 cm × 30 cm column of P6 DG, we found 0.95 molecules of PLP bound per subunit of enzyme. There was a complete separation of the enzyme from the excess PLP, and a second chromatography of the eluted enzyme on this column did not remove any of the tightly bound PLP.
The spectrum of the tightly bound PLP cannot be determined with holoPNPOx because the bound FMN masks the spectral properties of the PLP. To circumvent this problem, we removed the FMN from the human PNPOx to form apoPNPOx and incubated this with PLP as described above. Again we found that one PLP molecule binds tightly to each subunit of the apo oxidase. With the FMN removed, the spectrum of this apoPNPOx • PLP complex shows a peak at ∼410 nm (Fig. 2▶, curve 2). Addition of 0.5 mM NaCNBH3 does not alter the spectrum, indicating that the PLP is not bound at the surface as an aldimine.
The catalytic activity of the human PNPOx • PLP complex was tested with PNP as the substrate. If the PLP is bound at the active site, then it should greatly inhibit the activity of the enzyme because of its slow rate of dissociation. However, the initial velocity of the reaction is the same as enzyme without PLP bound.
Crystal structure of human PNPOx
Human PNPOx was crystallized without and with an excess of PLP. The structures were determined to 1.95 Å and 2.65 Å, respectively, by molecular replacement methods using the monomeric structure of E. coli PNPOx (PDB code 1G79). However, only the higher-resolution liganded structure will be reported here because the two structures are very similar. The final model of the human PNPOx structure has one monomer in the asymmetric unit containing 213 amino acid residues, one tightly bound FMN, one PLP, one phosphate, and 197 water molecules. The side chains of two residues, Glu 77 and Cys 156, were refined in two positions, whereas Cys 72 has a disulfide interaction with a mercaptoethanol molecule. Data statistical values are listed in Table 2▶.
Table 2.
Data collection and refinement statistics of the human PNPOx•PLP complex a
| Data collection statistics | |
| Space group | P3121 |
| Unit cell dimensions | a = b = 82.75 Å, c = 59.33 Å |
| Resolution (Å) | 71–1.95 (2.00–1.95) |
| No. of measurements | 94,811 (4746) |
| No. of unique reflections | 16,966 (1226) |
| I/Σ I | 14.7 (1.63) |
| Completeness (%) | 94.9 (95.8) |
| Rmerge (%)b | 6.5 (36.6) |
| Structure refinement | |
| Resolution limit (Å) | 45.7–1.95 (1.99–1.95) |
| No. of reflections | 16,953 (1094) |
| Rfactor for 95% working data set | 0.192 (0.311) |
| Rfree for 5% test data set | 0.252 (0.321) |
| Rmsd from standard geometry | |
| Bond (Å) | 0.014 |
| Angles (°) | 1.7 |
| Dihedral (°) | 24.1 |
| Improper (°) | 1.17 |
| Average B-values (Å2) | |
| For all 1991 non-hydrogen atoms | 44.3 |
| For the 31 FMN atoms | 28.6 |
| For the 16 PLP atoms | 61.1 |
| For the 5 phosphate atoms | 67.4 |
| For the 197 water molecules | 50.8 |
| Ramachandran plot (%) | |
| Most favored region | 90.1 |
| Additional allowed region | 7.7 |
| Generously allowed | 1.7 |
| Disallowed region | 0.6 |
| Estimated coordinate error (Å) | Rfactor |
| By Luzzati plot | 0.24 |
| By SigmaA plot | 0.31 |
a Numbers in parentheses refer to the outermost resolution bin.
bRmerge = ∑(≤I> − I)/∑I.
We have previously reported detailed sequence analysis of the PNPOx enzyme family based on the largest multiple alignments (Safo et al. 2000). The alignment shown in Figure 1▶ is for the human, yeast, and E. coli enzymes, the three enzymes with known crystal structures. The residue identities used in this paper correspond to the position numbers of the human PNPOx. Like the E. coli (PDB code 1G79) and yeast PNPOx (PDB code IC10) structures, which are missing the first 19 and 23 N-terminal amino acid residues, the first 48 N-terminal amino acid residues of human PNPOx are also absent in the structure because of disorder. Interestingly, the missing amino acids are truncated close to each other in the aligned sequences. A dissolved crystal of human PNPOx showed a single band on SDS-PAGE. Attempted amino acid sequence failed, indicating that the crystal contains only the slower moving of the two bands discussed above.
The 1.95 Å human PNPOx • PLP three-dimensional structure reveals a very similar protein fold to those of the E. coli (Safo et al. 2000, 2001; Di Salvo et al. 2002) and yeast structures. This is expected because of the 54% sequence similarity within the three PNPOx enzymes with 28% sequence identity. The monomer structure of human PNPOx shows the typical two-domain architecture (domain 1 and domain 2) previously observed for the E. coli and yeast structures (Fig. 3A▶). The larger domain 1 of the E. coli and yeast enzymes is formed by eight β-strands (β1–β8) and two α-helices (α1 and α2), with the human PNPOx having one additional β-strand, β7′. As shown in Figure 3A▶, the strand β7 flanks the C-terminal strand β8. The strand β7′ and part of the turn and loop regions associated with it are absent in both the E. coli and yeast PNPOx structures because of a 15- and 13-residue insertion at residue 238, respectively (Fig. 1▶). The presence of these additional structures in human PNPOx might be solely structural, because they have no direct catalytic role and are also not involved in dimer formation. These unique regions are open to the solvent, with the four-amino-acid residue loop that connects the strands β7 to β7′ highly disordered.
Figure 3.
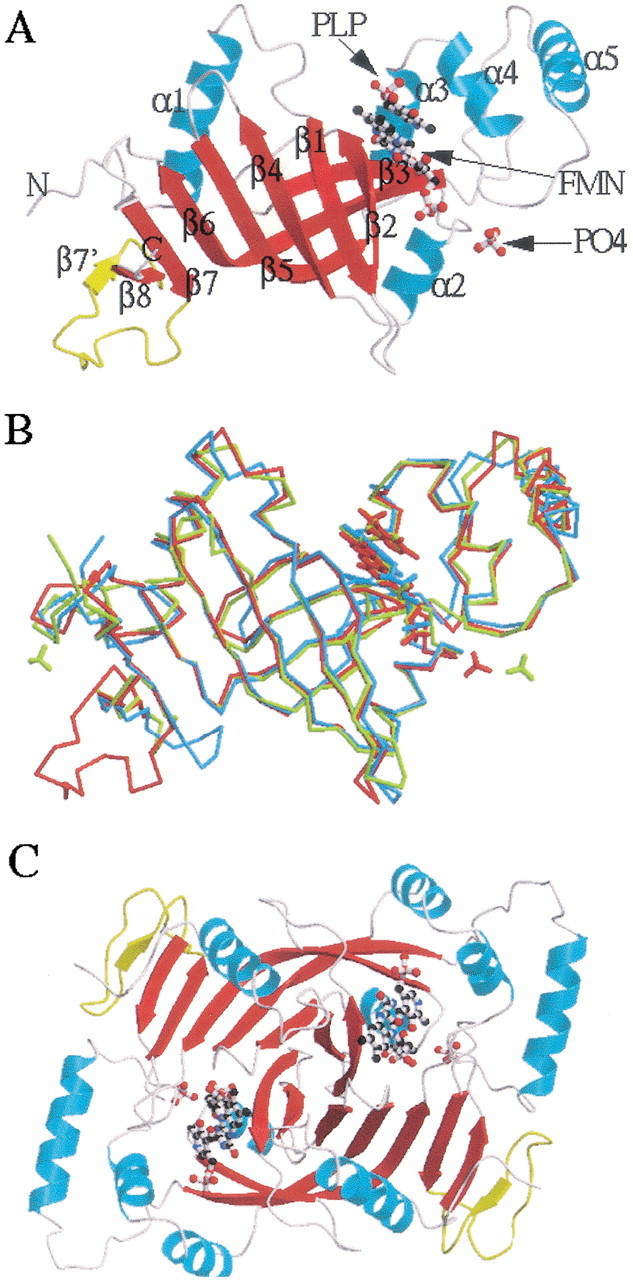
Structure of PNPOx enzyme complexed with FMN, PLP, and phosphate molecules. (A) Overall monomeric protein fold of human PNPOx enzyme. α-Helices are colored cyan, and β-strands are in red. The additional β-strand (β7′) observed in the human PNPOx structure, but missing in Escherichia coli and yeast PNPOx structures, is shown in yellow. (B) Least-squares superposition of human PNPOx • PLP structure (red) with those of the E. coli PNPOx • PLP (yellow) and yeast PNPOx (cyan) structures. The structures were superimposed as described in the text. The orientation is the same as the ribbon diagram. The figures were drawn using MOLSCRIPT (Kraulis 1991) and RASTER3D (Merritt and Murphy 1994), and labeled using SHOWCASE (Silicon Graphics, Inc.). (C) Overall dimer structure of the human PNPOx. The two monomers are colored red and cyan, respectively. The FMN and PLP molecules are surrounded by the two monomers.
The smaller domain 2 of all three PNPOx structures is made up of three α-helices (α3, α4, and α5). Major differences between the known structures occur mainly in turns, loops, the N-terminal segment, and the helix α5 (Fig. 3B▶). The monomeric structure of the human PNPOx superimposes on those of E. coli and yeast PNPOx structures with root mean square deviations (RMSDs) of 0.65 Å and 0.95 Å for 126 Cα atom pairs (from only strands and the helices), respectively. The corresponding values between the E. coli and the yeast PNPOx structures are 1.05 Å and 1.26 Å, respectively. The program LSQKAB as implemented in the CCP4 program suite (Collaborative Computing Project No. 4 1994) was used for the calculations. The larger values between the yeast and the other two structures are likely caused by the low resolution of the yeast PNPOx structure and changes in tertiary structures that result from PLP binding.
Two monomers, related by the crystallographic twofold axis, interact extensively along three regions of each subunit to constitute the functional dimer (Fig. 3C▶), as previously discussed for the E. coli PNPOx structure (Safo et al. 2000). The dimeric structure of the human PNPOx also closely resembles those of the E. coli and yeast PNPOx structures with RMSDs of 0.79 Å and 0.98 Å, for 252 Cα atom pairs from the strands and the helices, respectively. There are two pairs of intersubunit salt-bridge interactions, including Arg 116–Glu 143 and Arg 181–Asp 228. The latter is conserved in all three PNPOx structures, whereas the former occurs in the yeast but not in the E. coli enzyme. In E. coli, Glu 143 is replaced by isoleucine, necessitating the rearrangement of Arg 116 to make an intersubunit salt bridge with Glu 217. Interestingly, Glu 217 is strictly conserved in all three PNPOx structures, but the salt-bridge interaction between Arg 116 and Glu 217 is only observed in the E. coli structure. Unlike the strands β7 and β8 that are involved in extensive intersubunit interactions, the new strand β7′, as well as its associated turn and loop, make only one weak intersubunit hydrogen-bond interaction from Met 246 to Pro 179 (3.4 Å).
There are a total of six cysteine residues in the human PNPOx enzyme, but only four are modeled in the crystal structure. The other two are located in the missing N-terminal region. Two of the cysteine residues (Cys 82 and Cys 86) are located in the dimer interface, with each facing its symmetry-related counterpart. The —S—S— distances from Cys 82–Cys 82 and Cys 86–Cys 86 are 3.6 Å and 4.2 Å, respectively. These distances are significantly longer than an expected disulfide bond, but as discussed above there is evidence that some disulfide bonds are formed between the monomers. The two remaining cysteine residues (Cys 72 and Cys 156) are located outside the protein, and one (Cys 77), or possibly both of them, makes a disulfide interaction with 2-mercaptoethanol.
Active-site structure of bound FMN
The binding site of FMN is identical to those of the E. coli and yeast PNPOx structures, with the FMN located in a deep cleft formed by the two subunits with extensive hydrogen-bond interactions to the protein (Safo et al. 2000). These interactions involve both subunits (denoted with the letters A and B) and are recorded in Table 3▶. Most of the salt-bridge and hydrogen-bond interactions between the protein and the FMN are strictly conserved in all three PNPOx structures (Table 3▶). In particular, the human and E. coli structures have very similar FMN geometry, active-site environment, and bond distances. The differences between the yeast and the other two structures are probably caused by the poor resolution of the yeast structure and may not necessarily reflect any significant structural differences. One difference between the human and the E. coli PNPOx structures is the absence of the interaction between Arg 116 and the phosphate moiety in the human structure. In the human PNPOx structure, the side chain of Arg 116 has moved almost 3 Å to make an intersubunit salt-bridge interaction with Glu 143. As already pointed out, Glu 143 is substituted with isoleucine in E. coli. Another difference between the human and the E. coli structures is that Arg 141 of human PNPOx is replaced with methionine in E. coli PNPOx, and therefore the interactions between Arg 141 and the FMN are absent in the E. coli structure. In addition to the direct contacts between the FMN and the protein, there are four water-mediated interactions involving the FMN and the protein found in both the human and E. coli PNPOx structures, with three of them conserved. Only a few hydrophobic interactions of contacts <3.7 Å are observed between the FMN isoalloxazine ring and the protein residues Val 97 and Trp 219, and these are conserved in all three structures.
Table 3.
Salt-bridge and hydrogen-bond interactions between the protein and the PLP and FMN at the active site of three PNPOx enzymes
| Distancea |
|||||
|---|---|---|---|---|---|
| Ligand | Water | Proteinb | Human | Escherichia coli | Yeast |
| PLPc | |||||
| N1 | W1 | Trp206A NE1 | 3.3:3.2 | 3.4:3.3 | |
| O3′ | His227B NE2 | 2.7 | 2.6 | ||
| O4′ | W2 | Glu77A OE1 | 3.1:2.6 | 3.1:3.5 (Asp) | |
| OP1 | Lys100A NZ | 2.7 | 3.0 | ||
| OP2 | Tyr157A OH | 2.8 | 2.5 | ||
| OP2 | Arg161A NE | 2.7 | 2.6 | ||
| OP3 | Arg161A NH1 | 2.9 | 2.7 | ||
| OP3 | Arg225B NH2 | 2.7 | 2.9 | ||
| OP3 | Ser165A OG | 3.5 | — | ||
| OP3 | W3 | Ser165A OG | — | 2.7:2.5 | |
| FMN | |||||
| O2 | Thr111A OG1 | 2.8 | 2.8 | 3.1 (Ser) | |
| O2 | Glu174A NE2 | 3.1 | 3.1 | 3.1 | |
| N3 | Phe110A O | 2.9 | 2.8 (Tyr) | 3.6 (Tyr) | |
| O4 | Leu98A N | 3.3 | 3.2 | — | |
| O4 | W4 | Phe110A N | 2.7:2.7 | 2.7:2.8 (Tyr) | — |
| N5 | Leu98A N | 3.4 | 3.4 | — | |
| O2* | Met96A O | 2.7 | 2.7 (Ile) | 2.3 (Ile) | |
| O2* | Gln139B NE2 | 3.1 | 2.8 | 3.2 | |
| O3* | Trp219B NE1 | 3.4 | 3.5 | 3.5 | |
| O3* | W5 | Pro261B OXT | 2.7:3.0 | 2.8:2.8 | — |
| O3* | W6 | Trp219B NE1 | 2.8:2.5 | 2.8:2.8 | — |
| O3* | W7 | Glu217B OE1 | 2.7:2.5 | 2.7:3.1 | |
| O4* | Lys117A NZ | 2.7 | 2.8 | — | |
| O4* | Gln174A NE2 | 3.1 | 3.2 | — | |
| O4* | Arg138A NH2 | — | — | 2.7 | |
| O4* | Arg229A NH2 | — | — | 2.9 | |
| O5* | Lys117A NZ | 3.4 | 3.3 | 2.7 | |
| O5* | Arg141B NH1 | 3.3 | — | — | |
| O1P | Arg95A NE | 2.8 | 2.7 | 2.6 | |
| O1P | Arg141B NH2 | 2.8 | — (Met) | 2.9 | |
| O1P | W8 | Arg116A NH1 | — | 2.9:2.5 | — |
| O2P | Ser175A OG | 3.5 | 3.1 | 2.4 | |
| O2P | Arg116A N | 3.2 | 3.3 | — | |
| O2P | Lys117A NZ | 2.9 | 2.8 | — | |
| O3P | Arg229B NH2 | 2.8 | 3.0 | 2.9 | |
| O3P | Ser175A OG | 2.7 | 2.6 | — | |
| O3P | Arg116A NE | — | 3.1 | 2.9 | |
| O3P | Lys117A N | — | — | 2.6 | |
| O3P | W9 | Arg116A NH2 | 2.9:2.9 | — | — |
a Interactions that are less than 3.6 Å are listed.
b Nonidentical residues are shown in parentheses.
c Only the human and E. coli PNPOx have PLP bound at the active site.
Active-site structure of bound PLP
The human PNPOx was cocrystallized with PLP, which exhibits 80% occupancy in the electron density map. As previously observed for the E. coli PNPOx • PLP complex (Safo et al. 2001), C4′ of PLP is situated above the re face of N5 of FMN, with the phosphate group pointing out of the cavity mouth (Fig. 4▶). The bound PLP molecule occupies the same position in the active site and has essentially unaltered conformation in both the human and the E. coli PNPOx structures. In addition, the two crystals show similar interactions between the protein and the bound PLP, with almost all of the residues and interactions strictly conserved (Table 3▶). The interactions involve both protein subunits, denoted with the letters A and B in the table. The phosphate moiety makes salt-bridge and hydrogen-bond interactions with Lys 100A, Arg 161A, Arg 225B, Tyr 157A, and Ser 165A. The PLP pyridine ring is stacked parallel against the pyrazine and pyrimidine portions of the FMN isoalloxazine ring, with extensive van der Waals contacts between the two. As observed in the E. coli structure, C4′ of PLP and N5 of FMN are separated by ∼3.4 Å. The O3′-hydroxyl group of PLP makes a hydrogen-bond interaction with His 227B, whereas the carbonyl oxygen on C4′ makes a water-mediated interaction with Glu 77A. Lastly, the pyridine nitrogen makes a water-mediated hydrogen-bond interaction with Trp 206A. The phosphate moiety of PLP is oriented toward the N terminus of helix α4, which further compensates for the PLP negative charge in addition to the salt-bridge interactions. Among the active-site residues that make direct or water-mediated contact with the PLP, Glu 77 is the only nonconserved residue among the three structures. It is replaced by Asp 49 in E. coli PNPOx and Leu 54 in yeast PNPOx. In both the human and E. coli structures, the respective residues make water-mediated hydrogen-bond interactions with the PLP. The longer side chain of Glu 77 in the human structure has enabled water 2 (W2) to form a shorter hydrogen bond with the PLP and the protein compared with Asp 49 of the E. coli enzyme (Table 3▶). On the other hand, the side chain of Glu 77 occupies two alternate positions in the human structure as against one in the E. coli structure. The second conformer makes an intersubunit hydrogen-bond interaction with the hydroxyl of Tyr 132 (Fig. 4▶).
Figure 4.

Stereoview of the active site of human PNPOx, showing the bound PLP ligand (green) at the re face of the FMN cofactor (yellow). The protein residues are colored cyan and red for monomer A and monomer B, respectively. Atoms are shown in stick representation. Salt-bridge and hydrogen-bonding interactions between the PLP and protein are represented by dotted black lines. Water molecules are labeled W1 and W2, and are represented by red spheres. The figure was generated with INSIGHTII (INSIGHT/DISCOVER: Molecular Simulations, Inc.) and labeled with SHOWCASE.
We have previously discussed the active site in open, partially closed, and completely closed forms (Safo et al. 2001; Di Salvo et al. 2002). In the trigonal crystal forms of human PNPOx • PLP and E. coli PNPOx • PLP complex structures, the active site is partially closed because of the bound PLP (Safo et al. 2001). When the proteins are in the unliganded form as observed in the trigonal crystal form of E. coli PNPOx and the partially refined 2.65 Å human PNPOx structure (see Materials and Methods), the active site is in an open conformation because of the absence of PLP (Safo et al. 2000). In a monoclinic crystal form of the E. coli PNPOx • PLP complex (Di Salvo et al. 2002), the structure shows density for residues 5–19 that are missing in the trigonal crystal forms of both the E. coli and human (32–46 in the human) PNPOx structures. This structure shows a completely closed active site because of the presence of the 5–19 segment, which forms a lid over the active site, with Arg 14 and Tyr 17 forming hydrogen bonds to two phosphate oxygens and the aldehyde of the bound PLP. There is almost no sequence identity between the bacterial and human enzymes in this region, although there are considerable conservative replacements (Fig. 1▶). The residues Arg 14 and Tyr 17 of E. coli PNPOx are replaced by Tyr 41 and Asp 44, respectively, in human PNPOx. These two residues can form hydrogen bonds with the bound PLP, and it is likely that this sequence in the human enzyme also forms a lid over the active site.
Discussion
In mammals the role of PNPOx is not only in a salvage pathway but also to convert pyridoxine, the major vitamin form found in foods, to the active coenzyme following its phosphorylation to PNP.
There is a 39% sequence identity of the human enzyme to the one from E. coli, and it is clear from this study that the two enzyme forms share a remarkable similarity in structure and catalytic properties. From the crystal structure, human PNPOx is a homodimer, as found with the E. coli and yeast enzymes. E. coli and human PNPOx differ in substrate specificity, with PMP being a slightly better substrate than PNP for the human enzyme, whereas the E. coli enzyme has a greater preference for PNP (Table 1▶). PMP was also observed to be a good substrate for the rabbit liver enzyme (Kazarinoff and McCormick 1975). PLP is a product inhibitor of both enzymes, and studies with E. coli indicate that the cellular pool of free PLP is large enough in this organism to make product inhibition a significant method of regulation (Fu et al. 2001). Data on the pool size of free PLP in mammalian tissues are not yet available. These kinetic similarities between E. coli and human PNPOx are the result of the two active sites binding the substrate and product with a set of conserved residues in essentially identical environments (Table 3▶).
Another similarity between the E. coli and human enzymes is the property of each subunit binding a molecule of PLP tightly. The Ki of PLP for the human enzyme as a competitive inhibitor is 3.2 μM, which is not nearly high affinity enough to be tight binding, indicating that this tight binding of PLP occurs at an alternative noncatalytic site. There is a possibility that PLP is tightly bound at the active site in the absence of substrate and that an allosteric site binds the substrate PNP, which when occupied lowers the affinity of the active site for its product PLP. The observation that apoPNPOx also binds PLP tightly supports the argument against this model, however. The active-site structure shows that there is considerable hydrophobic interaction between the pyridine ring of PLP and the isoalloxazine ring of FMN. In the apo enzyme this interaction would not be present and should lower its affinity for PLP, but the apo enzyme also binds PLP tightly (Fig. 2▶).
Materials and methods
Materials
The following materials were purchased from Sigma Chemical Co.: kanamycin; 2-mercaptoethanol; FMN; PLP; sodium cyanoborohydride; protease inhibitor cocktail for bacterial cell extracts; and imidazole. All buffers were of the highest purity available. Ni-NTA agarose was purchased from QIAGEN, EMD TMAE-650 from EM Separations Technology, and Bio-Gel P6 DG from Bio-Rad. Enzymes used in cloning procedures were purchased from New England BioLabs Inc. PNP and PMP were prepared by reduction of PLP following the method of Kazarinoff and McCormick (1975). Rabbit liver and E. coli serine hydroxymethyltransferase (SHMT) and their apo forms were purified as previously described (Yang and Schirch 2000).
General cloning procedures
The cDNA coding for the hypothetical human PNPOx (NCBI nucleotide database entry AK001397) was inserted into the pET28a(+) expression vector. The pET28–human PNPOx construct was used to transform E. coli MDS00 cells, which lack E. coli PNPOx activity (Zhao and Winkler 1995), by the method of Sambrook et al. (1989). The pET28a(+) vector adds 20 residues to the N terminus of human PNPOx, including a sequence of six His residues for binding to an Ni-NTA column. The sequence of the DNA insert was verified in both directions.
Expression and purification
An overnight culture was used to inoculate 6 L of 1.5× LB medium containing 100 mM potassium phosphate (pH 7.2) and 40 mg of kanamycin per liter. Cells were grown at 37°C with rotary shaking for 8 h. After centrifugation, the cell pellet was suspended in 100 mL of cold 50 mM potassium phosphate (pH 7.4), and all subsequent purification steps were performed at 0 to 4°C. A 25-mL aliquot of protease inhibitor cocktail for bacterial extracts (Sigma) was added, and the cells were rapidly broken by osmotic shock in an Avestin Cell Disrupter. Ammonium sulfate was added with stirring to 25% of saturation (144 g/L), and then the mixture was centrifuged for 25 min at 17,000g. The pellet was discarded, ammonium sulfate was added to the supernatant to 55% of saturation (193 g/L), and the precipitated protein was collected by centrifugation as before. The protein pellet was dissolved in a minimal amount of 50 mM potassium phosphate buffer (pH 7.4), containing 1 mM dithiothreitol, and dialyzed with vigorous stirring for 4 h in 1 L of the same buffer, with a buffer change after each hour. A precipitate formed during dialyses, which was removed by a brief centrifugation. To the clear supernatant, a 10-mL solution of protamine sulfate (10 mg/mL) was added, and the resulting white precipitate was removed by a brief centrifugation.
The clarified protein solution was added to an Ni-NTA column (3 cm × 8 cm) equilibrated with 50 mM potassium phosphate (pH 7.4). The enzyme binds to the column as a yellow band. The column was washed with equilibration buffer until the absorbance at 260 nm was below 0.1. The enzyme was eluted with a linear gradient of 100 mL of equilibration buffer and 100 mL of equilibration buffer containing 50 mM imidazole. Fractions were collected, and those containing an absorbance at 445 nm >0.3 were pooled; the protein was precipitated with ammonium sulfate added to 55% saturation (351 g/L). The precipitate was collected by centrifugation, and the pellet was dissolved in 20 mM sodium N,N-bis[2-hydroxyethl]2-aminoethanesulfonic acid (BES; pH 7.4) and dialyzed overnight against two changes of this buffer. The dialyzed enzyme was added to an EMD TMAE column (1.5 cm × 8 cm) equilibrated with 20 mM NaBES (pH 7.4). A yellow band forms at the top of the column. The column was washed with equilibration buffer until the A280 nm was below 0.1, and the enzyme was eluted with a linear gradient of 75 mL of equilibration buffer and 75 mL of 75 mM potassium phosphate (pH 7.4). Fractions of 7 mL that had an A278 nm:A448 nm ratio of <7.5 were pooled and precipitated with 60% ammonium sulfate. After centrifugation, the pellet was dissolved in 20 mM potassium phosphate (pH 7.4), and the solution was dialyzed against an excess of the same buffer.
Determination of extinction coefficients for holo and apo forms of PNPOx
The determination of the molar extinction coefficient at 278 nm used the procedure as described by Gill and von Hippel (1989). The absorbance for the bound FMN at 278 nm was as described for the calculations of these constants for E. coli PNPOx (Di Salvo et al. 1998). Amino acid sequencing was performed as described (Maras et al. 1996). Preparation of human apoPNPOx, by removal of FMN, was done as described for E. coli apoPNPox (Yang and Schirch 2000).
Enzymatic assay and kinetic constants
PNPOx activity was assayed in 50 mM Tris neutralized with BES until the pH was 7.6, with dithiothreitol added to 1 mM just before kinetic studies were performed. Assays were done in a water-jacketed 10-cm pathlength cuvette, at 37°C. The progress of the reaction was followed at 414 nm, where the aldimine of the product PLP and Tris absorbs maximally with a molar absorbance coefficient of 5900 cm−1 M−1. A typical reaction contained in a final volume of 10 mL: 50 mM Tris-BES (pH 7.6); 52 μg of human PNPOx; and 2–20 μM PNP or PMP. Initial rates were determined from the increase in A414 nm during the first 60 sec of the reaction. Km and kcat values and the Ki value for PLP were determined from double reciprocal plots of initial rate versus substrate concentration.
Determination of —SH groups
The number of free sulfhydryl groups in human PNPOx was determined by titration of 10 nmole of enzyme with 200 nmole of 5,5′-dithio-bis-(2-nitrobenzoate) in 50 mM Tris-HCl buffer (pH 8.0) for the native enzyme or 50 mM Tris-HCl (pH 9.0) for the denatured enzyme.
Stoichiometry of PLP binding
PLP was incubated with both human holo- and apoPNPOx. After passage down a P6 DG column to remove excess PLP, the eluted enzyme was tested for tightly bound PLP by the method used for E. coli PNPOx (Yang and Schirch 2000).
Crystallography
Human PNPOx was crystallized both with and without PLP using hanging- and sitting-drop crystallization experiments, respectively. For the condition without PLP, large crystals (0.5 × 0.5 × 0.3 mm) of PNPOx grew in about a week in 20-μL sitting drops containing 7 mg/mL protein, 250 mM NaCl, 1.5% polyethyleneimine, 20 mM sodium citrate, 2.5 mM β-mercaptoethanol in 50 mM potassium phosphate (pH 7.5). These were equilibrated against a 1-mL reservoir solution containing 100 mM sodium citrate (pH 6.0), 0.5 M NaCl, and 3% polyethyleneimine. A solution containing 25% PEG4000, 160 mM sodium acetate, and 17% glycerol in 100 mM Tris-HCl (pH 8.0) was used as a cryoprotectant during data collection. Crystals diffracted to 2.65 Å resolution and belong to the space group P3121 or P3221 with typical unit cell parameters of a = b = 82.26 Å, c = 59.10 Å.
For the liganded experiment, a 4 M excess of PLP was added to a 29-mg/mL PNPOx stock solution just prior to crystallization and incubated on ice for 1 h. The subsequent hanging-drop solution contained 15 mg/mL protein, 0.35 M potassium/sodium tartrate, 4.2 mM PLP, 2.5 mM 2-mercaptoethanol, 50 mM potassium phosphate (pH 7.5), and was equilibrated at room temperature against a reservoir containing 0.7 M potassium/sodium tartrate. Like the unliganded crystals, the liganded crystals have trigonal shape; however, the liganded crystals grew slowly and only reached a maximum size of ∼0.2 × 0.15 × 0.3 mm in 2–3 wk. Prior to use for X-ray diffraction, crystals were first washed in cryoprotectant solution containing 0.7 M potassium/sodium tartrate, 2 mM PLP, and 14% ethylene glycol, followed by transfer to a similar solution with 26% ethylene glycol. The liganded crystal has the same space group as the unliganded crystal with similar unit cell dimensions: a = b = 82.75 Å, c = 59.33 Å, and diffracts to 1.95 Å. Diffraction data sets were collected at 100 K using a Molecular Structure Corporation (MSC) X-Stream Cryogenic Crystal Cooler System (Molecular Structure Corporation), an R-Axis II image plate detector equipped with OSMIC confocal mirrors, and a Rigaku RU-200 X-ray generator operating at 50 kV and 100 mA. All data sets were processed using the BIOTEX software of MSC.
Initially, only the 2.65 Å data set from the unliganded PNPOx crystal was available, and was therefore used with the molecular replacement method program AMoRe (Navaza 1994) of the CCP4 suite (Collaborative Computing Project No. 4. 1994) to determine the structure of human PNPOx enzyme. The monomeric structure of E. coli PNPOx (PDB code 1G79), omitting all PLP, FMN, phosphate, and solvent molecules, was used as a search model. One unique solution with a final correlation coefficient and Rfactor of 37.8 and 47.6%, respectively, was obtained using the space group P3121 and data in the resolution range of 8.0 to 4.0 Å. Structure refinements were performed with the CNS program (Brunger et al. 1998), with overall anisotropic B-factor correction and a bulk solvent correction. All amino acid residues that were not present in the human PNPOx model were mutated to alanine or to the corresponding glycine. Rigid body refinement of the molecular replacement model gave an Rfactor of 46% for all data set to 2.65 Å. In the initial Fo − Fc and 2Fo − Fc maps, the density for a bound FMN molecule was clearly visible, and the cofactor was included in the model. However, some loop, turn, and insertion regions showed ambiguous densities. Therefore, the following regions of the human PNPOx structure were deleted from the model: residues 47–52 (20–25 corresponding to E. coli sequence), 70–77 (43–49), 87–91 (59–63), 132–137 (104–109), 197–199 (169–171), and 233–253 (205–210). After subsequent rigid body, minimization, and annealing refinements, most of the deleted regions and the side chains that were mutated to alanine became apparent in the Fourier density maps and were built into the model. Further refinements with addition of water molecules interjected with model building resulted in a model with amino acid residues 52–234 and 246–261, an FMN and 81 water molecules, with Rfactor and Rfree values of 23% and 30%, respectively, at 2.65 Å resolution. Some extra densities were observed between the residues 234 and 246, but were not sufficient for interpretation except as solvent molecules.
During the course of the refinement, the isomorphous 1.95 Å data set from human PNPOx cocrystallized with PLP became available, and was therefore used to continue the refinement with the 2.65 Å model. One round of rigid body, minimization, and annealing refinements yielded initial Rfactor and Rfree of 26% and 31%, respectively, for all data at 1.95 Å. A PLP, phosphate, and more water molecules were added to the model. The Fourier maps also began to show connectivity between some "solvent" atoms, which actually correspond to the amino acid residues 235–245. Subsequent cycles of minimization and annealing allowed the residues 235–245 to be modeled into the structure. The final model contains residues 49–261, one each of FMN and PLP molecules, and 197 water molecules, with Rfactor and Rfree values of 19.2% and 25.2% at 1.95 Å resolution. Data collection, processing, and refinement statistics for the 1.95 Å structure are summarized in Table 2▶. The atomic coordinate and structure factor sets have been deposited in the RCSB Protein Data Bank with accession code 1NRG. Because the 2.65 Å unliganded structure was not refined to conclusion, data statistics for this structure are not reported in Table 2▶.
Acknowledgments
We thank Bruno Maras for performing the amino acid sequence analysis. This work was supported by United States Public Health Services Grant DK55648 (to V.S.) and a grant from the Italian Ministero dell’istruzione, Università e ricerca (MIUR).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0356203.
References
- Bahn, J.H., Kwon, O.-S., Joo, H.M., Jang, S.H., Park, J., Hwang, I.-K., Kang, T.-C., Won, M.H., Kwon, H.Y., Kwok, F., et al. 2002. Immunohistochemical studies of brain pyridoxine-5′-phosphate oxidase. Brain Res. 925 159–168. [DOI] [PubMed] [Google Scholar]
- Barton, G.L. 1993. ALSCRIPT: A tool to format multiple sequence alignments Protein Eng. 6 37–40. [DOI] [PubMed] [Google Scholar]
- Bowers-Komro, D. and McCormick, D.B. 1985. Pyridoxamine-5′-phosphate oxidase exhibits no specificity in prochiral hydrogen abstraction from substrate. J. Biol. Chem. 260 9580–9582. [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Choi, S.Y., Churchich, J.E., Zaiden, E., and Kwok, F. 1987. Brain pyridoxine-5′-phosphate oxidase: Modulation of its catalytic activity by reaction with pyridoxal 5′-phosphate and analogs. J. Biol. Chem. 262 12013–12017. [PubMed] [Google Scholar]
- Collaborative Computing Project No. 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Di Salvo, M., Yang, E., Zhao, G., Winkler, M.E., and Schirch, V. 1998. Expression, purification, and characterization of recombinant Escherichia coli pyridoxine 5′-phosphate oxidase. Protein Express. Purif. 13 349–356. [DOI] [PubMed] [Google Scholar]
- Di Salvo, M.L., Ko, T.-P., Musayev, J.N., Raboni, S., Schirch, V., and Safo, M. 2002. Active site structure and stereospecificity of Escherichia coli pyridoxine-5′-phosphate oxidase. J. Mol. Biol. 315 385–397. [DOI] [PubMed] [Google Scholar]
- Fu, T.-F., Di Salvo, M., and Schirch, V. 2001. Enzymatic determination of homocysteine in cell extracts. Anal. Biochem. 298 314–321. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Hill, R.E., Himmeldirk, K., Kennedy, I.A., Pauloski, R.M., Sayer, B.G., Wolf, E., and Spenser, I.D. 1996. The biogenetic anatomy of vitamin B6. J. Biol. Chem. 271 30426–30435. [DOI] [PubMed] [Google Scholar]
- Kazarinoff, M.N. and McCormick, D.B. 1975. Rabbit liver pyridoxamine (pyridoxine) 5′-phosphate oxidase: Purification and properties. J. Biol. Chem. 250 3436–3442. [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Maras, B., Greenblatt, H.M., Shoham, F., Spungin-Gialik, A., Blumberg, S., and Barra, D. 1996. Aminopeptidase from Streptomyces griseus: Primary structure and comparison with other zinc-containing aminopeptidases. Eur. J. Biochem. 236 843–846. [DOI] [PubMed] [Google Scholar]
- McCormick, D.B. 1996. Coenzymes, biochemistry of. In Encyclopedia of molecular biology and molecular medicine (ed. R.A. Meyers), Vol. 1, pp. 396–406. VCH, Weinheim, Germany.
- ———. 1997. Vitamins, structure and function of. In Encyclopedia of molecular biology and molecular medicine (ed. R.A. Meyers), Vol. 6, pp. 244–252. VCH, Weinheim, Germany.
- McCormick, D.B. and Chen, H. 1999. Update on interconversions of vitamin B-6 with its coenzyme. Rec. Adv. Nutr. Sci. 129 325–327. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3D version 2.0: A program for photorealistic molecular graphics. Acta Crystallogr. D 50 869–873. [DOI] [PubMed] [Google Scholar]
- Safo, M.K., Mathews, I.J.N., Musayev, F.N., di Salvo, M.L., Thiel, D.J., Abraham, D.J., and Schirch, V. 2000. X-ray structure of Escherichia coli pyridoxine 5′-phosphate oxidase complexed with FMN at 1.8 Å resolution. Structure 8 751–762. [DOI] [PubMed] [Google Scholar]
- Safo, M., Musayev, F.N., di Salvo, M.L., and Schirch, V. 2001. X-ray structure of Escherichia coli pyridoxine 5′-phosphate oxidase complexed with pyridoxal 5′-phosphate at 2.0 Å resolution. J. Mol. Biol. 310 817–826. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, T., and Maniatis, T. 1989. Molecular cloning. A laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Yang, E.S. and Schirch, V. 2000. Tight binding of pyridoxal 5′-phosphate to recombinant Escherichia coli pyridoxine 5′-phosphate oxidase. Arch. Biochem. Biophys. 377 109–114. [DOI] [PubMed] [Google Scholar]
- Zhao, G. and Winkler, M.E. 1995. Kinetic limitation and cellular amount of pyridoxine (pyridoxamine) 5′-phosphate oxidase of Escherichia coli K-12. J. Bacteriol. 177 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]