

Abstract
Binding of the product inhibitor p-nitrophenol to the monoclonal esterolytic antibody NPN43C9 has been investigated by performing NMR spectroscopy of the heterodimeric variable-domain fragment (Fv) of the antibody in the presence and absence of inhibitor. Structural information from changes in chemical shift upon binding has been related to the changes in local dynamics in the active site of the catalytic antibody using NMR relaxation measurements. Significant changes in the chemical shifts of the backbone resonances upon binding extend beyond the immediate vicinity of the antigen binding site into the interface between the two associated polypeptides that form the Fv heterodimer, a possible indication that the binding of ligand causes a change in the relative orientations of the component light (VL) and heavy (VH) chain polypeptides. Significant differences in backbone dynamics were observed between the free Fv and the complex with p-nitrophenol. A number of resonances, including almost all of the third hypervariable loop of the light chain (L3), were greatly broadened in the free form of the protein. Other residues in the antigen-binding site showed less broadening of resonances, but still required exchange terms (Rex) in the model-free dynamics analysis, consistent with motion on a slow timescale in the active site region of the free Fv. Binding of p-nitrophenol caused these resonances to sharpen, but some Rex terms are still required in the analysis of the backbone dynamics. We conclude that the slow timescale motions in the antigen-binding site are very different in the bound and free forms of the Fv, presumably due to the damping of large-amplitude motions by the bound inhibitor.
Keywords: Catalysis, NMR, relaxation measurements, anisotropy, model-free analysis
A great deal of structural work, by both X-ray crystallography and NMR spectroscopy, has been focused on antibody combining sites. In particular, the structure of the combining site in the presence and absence of ligands has been examined in detail by X-ray crystallography, and it appears that the incidence of structural change upon ligand binding is quite variable. For example, the structures of some antibodies reveal significant changes in the antibody-binding site upon complexation (Stanfield et al. 1993), whereas more recent studies of catalytic antibodies with bound hapten reveal no major structural changes upon binding (Gigant et al. 1998; Thayer et al. 1999). The complementarity-determining regions (CDRs) of antibodies consist of loops of variable length which might well be intrinsically rather disordered. Because crystal packing can influence the conformation of these loops, it is of interest to examine the location and type of structural changes in solution, and to relate these to changes in the flexibility and dynamics of the complementarity-determining loops upon binding of ligands.
Catalytic antibodies form a special class, where the specificity of antibody binding to ligands is utilized to catalyze a chemical or biochemical reaction. The thermodynamic and structural implications of this observation have been extensively discussed (Lerner et al. 1991; Schultz and Lerner 1993, 1995). Catalytic antibodies are generally raised against a strained transition state analog, and it appears that the specific binding energy of the antibody toward the immunogen can be harnessed in the analogous substrate to facilitate formation of the transition state. Catalytic antibodies generally show broader substrate specificity than natural enzymes and often have a lower turnover rate.
The catalytic monoclonal antibody NPN43C9 (Janda et al. 1988) is both an amidase and an esterase, catalyzing the hydrolysis of a variety of phenyl ester compounds and p-nitroanilides. Hydrolysis of the ester or amide bond is a multistep reaction that includes an acyl-antibody intermediate (Gibbs et al. 1992; Krebs et al. 1995). The rate-limiting step in the hydrolysis of the substrate p-nitrophenyl ester is the release of p-nitrophenol, for which the Fv has a strong affinity (Kd 0.6 μM; koff 40 sec−1; Gibbs et al. 1992). The structures of the original phosphonamidate immunogen, together with a representative substrate and the product inhibitor p-nitrophenol, are shown in Figure 1▶. The smallest portion of the NPN43C9 antibody molecule capable of catalysis is the ∼25-kD variable fragment (Fv), which consists of a heterodimer of a 117-residue heavy chain variable region (VH) and a 113-residue light chain variable region (VL).
Figure 1.

Structures of (A) the phosphoramidate immunogen used to elicit the antibody NPN43C9, (B) ester substrate of the catalytic antibody, and (C) product inhibitor p-nitrophenol.
The three-dimensional structures of the Fv NPN43C9 and its binary complex with p-nitrophenol have been solved (Thayer et al. 1999). The nitrophenyl group is positioned in a deep pocket between the CDRs L1 and H3, making contact with residues His L91 and Tyr H95 (numbering according to Kabat et al. 1991). In the free form, the residues involved in the binding of p-nitrophenol form part of an extensive H-bond network with four water molecules. Mutations that disrupt this network cause changes in catalysis, which were interpreted as due to additional motion in the binding pocket (Thayer et al. 1999). To obtain insights into the intrinsic flexibility of the active site region of NPN43C9 and its possible role in catalysis, we have undertaken NMR relaxation experiments to probe directly the dynamics of the polypeptide backbone in both free and p-nitrophenol-bound states.
Results
Preparation of ligand-free Fv
The final step in the purification of the catalytic Fv involves elution of the folded Fv heterodimer from an affinity column with excess p-nitrophenol. Because this yields the protein in complex with p-nitrophenol, a method had to be developed to remove the p-nitrophenol ligand from the Fv. This is not easy, as the protein must be maintained in its fully folded state during the process. Attempts to refold the protein from E. coli inclusion bodies showed that it cannot be reconstituted once it is unfolded. The high affinity of the p-nitrophenol makes it difficult to remove from the binding site without denaturation of the protein. Several methods, including elution of the protein from the affinity column at relatively low pH, gave samples of the free form that were prone to aggregation, presumably due to some local irreversible unfolding step. We were finally successful in obtaining stable samples of the free form by extensive dialysis of the purified p-nitrophenol complex, which resulted in complete removal of the p-nitrophenol ligand, according to UV absorbance measurements and HSQC NMR spectra. Samples prepared in this way were stable for a period of months.
Changes in chemical shifts upon binding of p-nitrophenol
Resonance assignments for the Fv bound to p-nitrophenol have been reported (Kroon et al. 1999). Backbone resonance assignments for the free Fv were made using 15N NOESY-HSQC and 15N TOCSY-HSQC spectra recorded at 800 MHz. Of the 219 backbone amides, 189 could be assigned for the free protein. The remaining resonances were absent from the spectrum, due either to rapid exchange with solvent, or to broadening of the resonance lines as a result of exchange processes on an intermediate timescale. The 22 unassigned residues in the VH subunit are found in the first β-strand (Leu H4 to Ser H7), in and near CDR H1 (Leu H29, His H35, Trp H36, and Ser H40), in CDR H2 (Glu H58 and Tyr H59), in CDR H3 (Gly H96, Tyr H97, Gly H99, and Ser H101), and in the region around CDR H3 (Tyr H90 to Val H93, Trp H103 to Gly H106). The eight unassigned residues in the VL subunit are found near the N-terminus (Leu L2), in the third β-strand (Met L21), in CDR L1 (Leu L27B and Ala L34), in the fifth β-strand (Leu L47), in the seventh β-strand (Thr L69 and Ile L75), and in CDR L3 (Arg L96).
A comparison of a region of the {1H}-15N HSQC spectrum for the Fv in the presence and absence of p-nitrophenol is shown in Figure 2▶. It is clear that a number of resonances, not just those in the antigen-binding site, are shifted upon binding of ligand. The changes in chemical shifts of 1H and 15N for the backbone amides are plotted in Figure 3▶. Differences larger than 0.05 ppm for proton or larger than 0.5 ppm for nitrogen were observed for Asn L31 and Tyr L32 in CDR L1, Trp L35 and Tyr L36 immediately following CDR L1, Ala L51, Ser L52, and Ser L56 in CDR L2, Gln L90, Tyr L92, Ala L94, and Thr L97 in CDR L3, Tyr H32 in CDR H1, Asp H100 and Phe H100B in CDR H3, Ser H28 in the loop between the second and third β-strand, and Asn H73 in the loop between β-strands six and seven. The largest differences in chemical shifts are observed for the residues in CDR L3, with a maximum of 7.3 ppm for the 15N chemical shift of Ala L94.
Figure 2.

Comparison of a portion of the 1H-15N HSQC spectra of the Fv free and bound to the product inhibitor p-nitrophenol. Free Fv, red; p-nitrophenol complex, black.
Figure 3.

Plot of sequence locations of differences in amide chemical shift (free minus bound) between the free and p-nitrophenol-bound forms of the Fv. Value of Δδ(15N) for residue 94 is 7.3 ppm.
The locations of amide resonances that are perturbed upon binding of p-nitrophenol are shown in Figure 4▶, plotted on the backbone trace of the crystal structure (Thayer et al. 1999). All of the assigned residues in CDR L3 show significant changes in chemical shift upon binding of p-nitrophenol. Four residues at the C-terminal part of CDR L1 also show a substantial difference in chemical shift upon binding. Only four assigned residues in CDR H1 and CDR H3 are influenced by the binding of p-nitrophenol. Most of the residues showing changes are found in the VL, suggesting that the position of the bound p-nitrophenol is closer to residues of the VL than the VH. The largest shifts are found in a part of the CDR L3 close to the residues involved in catalysis.
Figure 4.

Locations of the amide proton and 15N resonances that are perturbed upon complexation of p-nitrophenol, plotted onto the structure of p-nitrophenol-bound Fv (Thayer et al. 1999). Blue represents residues for which assignments could be made for both forms, and gray shows areas where resonances of the free and/or p-nitrophenol-bound forms could not be assigned. Red represents residues for which differences larger than 0.05 ppm for 1H and/or larger than 0.5 ppm for 15N were observed.
No major changes in NOE patterns were observed for those residues that show large changes in chemical shifts when comparing data from the NOESY-HSQC spectrum of the free Fv fragment to that obtained for the p-nitrophenol complex. This indicates that the changes in chemical shifts are caused not by major structural changes but rather by changes in the local chemical environment.
Diffusion experiments
In order to determine the oligomerization state of the Fv, water-suppressed longitudinal encode-decode (water-sLED) experiments were performed (Altieri et al. 1995). The diffusion coefficients for the Fv in the presence and absence of p-nitrophenol were 0.97 and 0.95 × 10−6 cm2s−1, respectively. These results are midway between values of 1.08 and 0.82 × 10−6 cm2s−1 obtained for lysozyme (14.1 kD) and IL-10 (37.2 kD), respectively (Altieri et al. 1995), consistent with the formation of a 25-kD heterodimer of the variable light chain VL and variable heavy chain VH in both the free and bound forms of the Fv. These measurements rule out dissociation of the Fv into monomers under the conditions of the NMR experiments, and show that further oligomerization does not occur.
Backbone relaxation measurements
Measurements of the T1, T2, and heteronuclear {1H}-15N NOE for the free Fv were made at 600 MHz, and those for the p-nitrophenol complex were made at 800 MHz. Figure 5▶ shows the values obtained for the VH and VL polypeptides of the free and bound forms of the Fv. Only well resolved cross-peaks in the 15N HSQC were used in the analysis, due to the complications introduced by resonance overlap. Of the 189 assigned backbone amides in the free Fv, 10 were excluded because of severe overlap, and 31 residues were not observed in the heteronuclear NOE experiment. A total of 148 HN vectors were used in further analysis. In the p-nitrophenol complex, 171 backbone resonances were used in the analysis of the relaxation data. Of the 195 assigned amides, 17 were excluded because of severe overlap, and four were too weak for reliable analysis in the heteronuclear NOE experiments.
Figure 5.

Per-residue plot of the T1, T2, and heteronuclear {1H}-15N NOEs measured for the Fv free (red) and in complex with p-nitrophenol (black). Axes labeled in black at the left of the figure refer to the 800 MHz data obtained for the Fv bound to p-nitrophenol. Axes labeled in red at the right of the figure refer to 600 MHz data obtained for the free Fv. The locations of the CDR loops on each chain of the heterodimer are indicated.
Diffusion tensor analysis
The inertia tensor for the free and bound forms of the Fv was calculated, based on the coordinates of the X-ray crystal structures of the free and bound Fv (Thayer et al. 1999). The values for the inertia tensor of both structures are 1.00 : 0.93 : 0.59, suggesting axial rotational anisotropy, which was accounted for in subsequent analysis of the backbone dynamics using ModelFree (Palmer 2000).
Before analysis of the diffusion tensor, residues with significant additional motions besides the overall tumbling must be eliminated. This was done using the method described by Tjandra et. al (1995), where residues with a heteronuclear NOE value lower than 0.65 were excluded, because this is an indication that these residues have high amplitude motions on a picosecond timescale.
After the first round of the diffusion tensor analysis for both the free Fv and the p-nitrophenol complex, some residues showed a poor fit in all three models (isotropic, axial, and anisotropic) resulting in a large value for χ2 (χ2 > 25). The total χ2 value for the diffusion tensor analysis decreased about 31% after removing residues Gly H26, Ala L12, Ser L14, and Ser L67 for the free Fv. Removing residues Ser H17, Lys H43, Gly H44, Ala L12, Ser L14, Ser L77, and Cys L88 for the p-nitrophenol complex resulted in a decrease of the total χ2 value of 57%. For the free Fv (600 MHz), the relaxation data were fit best by τc = 12.3 ns, D∥/D⊥ = 1.20 and θ = 11.7° (θ is the angle between the principal axis of the diffusion tensor and the principal axis of the inertia tensor). The corresponding values for the inhibitor-bound Fv (800 MHz) are: τc = 13.9 ns, D∥/D⊥ = 1.20 and θ = 8.6°.
Model-free analysis
The results of the model-free analysis of the relaxation data are presented in Figure 6▶, and these values are mapped onto the structures of the free and bound Fvs in Figure 7▶. The S2 values, which represent the nanosecond motions, indicate that the backbone of the protein is relatively rigid on a nsec timescale. The values do not differ greatly between the two forms of the protein. However, the τe (motion on a nsec timescale) and Rex (motion on a μsec-msec timescale) terms show significant differences, and where they occur in the same positions for the two forms of the protein, may differ markedly in amplitude. It should be noted that the apparent significant increase in Rex terms in the VL domain upon binding of p-nitrophenol is somewhat misleading. The resonances of the CDR loop L3 are broadened in the free form of the protein, to such an extent that relaxation data cannot reliably be obtained for these regions of the protein. This broadness is most likely due to slow timescale motions. Upon binding of p-nitrophenol, these motions are damped, to the extent that the resonances are now sharp enough to be observed. Nevertheless, motion on a msec-μsec timescale still occurs, giving rise to Rex terms for the CDR loops in the bound form.
Figure 6.

Calculated parameters S2, τe, and Rex from ModelFree 4.0 (Mandel et al. 1995) for the free (red) and p-nitrophenol-bound (black) Fv.
Figure 7.
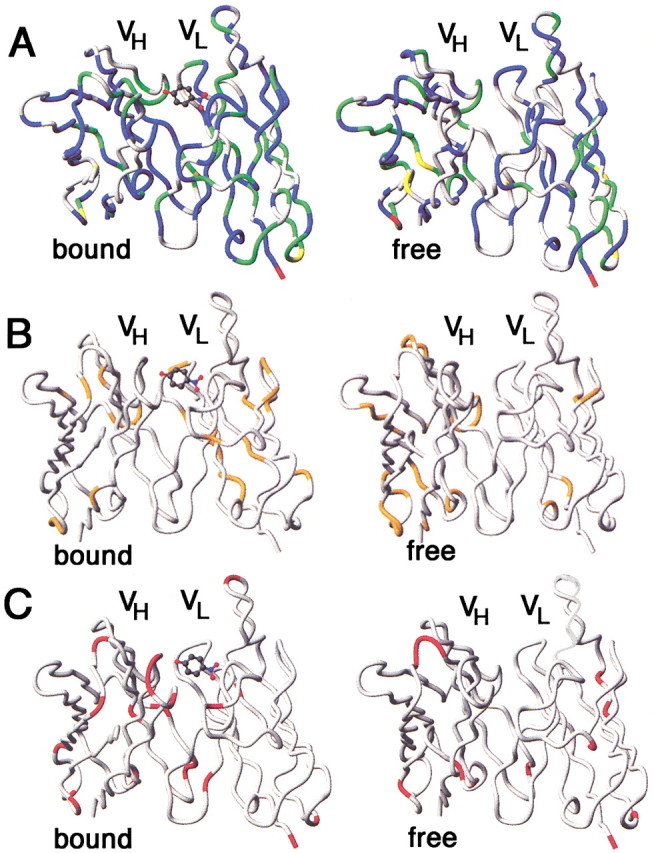
Mapping of model-free parameters onto structures of the free (right column) and p-nitrophenol-bound (left column) Fv (Thayer et al. 1999). (A) S2 (red, S2 < 0.7; yellow, 0.7 < S2 < 0.8; green, 0.8 < S2 < 0.9; blue, S2 > 0.9). (B) τe > 50 psec (orange) (C) Rex (red).
The model-free analysis of the Fv reveals the presence of different types of small-amplitude motions within local regions, even in the presence of a tightly bound inhibitor. Residues in the first loop close to the N-terminus of the VH, between the first two β-strands (residues Gly H10 to Gln H16) of both the free and bound protein showed a combination of motions on both millisecond and picosecond timescales (see Fig. 6▶). Motions on a millisecond timescale were observed in three regions of the heavy chain of the free Fv (see Fig. 6▶). One of these regions, Gly H55 to Lys H64, is part of the CDR H2. The other two regions include the residues Gln H77 to Leu H80 and Leu H82C to Ala H91. In the CDR H1 from Ser H30 to Tyr H32, only motions on a picosecond timescale were observed. Residues in CDR H3 showed no additional motions. However, in this region no peaks were observed for residues Tyr H95, Gly H96, Tyr H97, Gly H99, Asp H100, Phe H100B, and Ser H101.
In the presence of bound p-nitrophenol, two regions with motions in the pico- to nanosecond timescale were observed for the heavy chain of the Fv. One region includes residues Gln H83 to Asp H85, and the second region comprises residues Gly H98, Gly H99, and Arg H100A, which are part of CDR H3. The light chain of the Fv with bound inhibitor contains two distinct regions with additional motions. The first region consisting of residues Val L3 to Thr L7 shows additional motions on a picosecond timescale. The second region consisting of residues Arg L93 to Lys L103 shows additional motions on a millisecond timescale. Residue Thr L97 shows the highest rate, 16 s−1.
In the light chain of the free Fv, residues with additional motions are more scattered and no specific regions were observed (see Fig. 6▶). All observable residues in the three CDR regions of the VL fitted to the simplest model. The residues Gln L89, His L91, Arg L96, and Thr L97 in the CDR L3 were too weak to be included in the model-free analysis.
Discussion
Evidence for intermediate exchange in parts of the free Fv
In both the free Fv and the p-nitrophenol complex, no signals were observed for any of the backbone amides of Leu H4 to Ser H7 in the first β-strand and those of Tyr H95 to Tyr H97 in the N-terminal part of CDR H3. These regions of the VH are probably in intermediate exchange, and the lines are broadened beyond detection. There are no particular regions missing in the backbone assignments of the VL. Similar observations were reported by Freund et al. (1994) in an NMR study of the Fv of the phosphorylcholine-binding antibody McPC603. Both the CDR H3 and CDR L3 were poorly defined due to a lack of information, whereas in our case only assignments for residues of the N-terminal part of the CDR H3 are missing.
Other evidence for motions in the same region of an antibody comes from X-ray crystallographic analysis of a catalytic Fab, where weak electron density due to disorder was seen for residues H98 to H100B in CDR H3 (Buchbinder et al. 1998). In contrast, the electron density maps for the crystal structure of antibody 43C9 are well resolved (Thayer et al. 1999). In this case, crystal packing effects may restrict motions of the CDRs that are visible in solution.
Dynamics of the Fv
The model-free analysis of the free Fv shows differences in the dynamical properties between VL and VH. Compared to the light chain, the heavy chain contains more residues that fit to a model with additional motions on a millisecond timescale. The difference between the two monomers is especially noticeable when comparing the CDRs. In the VL, almost all observable residues in the CDRs fitted to the simplest model without any additional motions. Most of the observed residues in CDR H1 and H2 show additional motions on the picosecond and millisecond timescale, whereas all observed residues in CDR H3 show no additional motions. Despite the observed motions on the picosecond and millisecond timescale in both the VL and VH of the free Fv, the amplitudes of these motions are relatively small, as indicated by an overall large value for S2 (S2 > 0.8). One reason for the relative rigidity of the heterodimer could be explained by the formation of hydrogen bonds in the free Fv between residues of CDR H3 and L2 and between residues of CDR H2 and L3 (Thayer et al. 1999). The relatively high S2 values for most of the residues analyzed in the Fv dimer indicates that the amplitude of motions in the backbone is limited. Residues of the loop regions of Gly H10 to Ala H13, Ser H40 to Gly H44, and Gln H83 to Asp H86 of the Fv in the p-nitrophenol complex show additional effective correlation times for internal motions on a picosecond to nanosecond timescale and additional dynamics on a microsecond to millisecond timescale. Residues in the C-terminal part of CDR L3 and the N-terminal part of the last β-strand in the p-nitrophenol complex have high exchange rates (Figs. 6,7▶▶). In the free Fv, these residues fit to the simplest model without any additional motions. The koff rate constant for PNP is 40 s−1, which is too slow to account for the Rex terms observed in the bound form for these residues.
The only other relaxation data that have been reported for a complete Fv fragment are the heteronuclear NOE data on the phosphorylcholine-binding antibody McPC603 (Freund et al. 1994). No high-amplitude fast motions were observed in either the VH or VL, which is in good agreement with our results for the Fv 43C9. In the backbone dynamics study on the VL domain of the Fv fragment of an antidigoxin antibody (26–10 VL; Constantine et al. 1993), relatively high values for S2 were found, in good agreement with the values calculated for the VL domain of the NPN43C9 Fv. Of the 89 residues that were analyzed, 30 residues needed an additional exchange term in order to fit the relaxation data. Of the 67 residues of the free Fv 43C9, only four residues showed an additional Rex term. The large number of residues with Rex terms in the 26–10 VL is most likely the result of neglecting to calculate the parameters by fitting to an anisotropic model, as we have done for the 43C9 Fv. However, some of the regions of the 26–10 monomer that display Rex terms correspond to residues in the VL domain of the p-nitrophenol complex of Fv NPN43C9 that undergo μsec-msec time scale motions. These residues are located in the N-terminal part of the VL, the N-terminal part of CDR L1, and in the C-terminal part of CDR L3 (Fig. 7B▶).
Correlation of structural and dynamic changes upon ligand binding
Lindner et al. (1999) presented kinetic data for three catalytic antibodies with esterase activity that do not show the same severe product inhibition as the 43C9 antibody. These data suggest that some conformational flexibility provides an improved turnover rate through a mechanism termed "conformational isomerization" (Foote and Milstein 1994; Martinez-Yamout and McConnell 1994). The hypothesis is that there are multiple conformations of the antibody with different catalytic rates at pre-equilibrium, but during the catalysis only the most active conformation is retained.
In the case of Fv NPN43C9, no splitting of resonances was observed in the HSQC spectrum of the free form or the p-nitrophenol complex, indicating that multiple backbone conformations that interconvert slowly on the chemical shift timescale are absent. The NOE patterns observed in the 15N NOESY spectra of the free and bound forms of the Fv show no differences, indicating that there are no major conformational changes upon binding. If conformational isomerization is part of the catalytic process for this antibody, it must be on a fast timescale, or else it must involve only side-chain rearrangements, which are not observed in the 15N NOESY spectrum.
Residues Gln L89 to Thr L97 in the CDR L3 show the largest changes in chemical shift differences upon binding. These residues also show a significant change in the dynamics on a microsecond to millisecond timescale. In the free Fv, most of these residues were not observed in the T2 experiment and the heteronuclear NOE experiment because of the extensive line-broadening. In the p-nitrophenol complex, these residues still have more than average line-broadening but less than for the free form. This observation is an indication that the motions of the backbone in this region become more restricted upon binding of the p-nitrophenol.
Are dynamics important in catalysis by antibodies?
It is clear that the CDR loops, which form the catalytic sites of catalytic antibodies, are mobile in the absence of substrate or inhibitor and that these regions acquire more complex motions on a slower timescale upon ligand binding. It has been suggested (Cannon and Benkovic 1998; Miller and Benkovic 1998; Bruice and Benkovic 2000) that microsecond-millisecond time scale motions could be coupled to the catalytic mechanisms of enzymes, and an apparent connection has been made in the case of DHFR (Radkiewicz and Brooks 2000; Agarwal et al. 2002). For the NPN43C9 Fv, a difference is observed in the μsec-msec timescale motion in the CDR loops upon binding of inhibitor, which could be interpreted as indicating connection between polypeptide chain motions and catalysis in this system. Analysis of the X-ray structures of the free and bound Fv (Thayer et al. 1999) reveals a possible mechanism for this coupling.
Mutation of arginine H100A to a glutamine decreased the p-nitrophenol binding and increased the catalytic rate at high pH. This residue lies outside the binding pocket in the X-ray structure, and it was suggested that its effect on catalysis is due to the presence of a hydrogen bond between Arg H100A and the backbone of CDR H3 (Thayer et al. 1999). Loss of this hydrogen bond could increase the flexibility of the region, allowing more rapid loss of p-nitrophenol, and resulting in the increase in rate.
Coupling of polypeptide chain motion to catalysis remains an interesting hypothesis, which may be operative in certain enzymatic reactions, though probably not in all. Further investigation will be required to establish such connections unequivocally.
Materials and methods
Protein preparation
Protein samples uniformly labeled with 15N were used in the NMR experiments. The protein was overexpressed in Escherichia coli and isolated as described (Kroon et al. 1999). To obtain the free Fv, half of the purified Fv was taken after purification and dialyzed for 14 d against a total 28 L buffer containing 10 mM Tris and 100 mM NaCl at pH6.8 at 4°C.
The NMR samples were concentrated and exchanged into 10 mM d11 Tris, 100 mM NaCl at pH6.8 using an Amicon Centriprep membrane filter with a 3-kD cutoff. To the sample containing the complex, additional p-nitrophenol was added to obtain a final concentration of 80 μM. The final protein concentration in the NMR samples of the free Fv and the p-nitrophenol complex was 0.5 mM as determined from the A280 absorption (extinction coefficient 48,000 M−1 cm−1).
NMR experiments
The 15N-labeled samples of both the free Fv and the p-nitrophenol complex were used to collect 1H-15N HSQC, 1H-15N NOESY-HSQC, and 1H-15N TOCSY-HSQC spectra on a Bruker DRX-800 spectrometer. Both 3D data sets consist of 1024*(t1) × 256*(t2) × 128*(t3) data points. The experiments were collected with a spectral width of 7183.908 Hz (1H), 11904.762 Hz (1H), and 3846.154 Hz (15N) and the carrier at 8.4 ppm, 4.70 ppm, and 119.4 ppm, respectively. The mixing times were 100 msec in the NOESY and 35 msec in the TOCSY experiment. The 2D 15N HSQC on both samples were acquired with 2048*(t1) × 128*(t2) using a spectral width of 7183.908 Hz (1H) and 3846.154 Hz (15N) and the carrier at 8.4 ppm and 120.9 ppm, respectively. Water suppression was achieved using the ATERGATE (Piotto et al. 1992) in combination with water flip-back pulses (Grzesiek and Bax 1993).
The T1 and T2 relaxation data and steady-state heteronuclear 15N NOE data were recorded on Bruker DRX-600 and DRX-800 spectrometers, using the sensitivity-enhanced experiments described by Farrow et al. (1994). Water suppression was achieved using water flip-back pulses (Grzesiek and Bax 1993). All NMR relaxation experiments were recorded at 25.1°C calibrated using methanol. All relaxation data sets were acquired with 1024*(t1) × 256*(t2) data points and a spectral width of 4807.692 Hz (1H) and 2190.1011 Hz (15N), respectively. The carrier was set at 7.52 ppm (1H) and 118.8 ppm (15N). The same sample used for the backbone assignments of the free Fv was also used in the relaxation experiments.
The T1 relaxation rate was sampled using 10 different timepoints: 10, 80 (2x), 160, 1200, 520 (2x), 880, 2000 (2x), 3000, 4000, and 6000 msec for both the free Fv and the p-nitrophenol complex at 600 MHz. The T2 relaxation rate was sampled using 8 different timepoints: 6, 18, 34, 68 (2x), 106, 154 (2x), 210, and 322 msec for both the free Fv and the p-nitrophenol complex at 600 MHz. Data at the different time points in both T1 and T2 experiments were acquired in a random order to avoid systematic errors. For both samples, three sets of the steady-state 15N heteronuclear NOE experiments were recorded, and for each set the saturated and unsaturated data were collected in an interleaved manner.
Spectra were processed using NMRpipe/NMRDRAW (Delaglio et al. 1995) and analyzed using NMRVIEW (Johnson and Blevins 1994) on a Silicon Graphics O2 workstation.
Relaxation data analysis
The T1 and T2 values for each residue were determined using the Curvefit program (Mandel et al. 1995). Because all experiments sampled the full decay of the signals, a three-parameter fit was used on both the T1 and T2 curves. Overlapping peaks were not used in the analysis.
The relaxation data were used to determine the diffusion tensor using the crystal structures of the scFv and the scFv with bound p-nitrophenol (Thayer et al. 1999) and the quadric_diffusion program (Lee et al. 1997). Validation of the diffusion tensor calculation was performed using the program COPED (Osborne and Wright 2001). In order to exclude residues which show additional motions on a picosecond or millisecond timescale, a selection filter was applied prior to the diffusion tensor analysis (Tjandra et al. 1996).
Model-free analysis
The relaxation data were fit using the Model-free4.0 program (Mandel et al. 1995). This program is based upon the model-free formalism (Lipari and Szabo 1982a,b; Clore et al. 1990) and uses the correlation between relaxation rates and spectral density functions as described by Abragam (1961). During the analysis, the nitrogen-hydrogen bond length was set to 1.02 Å and the chemical shift anisotropy for the 15N nucleus was set to −170.0 ppm (Tjandra et al. 1996). A total of five different models were used during the analysis (S2, S2+τe, S2+Rex, S2+τe+Rex, and S2s+S2f+τe) as described by Mandel et al. 1995. The goodness of fit was determined by the value of the χ2 function with a critical value of α = 0.05. An F-test was applied with a critical value of α = 0.15 to decide whether the improvement of a fit using an extended model was statistically valid.
Diffusion experiments
The self-diffusion (Ds) coefficient for the free Fv and the Fv with p-nitrophenol was determined using a series of 1D water-sLED experiments (Altieri et al. 1995) with different gradient strengths (6.8, 8.9, 10.9, 12.9, 14.9, 17.0, 19.0, 21.0, 23.0, 25.0, and 27.1 G/cm). The values of Ds for the different Fv samples were compared to that determined for a 2-mM lysozyme sample, as well as with literature values (Altieri et al. 1995). The experiments were performed on a Bruker DRX-600 spectrometer at 25°C using the same Fv samples used in the relaxation experiments.
Acknowledgments
We thank Professor Stephen Benkovic for numerous helpful discussions, and for providing the original clone for the NPN43C9 Fv; Drs. Brendan Duggan, Ishwar Radhakrishnan, Michael Osborne, John Viles, James Huntley, Brian Lee, and Eduardo Zaborowski for helpful discussions and for making available programs and scripts; John Chung for help with NMR spectrometer setup; and Linda Tennant for technical assistance. This work was supported by grant CA 27489 from the NIH. The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0243303.
References
- Abragam, A. 1961. Principles of nuclear magnetism, Clarendon Press, Oxford.
- Agarwal, P.K., Billeter, S.R., Rajagopalan, P.T.R., Benkovic, S.J., and Hammes-Schiffer, S. 2002. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. 99 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri, A.S., Hinton, D.P., and Byrd, R.A. 1995. Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 117 7566–7567. [Google Scholar]
- Bruice, T.C. and Benkovic, S.J. 2000. Chemical basis for enzyme catalysis. Biochemistry 39 6267–6274. [DOI] [PubMed] [Google Scholar]
- Buchbinder, J.L., Stephenson, R.C., Scanlan, T.S., and Fletterick, R.J. 1998. A comparison of the crystallographic structures of two catalytic antibodies with esterase activity. J. Mol. Biol. 282 1033–1041. [DOI] [PubMed] [Google Scholar]
- Cannon, W.R. and Benkovic, S.J. 1998. Solvation, reorganization energy, and biological catalysis. J. Biol. Chem. 273 26257–26260. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Szabo, A., Bax, A., Kay, L.E., Driscoll, P.C., and Gronenborn, A.M. 1990. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 112 4989–4991. [Google Scholar]
- Constantine, K.L., Friedrichs, M.S., Goldfarb, V., Jeffrey, P.D., Sheriff, S., and Mueller, L. 1993. Characterization of the backbone dynamics of an antidigoxin antibody VL domain by inverse detected 1H-15N NMR: Comparisons with X-ray data for the fab. Proteins 15 290–311. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Guang, Z., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Foote, J. and Milstein, C. 1994. Conformational isomerism and the diversity of antibodies. Proc. Natl. Acad. Sci. 91 10370–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund, C., Ross, A., Plückthun, A., and Holak, T.A. 1994. Structural and dynamic properties of the Fv fragment and the single-chain Fv fragment of an antibody in solution investigated by heteronuclear three-dimensional NMR spectroscopy. Biochemistry 33 3296–3303. [DOI] [PubMed] [Google Scholar]
- Gibbs, R.A., Benkovic, P.A., Janda, K.D., Lerner, R.A., and Benkovic, S.J. 1992. Substituent effects on an antibody-catalyzed hydrolysis of phenyl esters: Further evidence for an acyl-antibody intermediate. J. Am. Chem. Soc. 114 3528–3534. [Google Scholar]
- Gigant, B., Charbonnier, J., Eshhar, Z., Green, B.S., and Knossow, M. 1998. Crossreactivity, efficiency and catalytic specificity of an esterase-like antibody. J. Mol. Biol. 284 741–750. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1993. The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115 12593–12594. [Google Scholar]
- Janda, K.D., Schloeder, D., Benkovic, S.J., and Lerner, R.A. 1988. Induction of an antibody that catalyzes the hydrolysis of an amide bond. Science 241 1188–1191. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 604–613. [DOI] [PubMed] [Google Scholar]
- Kabat, E.A., Wu, T.T., Perry, H.M., Gottesman, K.S., and Foeller, C. 1991. Sequences of proteins of immunological interest, National Institutes of Health, Bethesda, MD.
- Krebs, J.F., Siuzdak, G., Dyson, H.J., Stewart, J.D., and Benkovic, S.J. 1995. Detection of a catalytic antibody species acylated at the active site by electrospray mass spectrometry. Biochemistry 34 720–723. [DOI] [PubMed] [Google Scholar]
- Kroon, G.J.A., Martinez-Yamout, M.A., Krebs, J.F., Chung, J., Dyson, H.J., and Wright, P.E. 1999. Backbone resonance assignments for the Fv fragment of the catalytic antibody NPN43C9 with bound p-nitrophenol. J. Biomol. NMR 15 83–84. [DOI] [PubMed] [Google Scholar]
- Lee, L.K., Rance, M., Chazin, W.J., and Palmer, A.G. 1997. Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C α nuclear spin relaxation. J. Biomol. NMR. 9 287–298. [DOI] [PubMed] [Google Scholar]
- Lerner, R.A., Benkovic, S.J., and Schultz, P.G. 1991. At the crossroads of chemistry and immunology: Catalytic antibodies. Science 252 659–667. [DOI] [PubMed] [Google Scholar]
- Lindner, A.B., Eshhar, Z., and Tawfik, D.S. 1999. Conformational changes affect binding and catalysis by ester-hydrolysing antibodies. J. Mol. Biol. 285 421–430. [DOI] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- ———. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104 4559–4570. [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer, A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246 144–163. [DOI] [PubMed] [Google Scholar]
- Martinez-Yamout, M. and McConnell, H.M. 1994. Site-directed mutagenesis and 1H nuclear magnetic resonance of an antidinitrophenyl spin label antibody. J. Mol. Biol. 244 301–318. [DOI] [PubMed] [Google Scholar]
- Miller, G.P. and Benkovic, S.J. 1998. Stretching exercises—Flexibility in dihydrofolate reductase catalysis. Chem. Biol. 5 105–113. [DOI] [PubMed] [Google Scholar]
- Osborne, M.J. and Wright, P.E. 2001. Anisotropic rotational diffusion in model-free analysis for a ternary-DHFR complex. J. Biomol. NMR 19 209–230. [DOI] [PubMed] [Google Scholar]
- Palmer, A.G. 2000. ModelFree 4.10. http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/software.html
- Piotto, M., Saudek, V., and Sklenár, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Radkiewicz, J.L. and Brooks III, C.L. 2000. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J. Am. Chem. Soc. 122 225–231. [Google Scholar]
- Schultz, P.G. and Lerner, R.A. 1993. Antibody catalysis of difficult chemical transformations. Accounts Chem. Res. 26 391–395. [Google Scholar]
- ———. 1995. From molecular diversity to catalysis: Lessons from the immune system. Science 269 1835–1842. [DOI] [PubMed] [Google Scholar]
- Stanfield, R.L., Takimoto-Kamimura, M., Rini, J.M., Profy, A.T., and Wilson, I.A. 1993. Major antigen-induced domain rearrangements in an antibody. Structure 1 83–93. [DOI] [PubMed] [Google Scholar]
- Thayer, M.M., Olender, E.H., Arvai, A.S., Koike, C.K., Canestrelli, I.L., Stewart, J.D., Benkovic, S.J., Getzoff, E.D., and Roberts, V.A. 1999. Structural basis for amide hydrolysis catalyzed by the 43C9 antibody. J. Mol. Biol. 291 329–345. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Feller, S.E., Pastor, R.W., and Bax, A. 1995. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117 12562–12566. [Google Scholar]
- Tjandra, N., Szabo, A., and Bax, A. 1996. Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J. Am. Chem. Soc. 118 6986–6991. [Google Scholar]