

Abstract
NMR studies of the binding of a substrate to an inactive HIV-1 protease construct, containing an active site mutation PRD25N, are reported. Substrate titration measurements monitored by HSQC spectra and a 15N-edited NOESY experiment show that the chromogenic substrate analog of the capsid/p2 cleavage site binds to PRD25N with an equilibrium dissociation constant, KD, of 0.27 ± 0.05 mM, and upper limits of the association and dissociation rate constants, 2×104 M−1s−1 and 10 s−1, respectively, at 20°C, pH 5.8. This association rate constant is not in the diffusion limit, suggesting that association is controlled by a rare event, such as opening of the protease flaps. Analysis of 15N relaxation experiments reveals a slight reduction of S2 values in the flap region, indicating a small increase in the amplitude of internal motion on the sub-nsec timescale. In addition, several residues in the flap region are mobile on the conformational exchange timescale, msec–μsec. Flap dynamics of the protease-substrate complex are compared with those of protease-inhibitor complexes, and the implications of these results for substrate-binding models are discussed.
Keywords: Aspartic protease, enzyme, protein, AIDS, relaxation, slow exchange
Active human immunodeficiency virus-1 (HIV-1) protease is a homodimer composed of monomer subunits, each containing 99 amino acids with a single catalytic Asp residue. Because HIV-1 protease processes viral polyproteins to yield mature proteins required in the viral life cycle (Oroszlan and Luftig 1990), it has been a prime target of drugs directed against AIDS. Hundreds of crystal structures of protease-inhibitor complexes have been solved and have contributed to the development of drugs that bind to the active site (Erickson and Burt 1996; Vondrasek et al. 1997). Currently, at least six potent antiprotease drugs are in clinical use. Unfortunately, the long-term effectiveness of these drugs has been limited by the emergence of drug-resistant mutants (Condra 1998). Drug-resistant mutants of HIV protease have reduced sensitivity to specific inhibitors, but maintain sufficient enzymatic activity to accomplish polyprotein maturation. To obtain better understanding of the molecular mechanism of drug resistance, further structural characterizations of protease-ligand complexes are required, especially those involving substrates.
The Protein Data Bank (PDB) contains a wealth of structural information on the protease-inhibitor complexes. In contrast, structural studies of the protease bound to natural substrates have been limited, due to their rapid hydrolysis. For this reason, nonpeptide analogs are typically employed as substrate mimetics. These analogs generally exhibit higher binding affinities to the protease than natural substrates, suggesting that certain aspects of binding may be altered. Indeed, calorimetric studies have indicated different binding energetics for substrate analogs and small inhibitors (Luque et al. 1998; Todd and Freire 1999), emphasizing the need for structural studies of substrate binding. X-ray structures of protease-substrate complexes recently became available with the use of a protease construct that contained a single inactivating mutation of the active site (Prabu-Jeyabalan et al. 2000, 2002).
Nuclear magnetic resonance (NMR) spectroscopy provides information about protein structure and dynamics that complements the high-resolution structural information derived from crystallography. One unique feature of NMR is the ability to observe weakly binding systems in solution, free from potential perturbations due to the crystal lattice. In addition, information about the kinetics of binding and the difference in internal dynamics of the free and substrate-bound protein can be obtained.
Here we investigated the binding of an active protease (PR) and an inactive protease (PRD25N) to a highly soluble chromogenic peptide substrate (substrate C) in solution. We found that many amide signals are missing from the 15N-1H HSQC spectrum of PR in the presence of the substrate, presumably as a result of exchange broadening. We also found that high-quality spectra can be obtained using PRD25N in the presence of the substrate, and we obtained sequential backbone signal assignments of PRD25N, both free and bound to the substrate. These assignments together with substrate titration experiments were used to measure binding kinetics and the equilibrium dissociation constant of the PRD25N/substrate complex. The results are compared with complementary fluorescence studies of active protease-inhibitor complexes (Furfine et al. 1992; Rodriguez et al. 1993). Next, we characterized the internal dynamics of the substrate-bound PRD25N using 15N relaxation measurements. A question addressed here is whether flap dynamics relates to binding affinity. Finally, the information about binding kinetics and internal dynamics is used to discuss the nature of the binding of the protease to a substrate at the molecular level.
Results and Discussion
Addition of substrate strongly perturbs the spectrum of PR
The 15N-1H HSQC spectrum, Figure 1A▶, of free PR contains ∼90 well dispersed backbone amide signals, as expected for a symmetric homodimer. Although PR is fully active (Louis et al. 1999), its reduced autoproteolysis rate, particularly at pH 5.8, minimizes the appearance of signals arising from proteolytic fragments. Small cross-peaks in the random coil region of the spectrum, 8–8.5 ppm (1H) and 120–125 ppm (15N), can be noted, however. Assignments of PR are very similar to those reported for the free PR lacking the C67A/C95A mutations (Freedberg et al. 1998). For purposes of comparison with other spectra, selected signals of residues that are close to the substrate-binding site are labeled in Figure 1A▶.
Figure 1.

15N-1H HSQC spectra of (A) free active protease (PR) and (B) the active protease in the presence of 30-fold excess of substrate C. In (A), signals perturbed by the addition of substrate C are labeled. In (B), signals that do not show changes in chemical shifts upon addition of substrate C are labeled.
The spectrum recorded immediately after the addition of 6 mM substrate C to a 0.2 mM solution of PR (Fig. 1B▶) differs significantly from the spectrum of the free PR (Fig. 1A▶). The most pronounced effect of substrate addition is the disappearance of many of the signals outside the random coil region of the spectrum, presumably due to exchange broadening. Comparison of the two spectra (Fig. 1A,B) indicates that many of the perturbed signals arise from residues close to the substrate-binding region (Fig. 2▶). Signals labeled in Figure 1B▶ are those that are not perturbed by the addition of substrate, and are remote from the substrate-binding site, as shown in Figure 2▶.
Figure 2.

Ribbon drawing of the backbone of the protease, color-coded to depict the different responses of the HSQC signals upon addition of substrate C. The pink and blue regions of the ribbon identify residues whose chemical shifts were perturbed and unperturbed, respectively, by addition of substrate C. Others (cyan) are residues that could not be analyzed in crowded regions. Residues of the flap regions (43–58) and elbows (37–42) are indicated (Freedberg et al. 2002).
After the spectrum in Figure 1B▶ was recorded, the data were acquired two more times, without delay between the experiments. The two subsequently acquired spectra were essentially the same as the spectrum in Figure 1B▶. This observation suggests that the catalytic reaction was complete at an early stage in the acquisition of the spectrum in Figure 1B▶, and that the observed spectrum is therefore that of PR in the presence of hydrolysis products, rather than the intact substrate C. This notion is supported by the finding that products of the substrate bind to the protease (Rose et al. 1996). Thus, formation of PR/product complexes presumably has lifetimes on the msec timescale, resulting in severe broadening of PR signals for residues close to the substrate-binding site, accounting for their absence in Figure 1B▶.
Substrate binding to the inactive protease
Because rapid cleavage precludes the observation of spectra of active PR bound to the intact substrate, we investigated the binding of substrate C to inactive PR, PRD25N. Comparison of Figures 1A and 3A▶▶ shows that free PR and PRD25N exhibit very similar HSQC spectra, except for signals that arise due to autoproteolysis when using the active PR. Although the dimer dissociation constant of PRD25N is lower than that of PR, the monomer fraction was not significant under the solution conditions used for the NMR experiments. Essentially identical backbone 1H, 13C, and 15N chemical shifts, except for those of residue 25 and adjacent amino acids, are observed. In addition, the crystal structure of the PRD25N/CA-p2 complex is very similar to the crystal structure of wild-type PR bound to peptide substrate analogs (Prabu-Jeyabalan et al. 2000), supporting our current approach of using PRD25N for NMR studies of substrate binding to the protease.
Figure 3.

15N-1H HSQC spectra of (A) free PRD25N and (B) the PRD25N bound to substrate C. In the spectra, peaks are marked when the two signals at the identical amino-acid sequence position in two monomer subunits (indicated by a solid line in one subunit and a broken line in the other subunit) have significantly different chemical shifts in the bound form.
Figure 3B▶ shows well dispersed signals, including a large number of signals that were not observed when using the active PR in Figure 1B▶. As expected, binding of PRD25N to the asymmetric substrate eliminated the chemical shift degeneracy observed in a symmetric dimer, particularly of those residues close to the substrate-binding site. Despite the increased complexity of the spectrum, nearly complete backbone signal assignments were obtained, because only a single protease species was present (PRD25N bound to substrate C). Note that exchange broadening was not significant.
Distinctly different monomer chemical shifts were observed for four regions of the protease (Fig. 4▶). Residues in three of these regions—21–35, 42–59, and 76–91—interact with the substrate in the crystal structure of the PRD25N/CA-p2 substrate complex, whereas residues in the fourth region, 9–13, are adjacent to active site residues 21–23 (Prabu-Jeyabalan et al. 2000). Note that the PR residues whose amide signals were severely exchange-broadened in the presence of substrate C exhibit nondegenerate amide chemical shifts when PRD25N is bound to substrate C (Fig. 4▶, filled circles). This is consistent with the notion that line broadening for residues near the active site (Fig. 1B▶) arises from exchange between free and ligand-bound PR.
Figure 4.

Schematic diagram of the chemical shift perturbations of the PRD25N upon substrate C binding. Vertical lines indicate residues whose amide 15N and 1H chemical shifts differ in the two monomer subunits but whose 13Cα chemical shifts are indistinguishable. Open rectangles indicate residues in regions (I–IV) whose 15N, 1H, and 13Cα chemical shifts are differently assigned in each monomer subunit. Black dots indicate residues whose signals are perturbed in the active protease upon addition of substrate C (the residues described in Fig. 1A▶).
Substrate-binding kinetics of PRD25N
Knowing the signal assignments of both free and substrate-bound PRD25N permitted following the transition from the free to the substrate-bound form of PRD25N. This transition was monitored by recording a series of HSQC spectra as a function of increasing substrate concentration. Figure 5▶ displays selected regions of the spectra for residues L10 and L38, clearly illustrating the disappearance of the free form and appearance of the bound form as the substrate concentration increases. In contrast to the signal intensities, the line widths and chemical shifts of the signals were essentially independent of substrate concentration, as expected for the slow chemical exchange regime.
Figure 5.

Titration of substrate C to PRD25N. Portions of HSQC spectra are shown for the different substrate C-to-protein concentration ratios (A) 0:1, (B) 0.5:1, (C) 1:1, (D) 2:1, (E) 4:1, and (F) 8:1 (protein concentration, 0.3mM; subscripts indicate bound and free forms).
Assuming that substrate C binding to PRD25N is a reversible single-step transition, the dissociation constant, KD, is given by
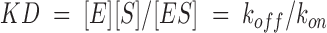 |
(1) |
Here, [E], [S], and [ES] are the respective concentrations of free PRD25N, free substrate C, and the protease-substrate complex, and koff and kon are the respective dissociation and association rate constants. [E]/[ES] ratios were determined as a function of [S] from free and bound peak intensities, yielding a KD of 0.27 ± 0.05mM. This is consistent with a previously reported Km value of 0.177 ± 0.018 mM for PR-catalyzed hydrolysis of substrate C in 100mM sodium acetate buffer, pH 5.0, at 25°C, given the differences in temperature and salt concentration (Louis et al. 1999).
For slow exchange, the difference in chemical shifts between free (E) and bound form (ES) peaks of the protease, and Δf, satisfies the condition
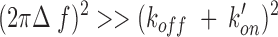 |
(2) |
where kon′ = kon[S] is a pseudo-first-order rate constant. The broadening of the peaks of the free and bound form of PRD25N are related to the rate constants according to
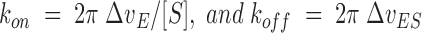 |
(3) |
respectively. Here, ΔνE = νE − νE° and ΔνES = νES − νES° where νE and νE° are the line widths of peaks of the free PRD25N in the presence and absence of substrate, respectively, whereas νES and νES° are the line widths of the peaks in the complex in the presence of intermediate and saturating levels of substrate, respectively. Comparing the line widths of the peaks throughout the titration, it was found that ΔνE and ΔνES were less than 1.5 Hz. From this result together with Equation 3, limiting values for kon and koff are found to be <2×104 M−1s−1 and <10 s−1, respectively.
An independent estimate of koff was derived from intensities of amide-amide exchange cross-peaks observed in a 3D 15N-edited NOESY experiment recorded with a mixing time of 80 msec. As has been noted in our discussion of Figure 3B▶, many amide residues, located at amino acid sequence positions i and i + 100 in the PRD25N/substrate C complex, exhibit distinct chemical shifts. Cross-peaks between the nondegenerate signals in the PRD25N/substrate C complex are observed in the NOESY spectrum. Because nearly all amide proton pairs connected by such cross-peaks are separated by distances much larger than 5 Å in the crystal structure of the PRD25N/CA-p2 substrate complex (Prabu-Jeyabalan et al. 2000), the cross-peaks are not due to cross-relaxation. Rather they result from an interchange of monomer chemical shifts caused by an exchange of the substrate orientation relative to the two monomers. Similar exchange cross-peaks have been observed for the protease bound to a potent asymmetric inhibitor, KNI529 (KD ∼1nM; Katoh et al. 1999). Assuming that the substrate dissociates and rebinds to the protease, either in its original or in a flipped orientation with equal probability, koff = 2k, where k is the exchange rate measured by the NOESY experiment (Katoh et al. 1999). Using a modified approximate two-site exchange equation (Katoh et al. 1999) and the ratios of diagonal and exchange cross-peak intensities observed in a NOESY spectrum (observed to be in the range 0.2–0.4), we calculate k in the range 2–10 s−1. The large uncertainty of the k is partly due to the difficulty of precisely correcting the two-site exchange equation for the effect of proton-proton dipolar NOEs. As we describe later, chemical exchange contributes significantly to transverse relaxation of residues in the flap region. Because this chemical exchange process did not produce cross-peaks in the NOESY spectrum, it did not affect the above estimation of koff, except by reducing the accuracy of intensity measurements due to exchange broadening of the flap signals.
The 15N zz exchange experiment (Farrow et al. 1994) was used to obtain an independent estimate of koff. Curve fitting the intensity of diagonal and cross-peak signals observed in the zz experiment yielded koff values in the range of 1–3 s−1. Therefore, the koff values estimated from the NOESY and 15N zz exchange experiments are in reasonable agreement with koff obtained from the line broadening observed in the titration experiments. This result implies that this substrate dissociates from PR when it reorients, otherwise koff estimated from the NOESY experiment (or zz-exchange) and line broadening would differ. In contrast, it was found that the inhibitor, KNI529, a small, more tightly bound ligand, reorients without dissociation from PR (Katoh et al. 1999).
It is interesting to compare values of kon and koff determined herein with rates obtained from fluorescence measurements (Furfine et al. 1992; Rodriguez et al. 1993). The fluorescence studies followed the binding of peptide inhibitors to the active protease, and therefore complement our investigation, which examined substrate binding to an inactive structural homolog of the active protease. In the fluorescence studies, kon and koff range from 0.1×106 to 3.0×106 M−1s−1 and from 0.02 to 0.4 s−1, respectively. These association and dissociation rate constants are 10–100-fold larger and 10–100-fold smaller, respectively, than observed here for the PRD25N/substrate C complex. This could be due to the higher temperature used in the fluorescence studies and to the fact that the NMR studies required protein and substrate concentrations that were nearly two orders of magnitude larger than in the fluorescence measurements. At these high concentrations, deviations from the law of mass action, upon which Equation 1 is based, may cause the value of kon determined by NMR to be underestimated. Nevertheless, the values of kon determined by both techniques are 3–5 orders of magnitude smaller than that expected if binding were diffusion-controlled (Johnson 1992; Fersht 1999). This observation suggests that a fast, diffusion-controlled collision of the protease and ligand is followed by a much slower structural change resulting in the final bound conformation of the complex, as has been discussed before (Furfine et al. 1992; Rodriguez et al. 1993).
Protein dynamics of PRD25N bound to substrate C
The crystal structure of PRD25N bound to the CA-p2 peptide substrate is very similar to those of wild-type protease bound to peptidomimetic inhibitors (Prabu-Jeyabalan et al. 2000). Our NMR results, chemical shifts, and NOESY cross-peaks also confirm this observation for the PRD25N bound to substrate C in solution. However, because the binding affinities of substrates are much lower than those inhibitors, the internal dynamics of the protease when bound to substrate are not necessarily similar to those bound to inhibitors. We therefore used the model-free approach to extract dynamics information from 15N spin relaxation measurements of PRD25N bound to substrate C. The amides of most residues in the substrate-bound PRD25N exhibit uniform relaxation values (T2∼70ms and T1∼0.8s) and NOEs larger than 0.7 (Fig. 6▶), indicative of a well ordered protein backbone. Relaxation data together with the orientations of amide NH bond vectors, derived from the crystal structure of PRD25N bound to the CA-p2 substrate (Prabu-Jeyabalan et al. 2000), were used to determine the average rotational correlation time, θc, and anisotropy, D∥/D⊥, of overall motion as described previously (Freedberg et al. 2002). The values obtained, θc = 12 ns and D∥/D⊥ = 1.4, are in good agreement with results obtained for the protease bound to inhibitors DMP323 and KNI272 (Tjandra et al. 1996; Freedberg et al. 1998).
Figure 6.

Comparison of backbone amide 15N (A) T2, (B) T1, and (C) NOE values and model-free parameters (D) S2 and (E) Rex values with residue number of PRD25N bound to substrate C. The open rectangles at the top of the figure indicate regions that exhibit different 1H/15N/13Cα backbone chemical shifts in each subunit. For these regions, the data are shown in two sets; for one, residues are numbered from 1 to 99, and for the other, residues are numbered from 101 to 199. Residues whose amide correlations were resolved, but whose 13Cα chemical shifts were indistinguishable in the two subunits yielded two relaxation data sets, both of which are plotted versus residues numbered 1–99. However, almost identical T2, T1, and NOE values were measured in each monomer for this set of residues, and therefore their relaxation rates are not distinguishable in the figure. Parameters for residues having degenerate amide chemical shifts in the two monomers are plotted versus residue numbers 1–99. For comparison, S2 values of the flap region for the free PRD25N, measured using the same experimental conditions, are shown in (D), by open circles.
As anticipated from inspection of the relaxation data in Figure 4▶, the model-free analysis yielded large values of S2 (>0.7) for nearly all sites in the bound protease, indicative of restricted backbone motion on the sub-nsec timescale. The only exceptions are residues in the flap elbows (residues 37–42/137–142). Internal mobility on the sub-nsec timescale for these residues has been reported previously in the protease bound to inhibitors (Nicholson et al. 1995; Freedberg et al. 1998). It is interesting that S2 values of residues 52, 53, 152, and 153 are slightly smaller (by ∼0.1) in the case of the protease bound to substrate C than for the protease bound to inhibitors (∼0.8∼0.95 in the previous study; Nicholson et al. 1995; Freedberg et al. 1998; R. Ishima, J.M. Louis, and D.A. Torchia, unpublished data measured at 20°C for the protease bound to DMP323). This observation suggests that flap motion on the sub-nsec timescale is not completely quenched by substrate C binding. However, the S2 values of these residues in the substrate C-protease complex are large (∼0.76) compared to those observed for residues in the flaps in the free protease (Freedberg et al. 2002), indicating that the flaps of the substrate-protease complex are in a closed conformation similar to that observed in inhibitor-protease complexes. Previous fluorescence studies of the quenching of sub-nsec timescale flap motions (Furfine et al. 1992; Rodriguez et al. 1993) and theoretical calculations (Collins et al. 1995; Rick et al. 1998) also indicate that the flaps close upon substrate binding. However, the KD of the substrate C-protease complex (0.27 ± 0.05mM) is at least 10−8-fold weaker than the KD of the complexes of the strongest inhibitors. The small-amplitude sub-nsec motion observed for residues 52 and 53 could be interpreted to relate to lower binding affinity of this substrate to the protease.
In contrast to this small-amplitude (based on a simple cone model) motion on the sub-nsec timescale for residues 52 and 53, the flaps of substrate-bound PRD25N exhibit significant chemical exchange, especially for residues 47 and 48. These residues have significantly smaller T2 values than residues 147 and 148 in the other flap tip (Fig. 6▶). In addition, residues 50, 51, and 151 have very weak signal intensity because of signal broadening. Therefore, although poor signal-to-noise precluded measuring the T2 values of these residues, their broad signals showed that their T2 values were very short. These results are evidence that both flaps undergo motion on the msec–μsec timescale, but that flexibility on this timescale is considerably more extensive in one flap than the other, suggesting that an asymmetric ligand packs more tightly with one flap than the other. Although the crystal structure of the substrate-bound form of PRD25N shows two conformations (having equal probability) for residues 50 and 51, and for 150 and 151, a single conformation is observed for residues 47 and 48, and for residues 147 and 148 (Prabu-Jeyabalan et al. 2000). Nevertheless, asymmetric contacts of residues 47 and 48 (and for 147 and 148) with the substrate P1–P4 (and P1′–P4′) positions were revealed by the crystal structure, consistent with the observation of asymmetric flap dynamics in solution.
To better understand substrate binding, the flap dynamics on msec–μsec motion of the substrate-protease complex was compared with dynamics of inhibitor-protease complexes. It is evident in Table 1▶ that the slow dynamics at the tip of each flap, residues 50 and 51 (also 150 and 151 in the protease bound to an asymmetric ligand) is common to all PR complexes. Although a significant chemical exchange contribution was not extracted by the model-free analysis of the relaxation data of residue I 150 in the PRD25N/substrate C complex, the I 150 signal was weak, presumably due to line-broadening in the proton dimension caused by chemical exchange. Previously, we suggested that chemical exchange at these sites 50/51 is due to interconverting β-I/β-II-type turns at the flap tips (Nicholson et al. 1995). The fact that the flap tip motion is common in ligand-protease complexes suggests that this motion does not interfere with flap-ligand interactions. Therefore, the dynamics of the flap tips is expected to result in favorable binding entropy without affecting enthalpy.
Table 1.
Flap residues that undergo slow dynamics on msec-μsec time-scale in protease ligand complexes detected using 15N relaxation data
| PR | Inhibitor | KDa | Residue numbers | |
|---|---|---|---|---|
| PRb | DMP325c | 10−9 M | 50, 51 | 50, 51 |
| PRb | P9941c | 10−8 M | 50, 51 | 50, 51 |
| PRb | KNI272d | 10−11 M | 48–51 | 148–153 |
| PRD25N | substrate C | 10−4 M | 46–48, 50, 51, 54e | 149, 151e |
a Ligand dissociation constant.
b The PR used in these studies did not contain the Q7K/L33I/L63I mutations.
e Relaxation data were not measured for residues 50, 51, and 151 because of severe signal broadening, presumably due to conformational exchange on the msec-μsec time scale.
Interestingly, in the case of the protease bound to symmetric inhibitors DMP323 and P9941, the slow dynamics in the flaps is restricted to the tip residues (Nicholson et al. 1995; Tjandra et al. 1996), which is clearly different from the motion extending beyond the flap tips seen in the protease bound to substrate C (Table 1▶). At first sight, it may be tempting to ascribe the reduced flap motion to the lower dissociation constants (less than 10−8 M) of the symmetric inhibitors. However, in protease bound to KNI272, a peptide-derived asymmetric inhibitor with a KD of ∼10−11 M, msec–μsec motion was detected for residues 48–51 and 148–153 (Freedberg et al. 1998). Thus, the results compiled in Table 1▶ demonstrate that slow flap dynamics is different between the peptide-type asymmetric ligands (substrate C and KNI272) and small synthetic inhibitors (DMP323 and P9941). It is interesting that slow flap dynamics is not simply related to ligand binding affinity, but appears to be very sensitive to the chemical structure of the ligand.
Recent calorimetric studies have shown that entropy and enthalpy contributions to ligand-binding energetics of the protease are strongly ligand-dependent (Luque et al. 1998; Velazquez-Campoy et al. 2001; Ohtaka et al. 2002). The reported variation of ligand-dependent energetics of the flap-ligand interaction parallels our observation of conformational exchange for residues beyond the flap tips when the protease is bound to peptide-type ligands but not when bound to small inhibitors. The observation of motion beyond the flap tips in complexes with peptide-type ligands (be they either a substrate C, KD = 0.27 ± 0.05 mM, or a peptide-type inhibitor, KNI272, KD = 10−11 M) shows that flap residues do not interact with peptide-type ligands in a unique manner, suggesting that they make minor contributions to binding specificity. On the other hand, sub-nsec motion in the flap region was observed only when the protease was bound to substrate C and was not detected in protease-inhibitor complexes, suggesting a relationship between sub-nsec flap dynamics and weak binding affinity. Nevertheless, the entropy gain in the substrate C-bound form compared to inhibitor-bound forms is estimated, <3 cal/mole K from a comparison of their S2 values (Yang and Kay 1996). Even if a tenfold larger contribution is attributed to the side chains in the substrate-bound complex, the entropy gain is apparently too small to contribute significantly to ligand-binding specificity. This conclusion is in agreement with reports that mutations of flap residues have little impact on protease specificity (Shao et al. 1997; Tozser et al. 1997), and may help explain how the protease can bind to nine different natural peptide substrates.
Flap motion and substrate binding
Because structures observed in crystals may be affected by crystal packing, it is important to perform NMR experiments in solution to better understand substrate binding to the protease at the molecular level. When the active site is occupied, crystal structures reveal that the flaps are closed and interact with the ligand, whereas, for free protease, the flaps are flexible on the sub-nsec timescale as revealed by NMR (Ishima et al. 1999; Freedberg et al. 2002). These observations have been supported by fluorescence measurements (Furfine et al. 1992; Rodriguez et al. 1993). In the present study, only a small-amplitude sub-nsec motion was detected in the flaps in the substrate C-protease complex, indicating that the flaps are closed when the protease binds to a substrate having low affinity, consistent with the results noted above.
It has long been recognized that the flexibility of the protease flaps must play a role in inhibitor/substrate binding. This is because of the early observation (Wlodawer and Erickson 1993) that the flaps undergo a large conformational change between free and ligand-bound protease forms. Furthermore, based on the model that semi-open form is the major conformation in solution, a further opening intermediate is expected to occur for substrate binding, because even the semi-open conformations block access to the active site (Collins et al. 1995). An alternative model for substrate binding was proposed based on an analysis of a molecular dynamics simulation and of the effect of lattice contacts on flap conformation observed in protease crystal structures (Scott and Schiffer 2000). In this model, the open conformation that allows access of the substrate to the active site was proposed as the predominant flap conformation in solution. Although the relative population of open conformations in the free state of the protease differs in these two models, both allow for a binding process in which diffusion of the ligand to the binding site of the protease is followed by a slow conformational change which involves closing of the flaps with the concomitant correct positioning of the ligand in the active site.
A mutagenesis study showed that substitution of solvent-exposed hydrophobic residues Met 46 and Phe 53 by hydrophilic residues resulted in reduction of enzymatic activity (Shao et al. 1997). If the major conformation in solution is assumed to be a semi-open form that must open for substrate binding, that study (Shao et al. 1997) suggests that interaction of the exposed hydrophobic side chains of Met 46 and Phe 53 with hydrophobic sites on the substrate triggers flap opening and enhances substrate binding to the protease. Although drug-resistant mutations of Met 46 and Phe 53 are common, they are highly conservative, and large hydrophobic residues are found at positions 46 and 53 in all drug-resistant mutant proteases (Parikh et al. 2000; Shafer et al. 2000).
Materials and methods
Proteins
The active HIV-1 protease construct, termed PR, contains five mutations compared to the wild-type protease: Q7K, L33I, L63I to suppress autoproteolysis, and C67A and C95A to prevent aggregation caused by formation of interprotein disulfide bonds (Louis et al. 1999). The enzymatic activity of PR is indistinguishable from that of wild type (Louis et al. 1999). A second construct, PRD25N, contains an additional mutation, D25N. This mutant permits the NMR studies of a substrate-bound form of the enzyme, because it is inactive and maintains a structure similar to that of PR. Both constructs were expressed in E. coli using minimal media containing either 15NH4Cl and 13C6-glucose for a 13C/15N-labeled sample, or 15NH4Cl alone for a 15N-labeled sample. Protein expression, purification, and folding were performed as described (Ishima et al. 1999; Louis et al. 1999). Purified, unfolded monomeric protein in 30mM formic acid, pH 2.8, was folded by rapid dilution into threefold excess of 10 mM acetate buffer, pH 6.0. The pH of this solution was changed to pH 5.8 by dialysis against 20 mM sodium phosphate at 4°C. Samples for NMR experiments were concentrated using centriprep/centricon centrifugal devices (Millipore) to yield 0.2–0.3 mM protein (as a dimer).
Substrate C (H-Lys-Ala-Arg-Val-Nle-(4-nitrophenylalanine)-Glu-Ala-Nle-NH2) was purchased from California Peptide Research. This substrate is a chromogenic analog of the CA-p2substrate (H-Lys-Ala-Arg-Val-Leu-Ala-Glu-Ala-Met-Ser-NH2), the capsid/p2 cleavage site in the Gag polyprotein.
NMR experiments
Experiments were performed using Bruker DMX500 and DMX750 spectrometers equipped with 5-mm triple-resonance probe heads and three-axis gradient coils, and using a DMX500 spectrometer with a 5-mm triple-resonance cold probe with a z-axis gradient coil. NMR spectra were recorded using 250 μL solutions in H2O/D2O (95%/5%), 20mM sodium phosphate buffer, at pH 5.8, 20°C, in Shigemi microcells.
15N-1H HSQC spectra of PR (0.2mM) were recorded before and after addition of 6 mM substrate C. Each spectrum was acquired in ∼40 min with eight scans, 256 and 1024 complex t1 and t2 increments with 2000 Hz F1 and 7000 Hz F2 spectral widths, respectively. Because the active protease rapidly cleaves the substrate at 20°C, each data acquisition was performed less than 30 min after the addition of substrate C to the NMR tube.
Backbone signal assignments of free and substrate C-bound PRD25N were carried out using HNCA, CBCACONH, and 15N-edited NOESY spectra at 20°C and acquisition parameters similar to those used to assign free PR (Freedberg et al. 2002). Protein concentrations in these experiments were 0.2–0.3 mM with substrate C concentrations of either 0.0 or 7.2 mM. In addition, a series of 15N-1H HSQC spectra of PRD25N were recorded, at a constant protein concentration (∼0.3 mM), increasing the amount of substrate C until the protease:substrate ratio became 1:8. 15N longitudinal relaxation times (T1), transverse relaxation times (T2), and hetero-nuclear Overhauser enhancements (NOE) of the PRD25N bound to substrate C were measured using pulse schemes described previously (Freedberg et al. 2002). Relaxation delays for T1 experiments were 0.016, 0.16, 0.32, 0.48, 0.65, 0.8, and 0.96 sec, and for T2 experiments 4.8, 9.6, 19.2, 38.4, 57.6, 76.8, and 105.6 msec. Two kinds of longitudinal relaxation experiments were performed with t1 evolution (1) before and (2) after relaxation delays to measure 15N zz-magnetization exchange and to measure 15N T1 values, respectively. The CPMG 180° pulses were applied every 1.2 msec. Because of their lower sensitivity, NOE experiments were performed three times, and the measured NOEs were averaged. All relaxation experiments were recorded at 20°C, and the protein and substrate concentrations were 0.3 mM and 7.2 mM, respectively. Model-free analysis was performed to obtain order parameters and the chemical exchange contribution to T2 as described (Freedberg et al. 1998). In order to include the effect of overall anisotropic diffusion in the analysis, the structure of PRD25N bound to substrate C was assumed to be the same as the X-ray structure of PRD25N bound to CA-p2 substrate (Prabu-Jeyabalan et al. 2000).
Electronic supplemental material
Backbone assignment tables and 15N relaxation data for the inactive protease (PRD25N) in its free and substrate C-bound forms are available.
Acknowledgments
We thank F. Delaglio and D. Garrett for data processing software. This work was supported by the Intramural AIDS Targeted Anti-Viral Program of the Office of the Director of the National Institutes of Health, and by the Bio-Oriented Technology Research Advancement Institution of Japan.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0300703.
References
- Collins, J.R., Burt, S.K., and Erickson, J.W. 1995. Flap opening in HIV-1 protease simulated by "activated" molecular dynamics. Nat. Struct. Biol. 2 334–338. [DOI] [PubMed] [Google Scholar]
- Condra, J.H. 1998. Resistance to HIV protease inhibitors. Haemophilia 4 610–615. [DOI] [PubMed] [Google Scholar]
- Erickson, J.W. and Burt, S.K. 1996. Structural mechanisms of HIV drug resistance. Annu. Rev. Pharmacol. Toxicol. 36 545–571. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Zhang, O., Forman-Kay, J.D., and Kay, L.E. 1994. A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J. Biomol. NMR 4 727–734. [DOI] [PubMed] [Google Scholar]
- Fersht, A. 1999. Structure and mechanism in protein science, pp. 158–168. W.H. Freeman and Company, New York.
- Freedberg, D.I., Wang, Y.X., Stahl, S.J., Kaufman, J.D., Wingfield, P.T., Kiso, Y., and Torchia, D.A. 1998. Flexibility and function in HIV protease—Dynamics of the HIV-1 protease bound to the asymmetric inhibitor kynostatin 272 (KNI-272). J. Am. Chem. Soc. 120 7916–7923. [Google Scholar]
- Freedberg, D.I., Ishima, R., Jacob, J., Wang, Y.X., Kustanovich, I., Louis, J.M., and Torchia, D.A. 2002. Rapid structural fluctuations of the free HIV protease flaps in solution: Relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 11 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furfine, E.S., D’Souza, E., Ingold, K.J., Leban, J.J., Spector, T., and Porter, D.J. 1992. Two-step binding mechanism for HIV protease inhibitors. Biochemistry 31 7886–7891. [DOI] [PubMed] [Google Scholar]
- Ishima, R., Freedberg, D.I., Wang, Y.X., Louis, J.M., and Torchia, D.A. 1999. Flap opening and dimer-interface flexibility in the free and inhibitor bound HIV protease. Structure 7 1047–1055. [DOI] [PubMed] [Google Scholar]
- Johnson, K.A. 1992. Transient-state kinetic analysis of enzyme reaction pathways. In The enzymes, Vol. 20, pp. 1–61. Academic Press, Orlando, FL.
- Katoh, E., Yamazaki, T., Kiso, Y., Wingfield, P.T., Stahl, S.J., Kaufman, J.D., and Torchia, D.A. 1999. Determination of the rate of monomer interchange in a ligand-bound homodimeric protein from NOESY cross peaks: Application to the HIV protease/KNI-529 complex. J. Am. Chem. Soc. 121 2607–2608. [Google Scholar]
- Louis, J.M., Clore, G.M., and Gronenborn, A.M. 1999. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 6 868–875. [DOI] [PubMed] [Google Scholar]
- Luque, I., Todd, M.J., Gomez, J., Semo, N., and Freire, E. 1998. Molecular basis of resistance to HIV-1 protease inhibition: A plausible hypothesis. Biochemistry 37 5791–5797. [DOI] [PubMed] [Google Scholar]
- Nicholson, L.K., Yamazaki, T., Torchia, D.A., Grzesiek, S., Bax, A., Stahl, S.J., Kaufman, J.D., Wingfield, P.T., Lam, P.Y., and Jadhav, P.K. 1995. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 2 274–280. [DOI] [PubMed] [Google Scholar]
- Ohtaka, H., Velazquez-Campoy, A., Xie, D., and Freire, E. 2002. Overcoming drug resistance in HIV-1 chemotherapy: The binding thermodynamics of Amprenavir and TMC-126 to wild-type and drug-resistant mutants of the HIV-1 protease. Protein Sci. 11 1908–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oroszlan, S. and Luftig, R.B. 1990. Retroviral proteinases. Curr. Top. Microbiol. Immunol. 157 153–185. [DOI] [PubMed] [Google Scholar]
- Parikh, U., Hammond, J., Calef, C., Larder, B., Schinazi, R., and Mellors, J.W. 2000. The 2000 HIV sequence compendium, reviews: Mutations in retroviral genes associated with drug resistance. Los Alamos, NM.
- Prabu-Jeyabalan, M., Nalivaika, E., and Schiffer, C.A. 2000. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J. Mol. Biol. 301 1207–1220. [DOI] [PubMed] [Google Scholar]
- Prabu-Jeyabalan, M., Nalivaika, E., and Schiffer, C.A. 2002. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 10 369–381. [DOI] [PubMed] [Google Scholar]
- Rick, S.W., Erickson, J.W., and Burt, S.K. 1998. Reaction path and free energy calculations of the transition between alternate conformations of the HIV-1 protease. Proteins 32 7–16. [DOI] [PubMed] [Google Scholar]
- Rodriguez, E.J., Debouck, C., Deckman, I.C., Abu-Soud, H., Raushel, F.M., and Meek, T.D. 1993. Inhibitor binding to the Phe53Trp mutant of HIV-1 protease promotes conformational changes detectable by spectrofluorometry. Biochemistry 32 3557–3563. [DOI] [PubMed] [Google Scholar]
- Rose, R.B., Craik, C.S., Douglas, N.L., and Stroud, R.M. 1996. Three-dimensional structures of HIV-1 and SIV protease product complexes. Biochemistry 35 12933–12944. [DOI] [PubMed] [Google Scholar]
- Scott, W.R.P. and Schiffer, C.A. 2000. Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure Fold. Des. 8 1259–1265. [DOI] [PubMed] [Google Scholar]
- Shafer, R.W., Jung, D.R., Betts, B.J., Xi, Y., and Gonzales, M.J. 2000. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 28 346–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, W., Everitt, L., Manchester, M., Loeb, D.D., Hutchison 3rd, C.A., and Swanstrom, R. 1997. Sequence requirements of the HIV-1 protease flap region determined by saturation mutagenesis and kinetic analysis of flap mutants. Proc. Natl. Acad. Sci. 94 2243–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra, N., Wingfield, P., Stahl, S., and Bax, A. 1996. Anisotropic rotational diffusion of perdeuterated HIV protease from 15N NMR relaxation measurements at two magnetic fields. J. Biomol. NMR 8 273–284. [DOI] [PubMed] [Google Scholar]
- Todd, M.J. and Freire, E. 1999. The effect of inhibitor binding on the structural stability and cooperativity of the HIV-1 protease. Proteins 36 147–156. [DOI] [PubMed] [Google Scholar]
- Tozser, J., Yin, F.H., Cheng, Y.S., Bagossi, P., Weber, I.T., Harrison, R.W., and Oroszlan, S. 1997. Activity of tethered human immunodeficiency virus 1 protease containing mutations in the flap region of one subunit. FEBS Lett. 244 235–241. [DOI] [PubMed] [Google Scholar]
- Velazquez-Campoy, A., Kiso, Y., and Freire, E. 2001. The binding energetics of first- and second-generation HIV-1 protease inhibitors: Implications for drug design. Arch. Biochem. Biophys. 390 169–175. [DOI] [PubMed] [Google Scholar]
- Vondrasek, J., van Buskirk, C.P., and Wlodawer, A. 1997. Database of three-dimensional structures of HIV proteinases. Nat. Struct. Biol. 4 8. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A. and Erickson, J.W. 1993. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 62 543–585. [DOI] [PubMed] [Google Scholar]
- Yang, D. and Kay, L.E. 1996. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: Application to protein folding. J. Mol. Biol. 263 369–382. [DOI] [PubMed] [Google Scholar]