

Abstract
We determined the 1.17 Å resolution X-ray crystal structure of a hybrid peptide based on sequences from coiled-coil regions of the proteins GCN4 and cortexillin I. The peptide forms a parallel homodimeric coiled-coil, with Cα backbone geometry similar to GCN4 (rmsd value 0.71 Å). Three stabilizing interactions have been identified: a unique hydrogen bonding–electrostatic network not previously observed in coiled-coils, and two other hydrophobic interactions involving leucine residues at positions e and g from both g-a‘ and d-e‘ interchain interactions with the hydrophobic core. This is also the first report of the quantitative significance of these interactions. The GCN4/cortexillin hybrid surprisingly has two interchain Glu-Lys‘ ion pairs that form a hydrogen bonding network with the Asn residues in the core. This network, which was not observed for the reversed Lys-Glu‘ pair in GCN4, increases the combined stability contribution of each Glu-Lys‘ salt bridge across the central Asn15-Asn15′ core to ∼0.7 kcal/mole, compared to ∼0.4 kcal mole−1 from a Glu-Lys‘ salt bridge on its own. In addition to electrostatic and hydrogen bonding stabilization of the coiled-coil, individual leucine residues at positions e and g in the hybrid peptide also contribute to stability by 0.7 kcal/mole relative to alanine. These interactions are of critical importance to understanding the stability requirements for coiled-coil folding and in modulating the stability of de novo designed macromolecules containing this motif.
Keywords: Coiled-coil, protein folding, protein stability, protein oligomerization, electrostatics, side-chain packing, circular dichroism spectroscopy
The rodlike α-helical coiled-coil is one of the simplest yet most common structural motifs occurring in proteins. Consisting of two to five α-helices twisted into a left-handed supercoil, the occurrence of this structure is well documented, occurring in a wide variety of proteins including motor proteins, DNA binding proteins, extracellular proteins, and viral fusion proteins (Lupas 1996; Kohn et al. 1997; Burkhard et al. 2001). The presence of a continuous interface of hydrophobic amino acids along the length of the helices provides a major source of stability to the fold as the hydrophobes pack in a knobs-into-holes fashion shielded from the bulk solvent (Crick 1953). The pattern of repeating hydrophobic residues at positions a and d of the heptad repeat (denoted abcdefg) that are responsible for coiled-coil formation was first identified by Hodges et al. (1972) from the amino acid sequence of tropomyosin. This 3–4 or 4–3 hydrophobic repeat allows for the prediction of coiled-coils based on statistical occurrence of residues in these positions (Berger et al. 1995; Berger and Singh 1997; Wolf et al. 1997; Singh et al. 1999), and more recently, with STABLECOIL, an algorithm based on experimentally derived stability data (Wagschal et al. 1999a; Tripet et al. 2000; Tripet and Hodges 2001). However, deletion and/or mutational analysis of some protein sequences (such as GCN4, cortexillin I, macrophage scavenger receptor, and intermediate filament chains) has revealed that the presence of heptad repeats does not always guarantee the formation of coiled-coil structure (Steinmetz et al. 1998; Frank et al. 2000; Wu et al. 2000; Kammerer et al. 2001; Lee et al. 2001). The absence of a key region of only one or two heptads (dubbed the ‘trigger sequence’) prevented folding, whereas its presence induced folding of the coiled-coil structure. We previously took a 31-residue peptide that did not fold even with an excellent hydrophobic core (two valine, one leucine, and one methionine residue at position a and four leucine residues in position d, shown by Wagschal et al. [1999a] and Tripet et al. [2000] to provide excellent stability) and then introduced stabilizing amino acid substitutions to induce dimeric coiled-coil folding without matching a proposed ‘consensus trigger sequence,’ to prove that such a sequence is not necessary for coiled-coil folding (Lee et al. 2001). Although trigger sequences may be important for folding in naturally occurring coiled-coils of substantial length, we proposed that any sequence with enough stability above a critical threshold will fold, whether the stability is distributed evenly along the coiled-coil sequence or concentrated heavily in distinct regions. Thus, the ability to induce coiled-coil folding is not limited to a consensus trigger sequence, but should be present in any peptide sequence with sufficient individual helix and/or coiled-coil stability (Burkhard et al. 2000a, 2002). This is further supported by the large number and variety of designed synthetic and recombinant peptide sequences that do not contain the consensus trigger sequence, yet still fold into two-stranded coiled-coils (Dieckmann et al. 1998; Harbury et al. 1998; Kohn et al. 1998; Micklatcher and Chmielewski 1999; Pandya et al. 2000; McClain et al. 2001; Acharya et al. 2002; Arndt et al. 2002; Campbell et al. 2002; Litowski and Hodges 2002; Phelan et al. 2002; Havranek and Harbury 2003).
Here we determined the crystal structure for the most stable folded peptide from our previous study, which did not contain a ‘consensus trigger sequence’. This stable peptide contained potentially stabilizing design elements over the nonfolding sequence, including increased helix propensity, elimination of ionic repulsions, and introduction of ionic attractions. These stabilizing elements are identified in the 1.17 Å crystal structure. We also quantify the previously unreported stability contribution of three interactions using thermodynamic analysis of urea denaturation curves of peptide analogs: a unique complex hydrogen bonding–electrostatic network involving i to i‘ + 5 electrostatic interactions and hydrogen bonding interactions with Asn in the hydrophobic core, and hydrophobic packing of leucine residues at positions e (d-e‘ interaction) and g (g-a‘ interaction).
Results
Peptide design
Our previous results showed that the native peptide (Hybrid 1) did not fold (Lee et al. 2001), even though it had an excellent hydrophobic core which makes a major contribution to overall stability because of the one leucine, one methionine, and two valine residues at position a (Wagschal et al. 1999a) and the four leucine residues at position d (Fig. 1▶; Tripet et al. 2000). This sequence was based primarily on the coiled-coil regions of the transcription factor, GCN4, and the actin bundling protein, cortexillin I. The N-terminal (residues 2 to 14) contains cortexillin I 270–282 (Burkhard et al. 2000b), whereas the C-terminal (residues 16–28) contains GCN4-p1 residues 3 to 15 (O’Shea et al. 1991). Both segments were inserted into the GCN4-p1 sequence 2–32, labeled as 1–31 in Figure 1▶. In the middle of this peptide sequence, residue 15 is a polar asparagine (a position) buried in the hydrophobic core. Polar residues in core a and d positions reduce coiled-coil stability (Wagschal et al. 1999a; Tripet et al. 2000; Zhu et al. 2000; Akey et al. 2001) versus more hydrophobic amino acids, but asparagine was left in the GCN4 template to specify dimerization over higher-order oligomerization states such as trimers or tetramers (Harbury et al. 1993). This central asparagine is analogous to the central Asn observed in the coiled-coil structure of GCN4-p1 (O’Shea et al. 1991).
Figure 1.

Sequences of peptides used in this study. Peptides are denoted by one-letter code and are labeled peptides 1 to 7. The sequence of GCN4-p1 (O’Shea et al. 1991) residues 2–32 is shown for comparison, where Met 2 of GCN4-p1 is labeled residue 1. Hybrid 1 is a GCN4-cortexillin I hybrid that did not fold into a coiled-coil, whereas Hybrid 2 formed a stable coiled-coil (Lee et al. 2001). The a and d positions of the heptad repeat abcdefg are boxed. Bolded residues indicate amino acid substitutions in Hybrid 2 relative to Hybrid 1. The circled and bolded residues in peptides 2 to 7 indicate amino acid substitutions relative to the Hybrid 2 peptide sequence. The residues 2–14 in the Hybrid 1 sequence are from cortexillin I residues 270–282, and residues 16–28 contain GCN4-p1 2–14, where both segments are inserted into the GCN4 template 1–31.
Because Hybrid 1 did not fold, we introduced amino acid substitutions into Hybrid 1 that would induce folding independently of a ‘consensus trigger sequence.’ The result was Hybrid 2, whose structure was determined in the present study. Hybrid 2 exhibited increased coiled-coil folding and stability over Hybrid 1 because of five amino acid substitutions: S6A, S13A, D20A, S27A, and E19K. Hybrid 2 contains four alanine residues in the f positions (A6, A13, A20, and A27) not found in GCN4 or cortexillin I, in order to increase α-helix propensity (Fig. 2▶). Also, a lysine replaced a glutamic acid at position 19 (e position), which eliminated a potential i to i‘ – 5 ionic repulsion with Glu 14‘ (g‘ position) in the original nonfolding peptide, and could instead form a potential interhelical i to i‘ – 5 salt bridge across the hydrophobic interface (Fig. 2▶).
Figure 2.

End-on view of the hybrid peptide analog, viewed from N- to C-terminus. Solid arrows denote predicted interchain (i to i‘ + 5) and intrachain (i to i + 3; i to i + 4) ion pairing between residues. Open arrows denote the hydrophobic interactions between residues a and a‘ or d and d‘.
Overall fold
The x-ray crystal structure of Hybrid 2 (Table 1▶) is a parallel two-stranded coiled-coil (Fig. 3▶). The dimer of the coiled-coil is built up from an internal two-fold crystal symmetry element. Two core residues at the dimer interface (Leu 8 and Asn 15), each in a positions of the heptad repeat, show two rotamer conformations (Figs. 4, 5▶▶). Asn 15 adopts the two most preferred rotamer conformations (Lovell et al. 2000) with χ1/χ2 torsion angles of −82°/14° and 169°/−90°, respectively. In contrast to the two-fold symmetry observed in Hybrid 2, the crystal structure of another coiled-coil, GCN4-p1 (sequence in Fig. 1▶; O’Shea et al. 1991) does not show two-fold symmetry but still shows Asn 15 in two different conformations (in and out).
Table 1.
Data collection and refinement statistics
| Space group | C2221 |
| Unit cell a, b, c (Å) | 18.098, 117.287, 2.209 |
| α, β, γ (°) | 90, 90, 90 |
| Monomers/au | 1 |
| Data collection statistics | |
| Resolution (Å) | 1.17 |
| Observed reflections | 263880 |
| Unique reflections | 8183 |
| Completeness (%) | 97.8 |
| Rsyma | 0.037 |
| Refinement statistics | |
| R factor (%)b | 17.9 |
| Rfree (%)b | 21.7 |
| Mean B factor protein atoms (Å2) | 15.1 |
| Mean B factor water atoms (Å2) | 31.3 |
| rmsd bond distances (Å)c | 0.009 |
| rmsd bond angles (°)c | 1.8 |
aRsym = ∑|l − ≤l>|/∑l.
bR factor = ∑||Fobs| − |Fcalc||/∑|Fcalc|.
c Root-mean-square deviation.
Figure 3.
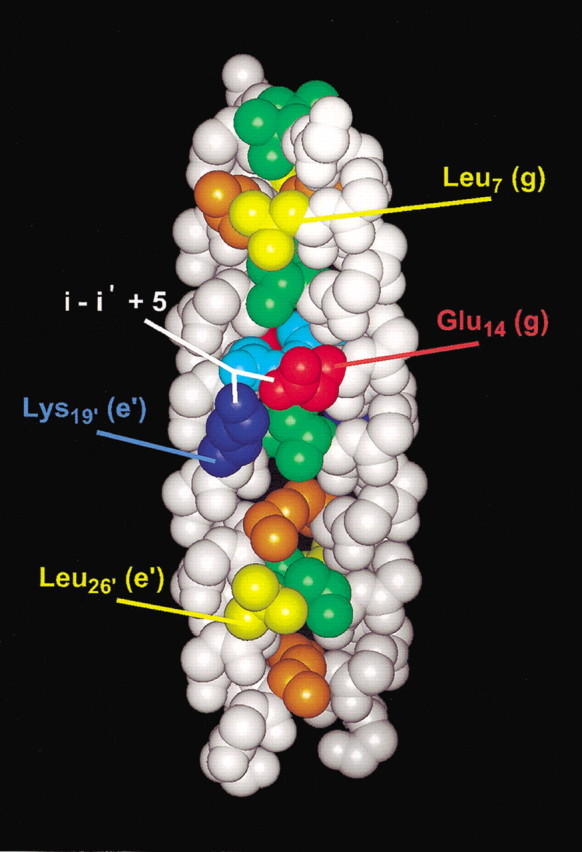
Space-filling representation of the GCN4/cortexillin Hybrid 2 structure. Residues in positions a and d along the hydrophobic core are displayed in orange and green, respectively, with the exception of Asn 15 (a position) in light blue. Leu 7 (g) and Leu 26 (e) side chains are displayed in yellow. Glu 14 (g) and Lys 19′ (e‘) side chains are displayed in red and dark blue, with the i to i‘ + 5 interchain interaction shown in white.
Figure 4.

Cross-sectional view of the region surrounding the Asn 15 (a) core in Hybrid 2. Hydrogen bonds are denoted by dashed yellow lines. Oxygen atoms are red; nitrogen atoms are blue. The Cα helix backbone is shown in white (ribbon representation). The two interconverting conformers of Asn 15 and Asn 15′ (‘in’ or ‘out’) are inside the pink boxes of panels A and B. (A) Asn 15 is ‘in’ (lower helix); Asn 15′ is ‘out’ (upper helix). (B) Asn 15 is ‘out’ (lower helix); Asn 15′ is ‘in’ (upper helix).
Figure 5.

Cross-sectional comparison of g-a packing and d-e packing of leucines in the hydrophobic core of the hybrid peptide analog, viewed from N-terminus to C-terminus. The Cα helix backbone is shown as white sticks, and hydrogen atoms are depicted as white balls. Panels A and B: Leu 7 (g) and Leu 7′ (g‘) side chains are shown in yellow; Leu 8 (a) and Leu 8′ (a‘) side chains are shown in orange. The two conformations of Leu 8 (a) and Leu 8′ (a‘), labeled from left to right, are (a) ‘out/in’ and (b) ‘in/out’. (C) Packing of Leu 25 (d), in green, and Leu 26 (e), in yellow, in the hydrophobic interface.
Interactions in the Asn 15 core region
The two asparagines in core a positions (Asn 15 and Asn 15′), together with the two Glu (14 and 14′) and two Lys (19 and 19′) residues, form an extended network of hydrogen bonds with a total of five (four interhelical and one intrahelical) hydrogen bonds not previously observed in the structures of coiled-coils (Fig. 4▶). The amide and carbonyl groups of the two Asn 15 side-chains are 3.1 Å apart from each other. The Nδ2 atom from asparagine with the ‘out’ conformation also forms a 2.8 Å hydrogen bond with the carboxyl group oxygen of Glu 14 (Fig. 4a▶) and Glu 14′ (Fig. 4b▶). Glu 14 – Lys 19′ and Glu 14′ – Lys 19 form the two interhelical salt bridges with an Nζ to Oɛ1 distance of 3.2 Å on both sides of the coiled-coil. The lone intrahelical hydrogen bond occurs between the Oδ of Asn 15 in the ‘out’ conformation and the Nζ of Lys 19.
In the GCN4-p1 coiled-coil, there is an interchain electrostatic interaction on only one side of the coiled-coil with an interatomic distance of 3.7 Å. However, there is no interaction with the nearest Asn residue in the hydrophobic core. On the other side of GCN4-p1, the side chains of Lys and Glu are 6.1 Å apart (Fig. 6b▶) and thus are too far apart to interact with each other, or with Asn 15 (indicated by the red X’s in Fig. 6b▶). This can be explained by the fact that in the GCN4-p1 peptide, the charged residues are in opposite positions, and due to the stereochemical restraints, the favorable hydrogen bonding pattern as observed in the Hybrid 2 structure cannot form. This is a clear indication that the configuration with Glu in the g position and Lys in the e‘ position (as it is in Hybrid 2) is more stabilizing to the coiled-coil than the opposite arrangement, if Lys is in the g position and Glu is in the e‘ position (as it is in the GCN4-p1 coiled-coil).
Figure 6.

Comparison of the region around the central Asn (a) in Hybrid 2 (A) and GCN4 (B) coiled-coils. The numbering of residues in GCN4 has been aligned to the hybrid sequence in Fig. 1▶. Oxygen atoms are red; nitrogen atoms are blue. Hydrogen bonds are denoted by dashed yellow lines. The Cα helix backbone is shown in white (ribbon representation). Solid yellow lines with a red ‘x’ denote a lack of noncovalent bonding between atoms.
Crystal contacts
Surprisingly, none of the proposed intrahelical salt bridges are formed in the crystal. Instead, all residues thought to form such interactions are involved in crystal lattice contacts. Lys 21 and Glu 24 (potential i to i + 3 ion pair) form salt bridges with the residues Glu 24 and Lys 21 from a symmetry-related molecule; the distance between these residues is approximately 4 Å. Glu 12 is involved in an interaction with Asn 15 of a symmetry-related molecule, and does not form an i to i + 4 intrahelical salt bridge with Lys 16. Glu 23 forms a salt bridge with Lys 28 from a symmetry-related molecule, despite the possibility that these residues could each form intrahelical salt bridges with Lys 19 (i to i + 4) and Glu 31 (i to i + 3), respectively. Also, three hydrophobic alanine residues in b and f positions—Ala 6(f), Ala 9(b), and Ala 13(f)—pack against identical residues from a crystallographically related molecule. Thus, crystal packing forces may disrupt intrachain interactions that would be very important in solution, in favor of intermolecular interactions in the crystal.
Comparison with GCN4
The peptide backbone can be superimposed upon the GCN4-p1 coiled-coil dimer, giving an rmsd of 0.71 Å for Cα atoms from residues 3 to 30. The largest differences in the positions of the Cα backbone atoms occur at the N-terminus. In contrast, the Cα atoms of the C-terminal portion of the coiled-coil (residues 15–29) can be superimposed with an rmsd of only 0.36 Å. Hybrid 2 has a leucine residue in an N-terminal a position (Leu 8) compared to a smaller valine residue in GCN4-p1 that may explain the difference in the Cα backbone between GCN4-p1 and Hybrid 2. The Cα–Cα distance between Leu 8 and Leu 8′ at position a is 6.3 Å, whereas in the GCN4-p1 structure the Val 8 and Val 8′ Cα carbons are separated by 5.5 Å (see Fig. 1▶ for sequences). According to the program TWISTER (Strelkov and Burkhard 2002), the coiled-coil radius of Hybrid 2 increases towards the N-terminus and also the coiled-coil pitch of Hybrid 2 increases, indicating a local unwinding of the coiled-coil (Fig. 7▶): the Cα–Cα distance between the two Leu 4 residues at position d gets as high as 8.4 Å, whereas in GCN4-p1 it is only 6.3 Å. Clearly, packing the larger Leu residue instead of Val at position a results in the increase in coiled-coil radius, as position 8 has the only residue that is different in the hydrophobic a and d positions of GCN4-p1 versus Hybrid 2.
Figure 7.

Coiled-coil pitch (solid lines) and coiled-coil radius (dashed lines) plotted against the residue number for the Hybrid 2 (•) and the GCN4-p1 peptide (○). The residue number is according to the Hybrid 2 peptide in both sequences, i.e., in GCN4-p1 they are shifted by one residue.
Biophysical characterization of peptides
To quantitatively determine the extent that residues in the e and g positions stabilize the coiled-coil structure of Hybrid 2, we synthesized a set of analogs substituted at various e and g positions along the sequence (peptides 2 to 7 in Fig. 1▶). We characterized the secondary structure, oligomerization state, and thermodynamic stability of the peptides by circular dichroism (CD), sedimentation equilibrium, and chemical unfolding experiments (Figs. 8, 9▶▶; Table 2▶). All peptides formed α-helices in benign medium and are folded as α-helical coiled-coils based on the following characteristics: high negative molar ellipticity values in benign medium, with double minima at 208 and 222 nm and a maximum at 190 nm; and a high α-helix content such that the addition of 50% trifluoroethanol (TFE) does not significantly increase the α-helix content (determined at 222 nm; Table 2▶). Furthermore, the ratio of molar ellipticities at 222 nm and 208 nm suggests that all of the peptides studied formed coiled-coils: the [θ]222/[θ]208 ratio for coiled-coils in benign medium is usually greater than 1.0 (Lau et al. 1984), and this value decreases to 0.90 in the presence of the monomeric helix-inducing solvent, trifluoroethanol. Also, results from sedimentation equilibrium experiments indicated that most of peptides in the present study were globally fit to a single dimeric species in benign medium, with observed molecular weights that are approximately double the calculated weight of the monomers (Table 2▶).
Figure 8.

CD spectra of the peptide K19E obtained at 20°C in 50 mM phosphate, 100 mM KCl, pH 7.0 buffer (benign). Peptide concentrations were 2592 μM (♦), 432 μM (▴), 86 μM (•), 22 μM (▾) 4 μM (▪) and in 1:1 (v:v) benign buffer and TFE at 432 μM (▵).
Figure 9.

Chemical unfolding curves of the Hybrid 2 peptide and analogs. Fraction folded was plotted against denaturant concentration for Hybrid 2 (•) and peptides E14A (▿); K19A (+); L7A (□); L26A (×); E14A, K19A (▵); and K19E (♦).
Table 2.
Biophysical characterization of hybrid peptide and analogs
| [θ]222 (×103 deg cm2 dmole−1)a |
||||||||
|---|---|---|---|---|---|---|---|---|
| Peptide | Benign | TFE | [θ]222/[θ]208 benignb | [θ]222/[θ]208 TFEc | Percent helixd benign | Helical residuese | mcalc (Da)f | mobs (Da)g |
| Hybrid 2 | −31.5 | −30.6 | 1.00 | 0.86 | 103 | 33 | 3439 | 6698 (D)h |
| L7A | −29.9 | −31.8 | 1.02 | 0.87 | 94 | 29 | 3397 | 6556 (D) |
| L26A | −29.8 | −31.1 | 1.03 | 0.90 | 96 | 30 | 3397 | 6266 (D) |
| E14A | −29.4 | −29.7 | 1.03 | 0.89 | 99 | 31 | 3381 | 6162 (M–D)* |
| K19A | −30.8 | −30.3 | 1.03 | 0.89 | 102 | 32 | 3382 | 6383 (D) |
| E14A, K19A | −31.3 | −33.0 | 1.02 | 0.89 | 95 | 29 | 3324 | 6524 (D)* |
| K19E | −27.7 | −30.3 | 1.01 | 0.88 | 91 | 28 | 3440 | 6618 (D)h |
a Molar ellipticity at 222 nm at 20°C in benign buffer (50 mM potassium phosphate, 100 mM KCl, pH 7.0), 400 μM peptide concentration. For samples containing trifluoroethanol (TFE), the above buffer was diluted 1:1 (v:v) with TFE.
b Ratio of molar ellipticities at 222 nm and 208 nm in benign buffer.
c Ratio of molar ellipticities at 222 nm and 208 nm in a mixture of 1:1 (v:v) benign buffer and trifluoroethanol.
d The percent α-helix was calculated by taking 100% α-helix as the [θ]222 value obtained in 50% TFE (the maximum inducible α-helical structure).
e The number of α-helical residues was calculated by multiplying the percent α-helix (column d) by the number of residues in the polypeptide chain, 31.
f Calculated molecular mass of monomeric peptides based on average isotopic masses.
g Observed molecular mass based on global fits of sedimentation equilibrium data (see Materials and Methods). Data was best fit to a monomer to dimer equilibrium (M–D), or dimer (D). Values labeled with an asterisk (*) were determined in the presence of 2 M urea to eliminate higher order oligomerization states observed in benign buffer. The sedimentation equilibrium data in benign buffer was best fit to a monomer-trimer equilibrium for E14A (MWobs 8087) and a monomer-tetramer equilibrium for E14A, K19A (MWobs 9896). See results for further details.
h Previously determined sedimentation equilibrium values (Lee et al. 2001).
Peptides E14A and E14A, K19A exhibited higher-order oligomerization in benign medium. E14A was best fit to a monomer-trimer equilibrium (MWobs 8087), and E14A, K19A was best fit to a monomer-tetramer equilibrium (MWobs 9896). These peptides showed essentially no change in molar ellipticities (or helicities) in the pretransition region (0 to 2 M urea) of the urea unfolding curves (Fig. 9▶). As shown previously by Wagschal et al. (1999b), the oligomerization state of coiled-coils can change from a higher-order state (in the absence of denaturant) to the two-stranded state at levels of denaturant that are below the denaturant concentration required for the start of the transition from the folded to unfolded state. Therefore, we performed the sedimentation equilibrium analyses of peptide E14A and peptide E14A, K19A in 2 M urea to show that the coiled-coil analogs are two-stranded prior to unfolding. Under these conditions, E14A existed as a monomer-to-dimer equilibrium, and E14A, K19A existed as a single dimeric species (Table 2▶). This is important because the unfolding curve must represent the transition from the same oligomerization state to the same unfolded state for all analogs in order for the calculated ΔΔGu values to be meaningful.
By monitoring the folded state of the coiled-coil as a function of denaturant concentration using CD spectroscopy, we obtained values for the urea concentration at the transition midpoint ([urea]½), the slope at the transition midpoint (m), and the stability contributions for Hybrid 2 relative to less stable analogs (ΔΔGuobs; Fig. 9▶, Table 3▶). Peptides were analyzed at ∼400 μM concentration to ensure that the total population of the entire set of peptides was essentially fully folded in the absence of denaturant; the effect of peptide concentration on the CD spectra of the least stable peptide in this study, K19E (peptide 7 in Fig. 1▶), is shown in Figure 8▶. Although K19E shows an increase in negative molar ellipticity from 432 μM to 2600 μM, indicating greater helicity at higher concentrations, it was calculated to be 91% helical at ∼400 μM by comparing [θ]222 values in benign conditions and in 50% TFE (Table 2▶). All the other peptides were more stable and more helical than K19E (Fig. 9▶) at 400 μM concentration in benign conditions (Table 2▶). Hybrid 2 was the most stable peptide in this study, so ΔΔGuobs was reported as a positive value, that is, a stabilizing contribution, by calculating the stability difference of Hybrid 2 over the less stable analog (Table 4▶).
Table 3.
Urea denaturation data
| Peptidea | Heptad positionb | [Peptide] (μM)c | [Urea]1/2 (M)d | m (kcal mole−1M−1)e | ΔΔGuobs (kcal mole−1)f |
|---|---|---|---|---|---|
| Hybrid 2 | — | 341 | 6.9 | 0.69 | — |
| L7A | g | 350 | 4.8 | 0.68 | 1.44 |
| L26A | e | 465 | 5.0 | 0.68 | 1.30 |
| E14A | g | 450 | 5.4 | 0.73 | 1.06 |
| K19A | e | 316 | 4.8 | 0.63 | 1.39 |
| E14A, K19A | g, e | 375 | 5.4 | 0.86 | 1.08 |
| K19E | e | 460 | 2.5 | 0.70 | 3.06 |
a The sequences are given in Fig. 1▶.
b The position of the substituted residue within the heptad repeat, abcdefg.
c Amino acid analysis of stock peptide solutions was used to determine the total peptide concentration in solutions analyzed by CD spectroscopy.
d Denaturant concentration required to achieve a 50% reduction of the folded state or loss of α-helical content as measured by [θ]222.
e Slope term in the equilibrium ΔGu([urea]) = ΔGuH2O − m[urea], where ΔGuH2O is the Gibbs free energy change of unfolding in the absence of denaturant (Santoro and Bolen 1988). The ΔGuH2O value for hybrid 2 was 9.4 kcal mole−1.
f ΔΔGuobs is the observed change in ΔGu, the free energy change of unfolding, between hybrid 2 and the peptide analog in column 1, and is calculated ΔGuobs = ([urea])1/2, hybrid 2 − [urea]1/2, analog)*(mhybrid 2 + manalog)/2 (Serrano and Fersht 1989).
Table 4.
Contributions of α-helix propensity, hydrophobic and electrostatic interactions to coiled-coil stability
| Peptide comparisons |
|||||
|---|---|---|---|---|---|
| More stable coiled-coil | Less stable coiled-coil | ΔΔGuobs coiled-coil (kcal mole−1) | Description of stabilizing contribution | ΔΔGuobs per substitution (kcal mole−1) | ΔΔGuobs correctedd (kcal mole−1) |
| Hybrid 2 | L7A | 1.44 | Hydrophobic contribution of Leu (g) vs. Ala | 0.74 | 0.74 |
| Hybrid 2 | L26A | 1.38 | Hydrophobic contribution of Leu (e) vs. Ala | 0.69 | 0.69 |
| Hybrid 2 | E14A | 1.06 | Electrostatic and H-bond attractions uncorrected for α-helix propensity | 0.53 | 0.75 |
| Hybrid 2 | K19A | 1.39 | Electrostatic and H-bond attractions uncorrected for α-helix propensity | 0.70 | 0.70 |
| Hybrid 2 | E14A, K19A | 1.08 | Electrostatic and H-bond attractions uncorrected for α-helix propensity | 0.54 | 0.76 |
| E14A, K19A | K19A | 0.45a | Helix propensity of Ala vs. Glu (g) | 0.22 | — |
| E14A, K19A | E14A | 0.00 | Helix propensity of Ala vs. Lys (e) | 0.00 | — |
| K19A | K19E | 1.53b | Removal of electrostatic repulsions uncorrected for α-helix propensity | 0.77 | 0.55 |
| Hybrid 2 | K19E | 3.06c | Removal of electrostatic repulsions and replacement with electrostatic and H-bond attractions | — | — |
a The α-helix propensity effect of E14 to A14 is calculated from the equation ΔΔGuobs = ([urea]1/2(E14A, K19A) − [urea]1/2(K19A))*(mE14A, K19A + mK19A)/2 (from Serrano and Fersht 1989).
b The stabilizing effects of removing two electrostatic repulsions is calculated from ΔΔGuobs = ([urea]1/2(K19A) − [urea]1/2(K19E)*(mK19A + mK19E)/2 (from Serrano and Fersht 1989).
c Observed values of ΔΔGu, coiled coil = 3.06 kcal mole−1 can be compared with the calculated corrected values from the loss of two electrostatic repulsions 2*(0.55), plus the gain of the average value for two electrostatic attractions and H-bonding 2*(0.73), = 2.56 kcal mole−1.
d ΔΔGuobs corrected is the ΔΔGuobs per substitution corrected for α-helix propensity effects. The α-helix propensity of Ala compared to Glu is 0.22 kcal mole−1, which means that the stabilizing effect of an electrostatic and H-bond attraction is the ΔΔGuobs value of 0.54 kcal mole−1 plus 0.22 kcal mole−1 to give 0.76 kcal mole−1.
The choice of substituting the residues by alanine to eliminate g to e‘ interchain effects such as ionic interactions or hydrophobic interactions also affects another parameter that influences coiled-coil stability: α-helix propensity. Alanine has the highest intrinsic helix propensity among the twenty naturally occurring amino acids (O’Neil and DeGrado 1990; Blaber et al. 1993; Monera et al. 1995; Myers et al. 1997; Pace and Scholtz 1998), and so the difference in helix propensities between Ala and the substituted residue was determined and corrected in the final column of Table 4▶. Alanine is an ideal residue to carry out substitutions in our case because it does not disrupt the α-helical coiled-coil structure, as it might if one were inserting it into an alternate nonhelical structure (Minor Jr. and Kim 1994). After correction, the average value for the strength of a g to e‘ ion pair was 0.74 kcal mole−1 (averaged over the ΔΔGuobs values of single substitutions E14A; K19A; and double substitutions E14A, K19A). Leucine was not corrected for helix propensity, although its value has previously been shown to be similar to Ala (Pace and Scholtz 1998).
Interactions of leucine residues at positions e and g
Peptides L7A and L26A contain destabilizing substitutions by replacing leucine residues at positions g and e respectively with the shorter, less hydrophobic alanine side chains. L7A reduced stability by 0.74 kcal mole−1, whereas L26A reduced stability by 0.69 kcal mole−1 per substitution. Overall, leucine residues at e and g each contributed ∼0.7 kcal mole−1 to stability relative to alanine, whereas the interchain Glu-Lys‘ salt bridge involved in the hydrogen bonding network contributed 0.74 kcal mole−1 to stability after correction for helix propensity.
Discussion
Unlike the a and d positions in the hydrophobic core, the environment of side chains located at the e and g positions in a two-stranded coiled-coil is partially solvent-excluded but also partially solvent-exposed (Fig. 2▶). Therein lies the controversy over the effects of interchain ion pairs located at these postions, as the stabilizing benefit of an electrostatic attraction must outweigh the energy penalty from both desolvation and the presence of charged residues in a hydrophobic environment. Similarly, hydrophobic residues such as the long aliphatic chains of leucine and isoleucine, while gaining a favorable energy contribution to stability in the hydrophobic environment, may suffer a penalty because of their solvent accessibility at the e and g positions.
Electrostatic interactions at positions e and g
In comparison with other values for interchain Glu-Lys‘ attractions (ranging from 0.4 to 0.5 kcal mole−1; Zhou et al. 1994; Krylov et al. 1998) and Glu-Glu‘ repulsions (0.45 kcal mole−1; Kohn et al. 1995) in g-e‘ positions of coiled-coils, our values for each electrostatic attraction in this study are greater by 0.2 to 0.3 kcal mole−1 (Table 4▶). The increased stability of the interchain Glu-Lys‘ attraction can be explained by the two hydrogen bonds (Fig. 4a▶) in the Asn core region (one from E14 to N15′, the other from K19‘ to N15′) which contribute the added stability.
Hydrophobic residues at positions e and g
Hydrophobic residues at positions a and d in the hydrophobic core and their contributions to stability have previously been studied in detail. Here we examined Leu packing and stability at positions e and g, and found similar stability values in both positions. The side-chain packing of Leu (Fig. 5▶) in both cases is adjacent to another Leu, either g-a‘ (Leu 8) or d-e‘ (Leu 25). Our values of 0.69 and 0.74 kcal mole−1 for leucine in positions e and g are less than half the stability contribution at the a and d positions (1.75 and 1.90 kcal mole−1; Wagschal et al. 1999a; Tripet et al. 2000). The stability differences between Leu at a or d versus e or g positions match the differences in solvent-accessible surface area: after calculating accessible surface area in the Hybrid 2 structure using GETAREA 1.1 (Fraczkiewicz and Braun 1998), we observed that Leu 26 and Leu 7 at positions e and g were 48% exposed to solvent, whereas Leu 8 and Leu 25 at positions a and d were only 7% exposed at position d, and 9% (‘in’ conformation) or 20% (‘out’ conformation) exposed at position a. Thus, the more that leucine was shielded from solvent (in positions a or d), the more it contributed to stability via the hydrophobic effect.
How does side-chain packing affect the stability of hydrophobes in positions e and g? Figure 5▶ shows the g-a packing of Leu 7 and Leu 8, and the d-e‘ packing of Leu 25 and Leu 26. With two conformations of Leu 8 in each helix of Hybrid 2, two possible combinations for the coiled-coil are shown in Figure 5▶, A and B: ‘out/in’ and ‘in/out’. These conformations would satisfy both the packing of the hydrophobic core as well as interactions with the neighboring Leu 7 at position g and could cause the coiled-coil radius to increase towards the N-terminal as observed. The neighboring Leu 7 (g) may stabilize Leu 8 (a) in the ‘out’ conformation, as none of the other hydrophobic residues in a or d positions (Val or Leu) show more than one rotamer conformation in the X-ray structure. For example, Leu 25 (d) and Leu 26 (e) adopt a stable packing conformation, where the side chains at position d are densely packed in the hydrophobic core and also make close contact with the side chains at position e (Fig. 5, e▶).
If a hydrophobic residue such as leucine at positions e and g contributes the same amount of stability to a two-stranded coiled-coil as an ion pair, should we expect to see the same statistical occurrence of hydrophobic residues as charged amino acids in these positions? The database of two- and three-stranded coiled-coil proteins from GenBank showed that at positions e and g, charged residues were heavily favored over hydrophobic residues by over a factor of four: 51.7% of residues at positions e and g were charged, whereas only 11.8% were hydrophobic (Lupas et al. 1991). What can explain the preference for charged residues over hydrophobes at these positions? First, having hydrophobes at positions e and g reduces protein solubility in an aqueous environment. Second, hydrophobes at positions e and g have been shown to affect oligomerization state, allowing higher orders of self-association (trimers and tetramers; Harbury et al. 1993; Potekhin et al. 1994). So, if one decides to incorporate a relatively small number of hydrophobes in the e and g positions of de novo designed coiled-coils, these residues can increase overall stability but possibly at the cost of reducing solubility and decreasing specificity for the dimeric state. Nevertheless, despite the relatively low occurrence of leucine at positions e and g in two-stranded coiled-coils, it would not be possible to predict overall protein stability or the variations of stability along the sequence of coiled-coils without understanding the quantitative contributions of the leucine residues at positions e and g.
Conclusions
The crystal structure of the GCN4/cortexillin I peptide (Hybrid 2) contains key details helping to explain its stability. In addition to the hydrophobic residues found in the core a and d positions are the alanine residues in the f positions, the leucine residues in the e and g positions of the hydrophobic interface, the interchain ionic Glu-Lys‘ salt bridges on both sides of the two central Asn 15 core residues, and the complex hydrogen bonding network in the surrounding region. Although structurally similar to GCN4, Hybrid 2 possesses more ionic and hydrogen bonds in the vicinity of the central asparagines (especially at the Asn in the ‘out’ conformation), and has a larger coiled-coil radius near the N-terminal due to the alternate packing conformers of leucine in an a position. Biophysical studies have shown the importance of Glu 14 and Lys 19 in establishing g-e‘ ionic interactions, as well as the contributions of Leu 26 and Leu 7 at positions e and g to coiled-coil stability. To our knowledge, this work is the first reported biophysical quantification of the stability contributions of d-e‘ or g-a‘ leucine packing in a two-stranded coiled-coil, as well as the first identification of a unique network of hydrogen bonds involved in the ion pairs from g-e‘ and the Asn residues in the hydrophobic core due to the reversed ion pairing compared to GCN4-p1. The results provide further information to assist in the de novo design of coiled-coils (Burkhard et al. 2000a, 2002) and understanding the folding and stability of coiled-coils, in ways that do not necessarily require the inclusion of a consensus trigger sequence.
Materials and methods
Peptide synthesis, purification, and analysis
Peptides were synthesized by conventional Boc solid-phase peptide synthesis methodology using methylbenzhydrylamine resin, as described (Litowski et al. 1999). Nαtert-butyloxycarbonyl amino acid side-chain protecting groups were Asn(β-xanthyl), Asp(β-cyclohexyl), Glu(O-benzyl), Ser(O-benzyl), and Lys(Nɛ-2-Cl-benzyloxycarbonyl). After completion of synthesis, resin was neutralized with 10:90 (v/v) diisopropylethylamine: N, N-dimethylformamide, and N-terminal amino groups were acetylated with 25:75 (v/v) acetic anhydride: dichloromethane. The peptides were cleaved from the resin using anhydrous liquid HF (20 mL/g resin) containing 10% (v/v) anisole and 1% (v/v) 1,2-ethanedithiol for 1 h at −4°C, extracted with 0.05% trifluoroacetic acid (v/v) in 50% aqueous acetonitrile, and purified by reversed-phase HPLC (Wagschal et al. 1999a). Peptide purity was verified by mass spectrometry, analytical reversed-phase HPLC, and amino acid analysis (Litowski et al. 1999).
Circular dichroism spectroscopy and equilibrium unfolding measurements
CD measurements were obtained on a JASCO J-810 Spectropolarimeter at 20°C using Spectra Manager software, Version 1.10.00 running on a Pentium III under Microsoft Windows 2000. Data were collected at 0.1-nm intervals at 20° C from 190 to 250 nm for wavelength scans, with the average of five scans reported. The CD buffer was 50 mM potassium phosphate, 100 mM potassium chloride, pH 7. Ellipticity was reported as mean residue molar ellipticity ([θ]) in deg • cm2 • dmole−1 using the following equation:
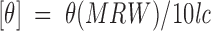 |
where θ is the observed ellipticity in degrees, MRW is the mean residue weight (molecular weight divided by number of residues), l is the optical path length of the cell in centimeters, and c is the peptide concentration in milligrams per milliliter.
Denaturation data were obtained by monitoring the ellipticity at 220 nm in 0.02 cm cells. Aliquots of ∼10 mg/mL peptide stock solution were diluted with the appropriate amount of CD buffer and 10 M urea solution in CD buffer to give final urea concentrations ranging from 0 to 8 M and ∼400 μM peptide concentration. For each peptide, ellipticity values were converted to fraction folded values, using the equation
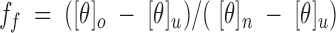 |
where ff is the fraction of the population folded, [θ]o is the observed molar ellipticity at a given urea concentration, [θ]u is the peptide molar ellipticity in the fully unfolded state, and [θ]n is the peptide molar ellipticity in the native (fully folded) state. For all peptides, the [θ]n value was assumed to be the same as [θ]o at 0 M, and the [θ]u value was taken to be −2400 deg cm2 dmole−1 at 8 M, obtained from peptide K19E, which appeared to approach a minimal [θ]u value at 8 M urea. Because peptide K19E was not fully folded at ∼400 μM peptide concentration in the absence of urea (Fig. 4▶), a value of −31,500 deg cm2 dmole−1 (taken from Hybrid 2 in benign, Table 2▶) was used for [θ]n in the calculation of the K19E fraction folded, to give a ff value less than 1.0 at 0 M urea (0.87, Fig. 9▶).
Thermodynamic analysis
For each urea denaturation curve, the midpoint of the unfolding curve ([urea]½), the slope of the linear portion of the transition (m), and the stability contribution of Hybrid 2 relative to the analog (ΔΔGuobs) were determined as described (Santoro and Bolen 1988; Serrano and Fersht 1989).
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed as described (Wagschal et al. 1999a), except that the concentrations used were 600, 150, and 50 μM, and rotor speeds were 42,000, 46,000, and 50,000 rpm.
Crystallization
Crystals of the peptide were grown in 24-well Falcon plates by vapor diffusion using the hanging-drop method (McPherson 1982). The 1-mL well solution contained 0.1–0.2 M ammonium acetate, 30% PEG4000, and 0.1 M sodium acetate buffer at pH 4.6. The 4-μL drop contained 2 μL peptide at a concentration of 20 mg/mL and 2 μL of well solution. Crystals grew within 2 d to dimensions up to 0.3 × 0.3 × 0.5 mm3. The crystals belong to the space group C2221 with unit cell dimensions a = 18.098 Å, b = 117.287 Å, and c = 22.209 Å and contain one monomer in the asymmetric unit (Table 1▶).
Data collection and processing
The X-ray diffraction data set from the peptide crystals was collected at 100 K on the X11 beam-line (EMBL, DESY Hamburg) at a wavelength of λ=0.8033 Å. The data set was processed and scaled using DENZO and SCALEPACK (Otwinowski and Minor 1997), respectively.
Model building and refinement
The structure was determined by molecular replacement methods using the program AMoRe (Navaza 1994). A polyalanine model of a 30-residue-long coiled-coil fragment based on the structure of the GCN4 coiled-coil (RSCB accession code 2zta) was used as a search model. The data set of the peptide was refined with the program SHELXL (Sheldrick and Schneider 1996) including anisotropic B-factor refinement to an R-factor of 17.9% and R-free calculated with 10% of the data set aside prior to refinement of 21.7%, at a resolution of 1.17 Å. R.m.s. deviations from ideality in bond lengths and angles are 0.009 Å and 1.8°, respectively (Table 1▶). The final model comprises one monomer, comprised of residues 3–31, and 32 water molecules in the asymmetric unit. The first two residues at the N-terminus appeared to be disordered in the crystal structure and displayed no interpretable electron density. The side chains of residues Leu 8, Ser 10, and Asn 15 were modeled in two alternative conformations. The overall quality of the model was good as judged by the low R values, the low deviations from stereochemical ideality, and the perfect appearance of the Ramachandran diagram with all residues in the most favored α-helical regions according to the program PROCHECK (Laskowsky et al. 1993). Graphical representations were performed using Insight II (Accelrys).
Calculation of solvent-accessible surface area
Models for L7A and L26A were built by modifying the PDB file for the Hybrid 2 peptide to substitute alanine residues for leucines. Accessible surface areas were calculated using GETAREA 1.1 (www.scsb.utmb.edu/getarea; Fraczkiewicz and Braun 1998).
Accession number
Coordinates have been deposited with the Research Collaboratory for Structural Bioinformatics under the accession code 1P9I.
Acknowledgments
We thank Leslie Hicks for sedimentation equilibrium experiments, Anthony Mehok for assistance with peptide synthesis, and Elsi Vacano for assistance with figures. We acknowledge the Natural Sciences and Engineering Research Council of Canada and the Alberta Heritage Foundation for Medical Research for providing scholarships to D.L.; NIH grant (RO1GM 61855); the John M. Stewart Fund for Peptide Research, and funds from the University of Colorado Health Sciences Center for R.S.H., and the Swiss National Science Foundation and the Maurice E. Müller Foundation of Switzerland for P.B.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
HPLC, high-performance liquid chromatography
TFE, 2, 2, 2-trifluoroethanol
PEG, polyethylene glycol
rmsd, root-mean-square deviation
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0241403.
References
- Acharya, A., Ruvinov, S.B., Gal, J., Moll, J.R., and Vinson, C. 2002. A heterodimerizing leucine zipper coiled coil system for examining the specificity of a position interactions: Amino acids I, V, L, N, A, and K. Biochemistry 41 14122–14131. [DOI] [PubMed] [Google Scholar]
- Akey, D.L., Malashkevich, V.N., and Kim, P.S. 2001. Buried polar residues in coiled-coil interfaces. Biochemistry 40 6352–6360. [DOI] [PubMed] [Google Scholar]
- Arndt, K.M., Pelletier, J.N., Muller, K.M., Pluckthun, A., and Alber, T. 2002. Comparison of in vivo selection and rational design of heterodimeric coiled coils. Structure 10 1235–1248. [DOI] [PubMed] [Google Scholar]
- Berger, B. and Singh, M. 1997. An iterative method for improved protein structural motif recognition. J. Comput. Biol. 4 261–273. [DOI] [PubMed] [Google Scholar]
- Berger, B., Wilson, D.B., Wolf, E., Tonchev, T., Milla, M., and Kim, P.S. 1995. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. 92 8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaber, M., Zhang, X.J., and Matthews, B.W. 1993. Structural basis of amino acid α helix propensity. Science 260 1637–1640. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Meier, M., and Lustig, A. 2000a. Design of a minimal protein oligomerization domain by a structural approach. Protein Sci. 9 2294–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhard, P., Kammerer, R.A., Steinmetz, M.O., Bourenkov, G.P., and Aebi, U. 2000b. The coiled-coil trigger site of the rod domain of cortexillin I unveils a distinct network of interhelical and intrahelical salt bridges. Structure Fold. Des. 8 223–230. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Stetefeld, J., and Strelkov, S.V. 2001. Coiled coils: A highly versatile protein folding motif. Trends Cell Biol. 11 82–88. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Ivaninskii, S., and Lustig, A. 2002. Improving coiled-coil stability by optimizing ionic interactions. J. Mol. Biol. 318 901–910. [DOI] [PubMed] [Google Scholar]
- Campbell, K.M., Sholders, A.J., and Lumb, K.J. 2002. Contribution of buried lysine residues to the oligomerization specificity and stability of the fos coiled coil. Biochemistry 41 4866–4871. [DOI] [PubMed] [Google Scholar]
- Crick, F.H.C. 1953. The packing of α-helices: Simple coiled-coils. Acta Crystallog. 6 689–697. [Google Scholar]
- Dieckmann, G.R., McRorie, D.K., Lear, J.D., Sharp, K.A., DeGrado, W.F., and Pecoraro, V.L. 1998. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J. Mol. Biol. 280 897–912. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz, R. and Braun, W. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 19 319–333. [Google Scholar]
- Frank, S., Lustig, A., Schulthess, T., Engel, J., and Kammerer, R.A. 2000. A distinct seven-residue trigger sequence is indispensable for proper coiled-coil formation of the human macrophage scavenger receptor oligomerization domain. J. Biol. Chem. 275 11672–11677. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Zhang, T., Kim, P.S., and Alber, T. 1993. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262 1401–1407. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Plecs, J.J., Tidor, B., Alber, T., and Kim, P.S. 1998. High-resolution protein design with backbone freedom. Science 282 1462–1467. [DOI] [PubMed] [Google Scholar]
- Havranek, J.J. and Harbury, P.B. 2003. Automated design of specificity in molecular recognition. Nat. Struct. Biol. 10 45–52. [DOI] [PubMed] [Google Scholar]
- Hodges, R.S., Sodek, J., Smillie, L.B., and Jurasek, L. 1972. Tropomyosin: Amino acid sequence and coiled-coil structure. Cold Spring Harbor Symp. Quant. Biol. 37 299–310. [Google Scholar]
- Kammerer, R.A., Jaravine, V.A., Frank, S., Schulthess, T., Landwehr, R., Lustig, A., Garcia-Echeverria, C., Alexandrescu, A.T., Engel, J., and Steinmetz, M.O. 2001. An intrahelical salt bridge within the trigger site stabilizes the GCN4 leucine zipper. J. Biol. Chem. 276 13685–13688. [DOI] [PubMed] [Google Scholar]
- Kohn, W.D., Kay, C.M., and Hodges, R.S. 1995. Protein destabilization by electrostatic repulsions in the two-stranded α-helical coiled-coil/leucine zipper. Protein Sci. 4 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1998. Orientation, positional, additivity, and oligomerization-state effects of interhelical ion pairs in α-helical coiled-coils. J. Mol. Biol. 283 993–1012. [DOI] [PubMed] [Google Scholar]
- Kohn, W.D., Mant, C.T., and Hodges, R.S. 1997. α-helical protein assembly motifs. J. Biol. Chem. 272 2583–2586. [DOI] [PubMed] [Google Scholar]
- Krylov, D., Barchi, J., and Vinson, C. 1998. Inter-helical interactions in the leucine zipper coiled coil dimer: pH and salt dependence of coupling energy between charged amino acids. J. Mol. Biol. 279 959–972. [DOI] [PubMed] [Google Scholar]
- Laskowsky, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lau, S.Y., Taneja, A.K., and Hodges, R.S. 1984. Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded α-helical coiled-coils. J. Biol. Chem. 259 13253–13261. [PubMed] [Google Scholar]
- Lee, D.L., Lavigne, P., and Hodges, R.S. 2001. Are trigger sequences essential in the folding of two-stranded α-helical coiled-coils? J. Mol. Biol. 306 539–553. [DOI] [PubMed] [Google Scholar]
- Litowski, J.R. and Hodges, R.S. 2002. Designing heterodimeric two-stranded α-helical coiled-coils. Effects of hydrophobicity and α-helical propensity on protein folding, stability, and specificity. J. Biol. Chem. 277 37272–37279. [DOI] [PubMed] [Google Scholar]
- Litowski, J.R., Semchuk, P.D., Mant, C.T., and Hodges, R.S. 1999. Hydrophilic interaction/cation-exchange chromatography for the purification of synthetic peptides from closely related impurities: Serine side-chain acetylated peptides. J. Pept. Res. 54 1–11. [DOI] [PubMed] [Google Scholar]
- Lovell, S.C., Word, J.M., Richardson, J.S., and Richardson, D.C. 2000. The penultimate rotamer library. Proteins 40 389–408. [PubMed] [Google Scholar]
- Lupas, A. 1996. Coiled coils: New structures and new functions. Trends Biochem. Sci. 21 375–382. [PubMed] [Google Scholar]
- Lupas, A., Van Dyke, M., and Stock, J. 1991. Predicting coiled coils from protein sequences. Science 252 1162–1164. [DOI] [PubMed] [Google Scholar]
- McClain, D.L., Woods, H.L., and Oakley, M.G. 2001. Design and characterization of a heterodimeric coiled coil that forms exclusively with an antiparallel relative helix orientation. J. Am. Chem. Soc. 123 3151–3152. [DOI] [PubMed] [Google Scholar]
- McPherson, A. 1982. Preparation and analysis of protein crystals. Wiley, New York.
- Micklatcher, C. and Chmielewski, J. 1999. Helical peptide and protein design. Curr. Opin. Chem. Biol. 3 724–729. [DOI] [PubMed] [Google Scholar]
- Minor Jr., D.L. and Kim, P.S. 1994. Context is a major determinant of β-sheet propensity. Nature 371 264–267. [DOI] [PubMed] [Google Scholar]
- Monera, O.D., Sereda, T.J., Zhou, N.E., Kay, C.M., and Hodges, R.S. 1995. Relationship of sidechain hydrophobicity and α-helical propensity on the stability of the single-stranded amphipathic α-helix. J. Pept. Sci. 1 319–329. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1997. A direct comparison of helix propensity in proteins and peptides. Proc. Natl. Acad. Sci. 94 2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil, K.T. and DeGrado, W.F. 1990. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250 646–651. [DOI] [PubMed] [Google Scholar]
- O’Shea, E.K., Klemm, J.D., Kim, P.S., and Alber, T. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254 539–544. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. In Methods Enzymology (eds. C.W. Carter and R.M. Sweet), Vol. 277, pp. 307–326. Academic Press, Inc., New York. [DOI] [PubMed]
- Pace, C.N. and Scholtz, J.M. 1998. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya, M.J., Spooner, G.M., Sunde, M., Thorpe, J.R., Rodger, A., and Woolfson, D.N. 2000. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry 39 8728–8734. [DOI] [PubMed] [Google Scholar]
- Phelan, P., Gorfe, A.A., Jelesarov, I., Marti, D.N., Warwicker, J., and Bosshard, H.R. 2002. Salt bridges destabilize a leucine zipper designed for maximized ion pairing between helices. Biochemistry 41 2998–3008. [DOI] [PubMed] [Google Scholar]
- Potekhin, S.A., Medvedkin, V.N., Kashparov, I.A., and Venyaminov, S. 1994. Synthesis and properties of the peptide corresponding to the mutant form of the leucine zipper of the transcriptional activator GCN4 from yeast. Protein Eng. 7 1097–1101. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Serrano, L. and Fersht, A.R. 1989. Capping and α-helix stability. Nature 342 296–299. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G.M. and Schneider, T.R. 1996. SHELXL: High resolution refinement. In Methods enzymology (eds. C.W. Carter and R.M. Sweet), Vol. 276, pp. 319–343. Academic Press, Inc., New York. [PubMed]
- Singh, M., Berger, B., and Kim, P.S. 1999. LearnCoil-VMF: Computational evidence for coiled-coil-like motifs in many viral membrane-fusion proteins. J. Mol. Biol. 290 1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz, M.O., Stock, A., Schulthess, T., Landwehr, R., Lustig, A., Faix, J., Gerisch, G., Aebi, U., and Kammerer, R.A. 1998. A distinct 14 residue site triggers coiled-coil formation in cortexillin I. EMBO J. 17 1883–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelkov, S.V. and Burkhard, P. 2002. Analysis of α-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 137 54–64. [DOI] [PubMed] [Google Scholar]
- Tripet, B.T. and Hodges, R.S. 2001. STABLECOIL: An algorithm designed to predict the location and relative stability of coiled-coils in native protein sequences. In Peptides: The wave of the future, Proceedings of the Second International/Seventeenth American Peptide Symposium (eds. M. Lebl and R.A. Houghten), pp. 365–366. Kluwer Academic Publishers, San Diego, CA.
- Tripet, B., Wagschal, K., Lavigne, P., Mant, C.T., and Hodges, R.S. 2000. Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil: 20 amino acid substitutions in position "d". J. Mol. Biol. 300 377–402. [DOI] [PubMed] [Google Scholar]
- Wagschal, K., Tripet, B., Lavigne, P., Mant, C., and Hodges, R.S. 1999a. The role of position a in determining the stability and oligomerization state of α-helical coiled coils: 20 amino acid stability coefficients in the hydrophobic core of proteins. Protein Sci. 8 2312–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagschal, K., Tripet, B., and Hodges, R.S. 1999b. De novo design of a model peptide sequence to examine the effects of single amino acid substitutions in the hydrophobic core on both stability and oligomerization state of coiled-coils. J. Mol. Biol. 285 785–803. [DOI] [PubMed] [Google Scholar]
- Wolf, E., Kim, P.S., and Berger, B. 1997. MultiCoil: A program for predicting two- and three-stranded coiled coils. Protein Sci. 6 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, K.C., Bryan, J.T., Morasso, M.I., Jang, S.I., Lee, J.H., Yang, J.M., Marekov, L.N., Parry, D.A., and Steinert, P.M. 2000. Coiled-coil trigger motifs in the 1B and 2B rod domain segments are required for the stability of keratin intermediate filaments. Mol. Biol. Cell 11 3539–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, N.E., Kay, C.M., and Hodges, R.S. 1994. The net energetic contribution of interhelical electrostatic attractions to coiled-coil stability. Protein Eng. 7 1365–1372. [DOI] [PubMed] [Google Scholar]
- Zhu, H., Celinski, S.A., Scholtz, J.M., and Hu, J.C. 2000. The contribution of buried polar groups to the conformational stability of the GCN4 coiled coil. J. Mol. Biol. 300 1377–1387. [DOI] [PubMed] [Google Scholar]