

Abstract
The structure of Vibrio cholerae protein VC0424 was determined by NMR spectroscopy. VC0424 belongs to a conserved family of bacterial proteins of unknown function (COG 3076). The structure has an α-β sandwich architecture consisting of two layers: a four-stranded antiparallel β-sheet and three side-by-side α-helices. The secondary structure elements have the order αβαββαβ along the sequence. This fold is the same as the ferredoxin-like fold, except with an additional long N-terminal helix, making it a variation on this common motif. A cluster of conserved surface residues on the β-sheet side of the protein forms a pocket that may be important for the biological function of this conserved family of proteins.
Keywords: VC0424, COG 3076, YjgD, structural genomics, NMR structure, Vibrio cholerae
The structure of Vibrio cholerae protein VC0424 (GI: 11282637 / PIR: G82323 / SPTR: Q9KUU1 / EMBL: AE004130) was determined by NMR spectroscopy. VC0424 belongs to a conserved family of bacterial proteins of unknown function (COG 3076). VC0424 is protein target OP3 of the Northeast Structural Genomics Consortium (NESG). It was chosen from the recently sequenced V. cholerae genome (Heidelberg et al. 2000) for structural characterization because it has no sequence homologs with structures in the Protein Data Bank (PDB). A protein-protein BLAST (Altschul et al. 1990) search identified eight bacterial homologs of approximately the same length as VC0424 (134–153 residues) with 51%–88% sequence identity. The aligned sequences are shown in Figure 1▶. Examination of conserved residues and their location on the structure of VC0424 provides information about possible functional residues for this family of proteins.
Figure 1.

Sequence of alignment of VC0424 (gi│11282637) from Vibrio cholerae with eight other conserved bacterial proteins: gi│27364852 (Vibrio vulnificus), gi│586747 (Salmonella typherium), gi│732037 (Escherichia coli), gi│16123593 (Yersinia pestis), gi│24372161 (Shewanella oneidensis), gi│1176470 (Haemophilus influenza), gi│23468052 (Haemophilus somnus), and gi│15603430 (Pasteurella multocida). Secondary structure of VC0424 is shown above: b, β-bulge; c, coil; e, extended β strand; h, α helix; t, turn. Conserved residues are highlighted in black and gray.
Results and Discussion
VC0424 has an open-faced (or two-layer) α-β sandwich architecture. Secondary structural elements are ordered αβαββαβ along the sequence (Fig. 2▶). One layer consists of the three α-helices, and the other layer has the four β-strands. The strands are antiparallel with the order 2-3-1-4, and the three helices are also aligned with opposite N- to C-terminal orientation with respect to each other, with the order 1-3-2. Excluding the N-terminal helix, the structure has the ferredoxin-like fold βαββαβ topology, which consists of two intercalated right-handed βαβ motifs. The N-terminal helix is the longest with 22 residues, and the other two have 14 and 18 residues each. The amide proton of Q17 in the middle of α1 was doubled in the 1H dimension, suggesting two chemical environments. However, the NOEs were the same in both environments, so they could not be distinguished. The four β-sheets have the usual right-handed twist, and strands 2 and 4 have β-bulges at A70 and G117, respectively.
Figure 2.

Stereo diagram of backbone atoms of residues 8–124 for 20 NMR structures of VC0424 optimally superimposed with respect to the average coordinates of the backbone atoms of residues 10–74 and 81–121 (N, Cα, C‘, and O atoms). Figure was created using MOLMOL (Koradi et al. 1996).
A 3D structure search using Dali (Holm and Sander 1993), CE (Shindyalov and Bourne 1998), and PrISM (Yang and Honig 2000) showed that VC0424 has structural similarity with hundreds of proteins containing the ferredoxin-like fold. The best Dali match was subunit A of the carboxypeptidase G2 biological unit (PDB id 1CG2, Z-score 8.6 for 89 residues). This protein is a homodimer, with the ferredoxin-like fold region forming a dimer interface with the corresponding fold on the other subunit. In many cases, the ferredoxin-like fold is found in large multidomain proteins. Presently there are 40 superfamilies in this SCOP classification (Murzin et al. 1995). VC0424 represents a new example of a monomeric domain of a ferredoxin-like fold with an N-terminal helix. The ferredoxin-like fold is found in numerous proteins with varying biological functions, including the ACT and RAM domains, ribosomal proteins, RNA binding domains, metallo-copper chaperone proteins, transcriptional regulators, and several enzymes.
A phylogenetic analysis of the nine bacterial homologs was performed with the program ConSurf (http://consurf.tau.ac.il; Armon et al. 2001; Glaser et al. 2003), which evaluates the degree of conservation of each amino acid and maps this to the tertiary structure to identify surface clusters of conserved residues, which may have a functional role. The conserved residues (Fig. 3▶, in dark pink) cluster to a discrete region on the β-sheet face of VC0424. The conserved residues are found primarily in the C-terminus of α1, the α1–β1 loop, β1, and β4 (Fig. 3A,B▶), and form a shallow concave surface. In addition, the electrostatic surface of VC0424 (Fig. 3C▶) is negatively charged on the same β-sheet face due to many conserved, solvent-exposed Asp and Glu residues. Interestingly, the β-sheet face of the ferredoxin-like fold has been identified as a "supersite," a region of superfold (a fold common in analogous proteins) that has statistical significance for ligand binding, based on analyses of ligand-bound superfolds (Russell et al. 1998). The localization of conserved residues in VC0424 to the β-sheet face may represent another example of this supersite. Taken together, these observations suggest that this β-sheet surface may be involved in mediating the functional activity for this protein.
Figure 3.
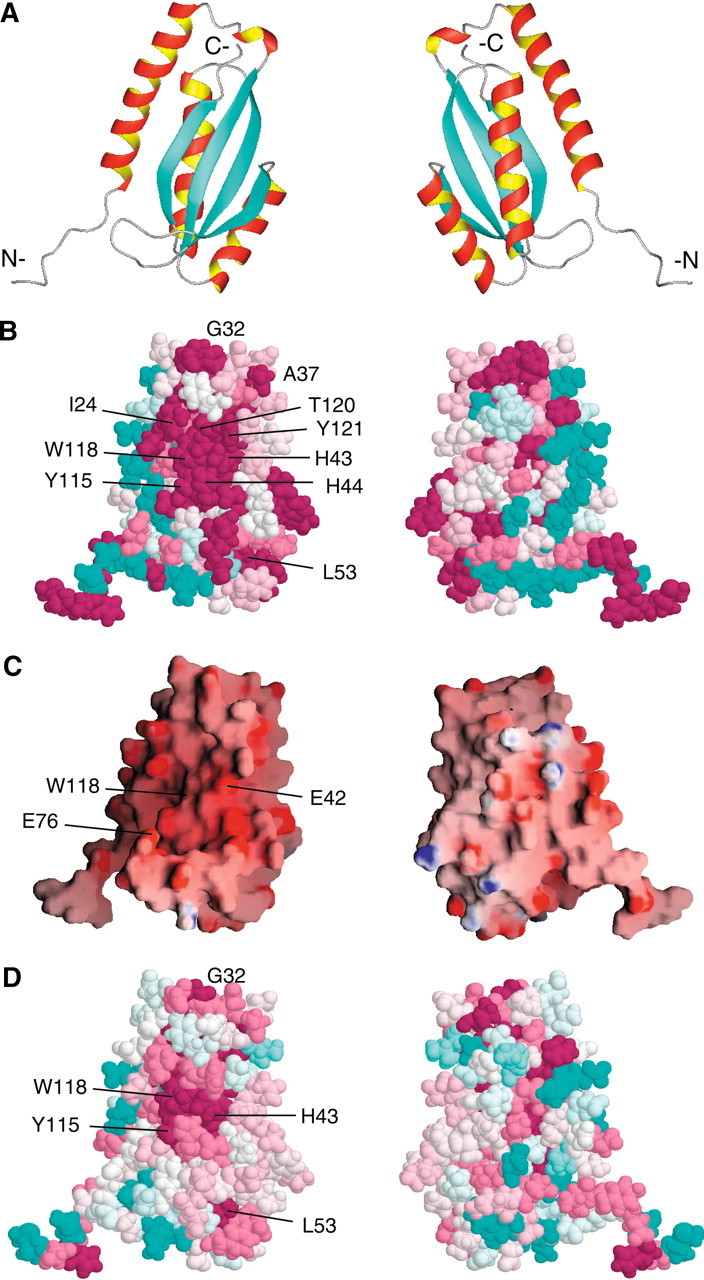
(A) Results of ConSurf analysis mapped onto VC0424 structure (residues 1–124) using the maximum likelihood method (Armon et al. 2001; Glaser et al. 2003). Shown on the left is the β-sheet side of the protein; right, the opposite side following a 180° rotation about the y-axis. All figures in columns have the same orientation. Conserved residues are darkest pink, variable residues are cyan, and others are white. (B) ConSurf analysis of VC0424 and eight bacterial homologs (see Fig. 1▶). Six N-terminal residues are present in only two of the alignments and appear conserved, probably as an artifact of the alignment and the way ConSurf treats gaps (Glaser et al. 2003). (C) GRASP (Nicholls et al. 1991) representation of the VC0424 electrostatic surface potential. (D) ConSurf analysis including seven "distant homologs" of VC0424: FN0296 gi│19703641 (Fusobacterium nucleatum), PM0690 gi│15602555 (Pasteurella multocida), HI0040 gi│16272015 (Haemophilus influenza), orf1 gi│7592806, unnamed gi│1944176, ORF19 gi│7467535, and ORF17 gi│5545328 (Actinobacillus actinomycetemcomitans).
In order to gain insights into putative functional residues in VC0424, structural alignments between VC0424 and other ferredoxin-like fold domains were examined to look for conservation of functional residues. VC0424 does not share sequence similarity with ligand-binding regions of example proteins in the ACT or RAM domains, which are domains of proteins involved in amino acid biosynthesis and metabolic regulation (Chipman and Shaanan 2001; Ettema et al. 2002). In addition, although there is overlap between the cluster of conserved residues in VC0424 and the regions that interact with RNA in the RNA-binding domains (1), it is unlikely that VC0424 is an RNA-binding protein like the ribosomal protein S6, due to the negative electrostatic potential of this surface (Fig. 3C▶).
The E. coli homolog of VC0424, YjgD (60% identical), is a hypothetical protein that appears to have a link to proteins involved in tryptophan metabolism (Khodursky et al. 2000). Expression of the yjgD gene increased in response to tryptophan starvation as detected by measurements of mRNA abundance in DNA microarray analysis (group I; Khodursky et al. 2000). Regulation of the yjgD gene by the transcription factor ArgR is proposed based on analysis of potential ArgR binding sites. The expression profile of yjgD was found to be correlated with argI expression. Putative transcription factor binding sites for ArgR have been identified between argI and yjgD, which are adjacent genes on opposite DNA strands arranged in a head-to-head alignment (McCue et al. 2002). The gene argI codes for ornithine carbamoyltransferase I (OCT), which is involved in arginine biosynthesis. However, an equivalent of argI is not found near VC0424 in the V. cholerae genome. Interestingly, the genes VC0423 and VC0424 are in a similar head-to-head alignment, and the gene VC0423 codes for an arginine deiminase (ADI) that is involved in arginine catabolism. Taken together, these comparisons suggest that VC0424 and YjgD could both be involved in amino acid metabolic pathways such as that for arginine.
A PSI-BLAST (Altschul et al. 1997) search revealed remote sequence similarity to the C-terminal domains of a group of longer bacterial proteins (22%–25% for 110 residues with two gaps). Four of the proteins are found in Actinobacillus actinomycetemcomitans and belong to gene clusters involved in synthesis of serotype-specific polysaccharide antigens b–e, respectively. However, the two proteins in the serotype c- and serotype d-specific polysaccharide antigen gene clusters were not essential for the synthesis of these antigens (Nakano et al. 1998, 2000). Their functions and whether they even play a role in polysaccharide synthesis are not known. Secondary structure prediction using the program PHD (Rost et al. 1994) for these domains revealed predicted structure that is consistent with that of VC0424 and aligns with it, suggesting that these distantly related proteins have a similar fold for their C-terminal domains. Seven of these longer bacterial homologs were aligned with VC0424, along with the previous eight identified homologs, using CLUSTAL W (Thompson et al. 1994) and mapped onto the surface of VC0424 using the program ConSurf, as before. The analysis yields similar results, but with a smaller patch of conserved residues (Fig. 3D▶). The conserved residues include the aromatic residues that align with H43, Y115, and W118 in VC0424.
Materials and methods
The VC0424 gene was PCR-amplified from genomic DNA (ATCC no. 51394D) and inserted into a pET28b vector (Novagen). Constructs were made expressing the full-length protein (140 residues) with a C-terminal His6-tag (residues LEHHHHHH) and a truncated form with the low-complexity 16 C-terminal residues of VC0424 removed (EDALYSDEDDEDDEH), again with a C-terminal His6 tag. Proteins were expressed in Escherichia coli strain Rosetta (λDE3; Novagen) and grown in M9 minimal media at 25°C. VC0424 was purified by Ni2+ affinity chromatography followed by size exclusion chromatography. NMR samples of approximately 1.5 mM [U-15N, U-13C] VC0424 and the truncated protein were prepared in 20 mM Tris HCl, 500 mM NaCl, 5 mM DTT in 10% (v/v) 2H2O/H2O at pH 7.2. Samples were placed in 5-mm Shigemi susceptibility matched NMR tubes.
Two-dimensional 1H-15N-HSQC (Kay et al. 1992; Zhang et al. 1994) spectra were recorded on both full-length and truncated VC0424. Both spectra looked essentially the same with isolated cross-peaks at the same location. However, the center of the spectrum, where cross-peaks for unstructured residues would be expected to occur, was less crowded in the truncated protein. This suggests that the 16 C-terminal residues of full-length VC0424 are unstructured in solution. In order to work with simplified spectra, we solved the structure of the truncated protein, which we refer to as VC0424 in this paper.
NMR spectra were recorded at 25°C on 600, 750, and 800 MHz Varian Inova spectrometers. Chemical shifts were referenced to external DSS (Wishart et al. 1995). Backbone and side chain assignments were made using the following triple resonance experiments recorded on [U-15N, U-13C]-VC0424: 3D HNCO, HNCACB, CBCA(CO)NNH (Kay et al. 1992; Muhandiram and Kay 1994), HNHA (Vuister and Bax 1993; Grzesiek et al. 1995; Zhang et al. 1997), HCCH-TOCSY (Kay et al. 1993), HCC-TOCSY-NNH (Montelione et al. 1992; Lyons and Montelione 1993), and CC-TOCSY-NNH (Montelione et al. 1992; Grzesiek et al. 1993; Logan et al. 1993). NOE restraints were derived from 3D 15N-edited NOESY-HSQC (τm=150 msec; Kay et al. 1992; Zhang et al. 1994), 13C-edited NOESY-HSQC (τm=120 msec; Pascal et al. 1994), and 4D CC-NOESY (2H2O, τm=100 msec; Vuister et al. 1993) experiments. Two-dimensional 1H-15N-HSQC (Kay et al. 1992; Zhang et al. 1994) and 1H-13C-HSQC (Kay et al. 1992; John et al. 1993) spectra were recorded in H2O, and 1H-15N-HSQC spectra were recorded at 20, 35, and 60 min, 15 h, and 4 d and after a lyophilized sample was redissolved in 2H2O.
Spectra were processed with Felix (MSI) and analyzed with Sparky (http://www.cgl.ucsf.edu/home/sparky). No chemical shifts were obtained for residues M1–H3 and residues H129–H132 in the His6-tag. Chemical shifts of 1H, 15N, and 13C resonances have been deposited in BioMagResBank (BMRB) under accession code 5589.
Sequential NOE cross-peaks were characterized as strong (1.8–2.5 Å), medium (1.8–3.5 Å), or weak (1.8–5 Å) and given the corresponding distance restraints. Medium and long-range NOEs were given the restraint of 1.8–5.0, unless they were very intense (1.8–4 Å). Pseudoatom correction of 1 Å for methyls and methylenes, 2.4 Å for nonstereospecifically assigned Leu and Val methyl groups, or 2.0 Å for equivalent aromatic protons were added to the upper bounds. Dihedral angle restraints for φ were derived from the HNHA experiment (57 ± 25° for J < 6 Hz and −139 ± 40° for J > 8 Hz). Dihedral angle restraints for Ψ were added during the later stages of refinement for residues in helix and strand regions where chemical shift and αiNi and αiNi+1 NOESY peak intensities indicated they were appropriate. In addition, 11 χ1 restraints were derived from the HNHB experiment (Archer et al. 1991; Bax et al. 1994). Hydrogen bond restraints were added for 58 slowly exchanging amide protons for which an acceptor could be identified from preliminary structures (dNO = 2.8–3.3 Å and dHN=1.8–2.3 Å). NOE and dihedral angle restraint tables in Xplor format are deposited in the BMRB (code 5589). A total of 1148 distance restraints and 117 dihedral angle restraints were used in the structure calculations using the Xplor-NIH software (Schwieters et al. 2003) with the Xplor-NIH routines mkpsf.inp and generate_ template.inp and the standard Xplor-3.84 (Brünger 1992) routines dg_subembed.inp, dg_full_embed.inp, and dgsa.inp. The program AutoStructure was used to add 160 medium and long-range restraints to the manual assignments (Huang 2001; Huang et al. 2003). The 20 calculated structures were deposited in the PDB with accession code 1NXI. The backbone trace of the ensemble of 20 lowest energy structures is shown in Figure 2▶, and the structural statistics are compiled in Table 1▶. Ramachandran statistics were generated with PROCHECK-NMR (Laskowski et al. 1996).
Table 1.
Structural statistics for VC0424 ensemble of 20 structures
| Distance restraints | |
| All | 1148 |
| Intraresidue | 5 |
| Sequential (|i − j| = 1) | 252 |
| Medium (1 < |i − j| < 5) | 329 |
| Long-range (|i • j| ≥ 5) | 446 |
| Hydrogen bonds (2 per hydrogen bond) | 58 × 2 |
| Dihedral restraints | |
| All | 117 |
| φ | 44 |
| Ψ | 62 |
| χ1 | 11 |
| Restraints per residue (res. 9–123) | 11 |
| Distance restraint violations | |
| Mean number of violations | 27.7 ± 4.7 |
| Mean r.m.s. violation (Å) | 0.0023 ± 0.0003 |
| Dihedral restraint violations | |
| Mean number of violations | 2.0 ± 1.5 |
| Mean r.m.s. violation (°) | 0.03 ± 0.02 |
| R.m.s. deviation from the average coordinates (Å) (residues 10–74, 81–121) | |
| Backbone atoms (N, Cα, C′, O) | 0.51 ± 0.08 |
| All heavy atoms | 1.03 ± 0.04 |
| Ramachandran statistics (residues 10–74, 81–121) | |
| Residues in most favored region | 82% |
| Residues in additional allowed regions | 16% |
| Residues in generously allowed regions | 1% |
| Residues in disallowed regions | 1% |
Acknowledgments
We thank Janet Huang for her help with the program AutoStructure. Acquisition and processing of NMR spectra and structure calculations were performed at the Environmental Molecular Sciences Laboratory (a national scientific user facility sponsored by the U.S. Department of Energy Office of Biological and Environmental Research) located at Pacific Northwest National Laboratory and operated by Battelle for the Department of Energy (contract KP130103). This work was supported by the NIH Protein Structure Initiative Northeast Structural Genomics Consortium (grant P50-GM62413) and the grant NSF# DBI-9601463. VC0424 from V. cholerae is target OP3 of the NESG.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03108103.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer, S.J., Ikura, M., Torchia, D.A., and Bax, A. 1991. An alternate 3D NMR technique for correlating backbone 15N with side chain Hb resonances in larger proteins. J. Magn. Reson. 94 636–641. [Google Scholar]
- Armon, A., Graur, D., and Ben-Tal, N. 2001. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface-mapping of phylogenetic information. J. Mol. Biol. 307 447–463. [DOI] [PubMed] [Google Scholar]
- Bax, A., Vuister, G.W., Grzesiek, S., Delaglio, F., Wang, A.C., Tschudin, R., and Zhu, G. 1994. Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol. 239 79–105. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. X-PLOR Version 3.1: A system for X-ray crystallography and NMR. Yale University Press, New Haven, CT.
- Chipman, D.M. and Shaanan, B. 2001. The ACT domain family. Curr. Opin. Struct. Biol. 11 694–700. [DOI] [PubMed] [Google Scholar]
- Ettema, T.J., Brinkman, A.B., Tani, T.H., Rafferty, J.B., and Van Der Oost, J. 2002. A novel ligand-binding domain involved in regulation of amino acid metabolism in prokaryotes. J. Biol. Chem. 277 37464–37468. [DOI] [PubMed] [Google Scholar]
- Glaser, F., Pupko, T., Paz, I., Bell, R.E., Bechor-Shental, D., Martz, E., and Ben-Tal, N. 2003. ConSurf: Identification of functional regions in a proteins by surface-mapping of phylogenetic information. Bioinformatics 19 163–164. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S., Anglister, J., and Bax, A. 1993. Correlation of backbone and amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. B 101 114–119. [Google Scholar]
- Grzesiek, S., Kuboniwa, H., Hinck, A.P., and Bax, A. 1995. Multiple-quantum line narrowing for measurement of Hα-Hβ J-couplings in isotopically enriched proteins. J. Am. Chem. Soc. 117 5312–5315. [Google Scholar]
- Heidelberg, J.F., Eisen, J.A., Nelson, W.C., Clayton, R.A., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Umayam, L., et al. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Huang, Y.J. 2001. Automated determination of protein structures from NMR data by iterative analysis of self-consistent contact patterns. Ph.D. thesis, Rutgers University, New Brunswick, NJ.
- Huang, Y.J., Swapna, G.V.T., Rajan, P.K., Ke, H., Xia, B., Shukla, K., Inouye, M., and Montelione, G.T. 2003. Solution NMR structure of ribosomo-binding factor A (RbfA), a cold-shock adaptation protein from Escherichia coli. J. Mol. Biol. 327 521–536. [DOI] [PubMed] [Google Scholar]
- John, B.K., Plant, D., and Hurd, R.E. 1993. Improved proton-detected heteronuclear correlation using gradient-enhanced Z and ZZ filters. J. Magn. Reson. 101 113–117. [Google Scholar]
- Kay, L.E., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114 10663–10665. [Google Scholar]
- Kay, L.E., Xu, G.Y., Singer, A.U., Muhandiram, D.R., and Forman-Kay, J.D. 1993. A gradient-enhanced HCCH TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. B 101 333–337. [Google Scholar]
- Khodursky, A.B., Peter, B.J., Cozzarelli, N.R., Botstein, D., Brown, P.O., and Yanofsky, C. 2000. DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Eschericia coli. Proc. Natl. Acad. Sci. 97 12170–12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Rullmann, J.A., MacArthur, M.W., Kaptein, R., and Thornton, J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 447–486. [DOI] [PubMed] [Google Scholar]
- Logan, T.M., Olejniczak, E.T., Xu, R.X., and Fesik, S.W. 1993. A general method for assigning NMR spectra of denatured proteins using 3D HC(CO)NH-TOCSY triple resonance experiments. J. Biomol. NMR 3 225–231. [DOI] [PubMed] [Google Scholar]
- Lyons, B.A. and Montelione, G.T. 1993. An HCCNH triple-resonance experiment using 13C isotropic mixing for correlating backbone amide and side-chain aliphatic resonances in isotopically enriched proteins. J. Magn. Reson. B. 1993 206–209. [Google Scholar]
- McCue, L.A., Thompson, W., Carmack, C.S., and Lawrence, C.E. 2002. Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res. 10 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montelione, G.T., Lyons, B.A., Emerson, S.D., and Tashiro, M. 1992. An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically enriched proteins. J. Am. Chem. Soc. 114 10974–10975. [Google Scholar]
- Muhandiram, D.R. and Kay, L.E. 1994. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. B 103 203–216. [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T.J.P., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Nakano, Y., Yoshida, Y., Yamashita, Y., and Koga, T. 1998. A gene cluster for 6-deoxy-L-talan synthesis in Actinobacillus actinomycetemcomitans. Biochim. Biophys. Acta 1442 409–414. [DOI] [PubMed] [Google Scholar]
- Nakano, Y., Yoshida, Y., Suzuki, N., Yamashita, Y., and Koga, T. 2000. A gene cluster for the synthesis of serotype d-specific polysaccharide antigen in Actinobacillus actinomycetemcomitans. Biochim. Biophys. Acta 1493 259–263. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. GRASP—Graphical representation and analysis of surface-properties. Struct. Funct. Genet. 11 281–296. [Google Scholar]
- Pascal, S.M., Muhandiram, D.R., Yamazaki, T., Forman-Kay, J.D., and Kay, L.E. 1994. Simultaneous acquisition of 15N-edited and 13C-edited NOE spectra of proteins dissolved in H2O. J. Magn. Reson. B 103 197–201. [Google Scholar]
- Rost, B., Sander, C., and Schneider, R. 1994. PHD—An automatic mail server for protein secondary structure prediction. Comput. Appl. Biosci. 10 53–60. [DOI] [PubMed] [Google Scholar]
- Russell, R.B., Sasieni, P.D., and Sternber, M.J. 1998. Supersites within superfolds. Binding site similarity in the absence of homology. J. Mol. Biol. 282 903–918. [DOI] [PubMed] [Google Scholar]
- Schwieters, C.D., Kuszewski, J.J., Tjandra, N., and Clore, G.M. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160 65–73. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuister, G.W. and Bax, A. 1993. Quantitative J correlation: A new approach for measuring homonuclear three-bond JHNHa coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 115 7772–7777. [Google Scholar]
- Vuister, G.W., Clore, G.M., Gronenborn, A.M., Powers, R., Garrett, D.S., Tschudin, R., and Bax, A. 1993. Increased resolution and improved spectral quality in 4-dimensional 13C/13 C-separated HMQC-NOESY-HMQC spectra using pulsed-field gradients. J. Magn. Reson. B 101 210–213. [Google Scholar]
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C, and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6 135–140. [DOI] [PubMed] [Google Scholar]
- Yang, A.-S. and Honig, B. 2000. An integrated approach to the analysis and modeling of protein sequences and structures. I. Protein structural alignment and quantitative measure for protein structural distance. J. Mol. Biol. 301 665–678. [DOI] [PubMed] [Google Scholar]
- Zhang, O., Kay, L.E., Olivier, J.P., and Forman-Kay, J.D. 1994. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of Drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 4 845–858. [DOI] [PubMed] [Google Scholar]
- Zhang, W.X., Smithgall, T.E., and Gmeiner, W.H. 1997. Three-dimensional structure of the Hck SH2 domain in solution. J. Biomol. NMR 10 263–272. [DOI] [PubMed] [Google Scholar]