

Abstract
Glutamine synthetase (GS) is the key enzyme responsible for the primary assimilation of ammonium in all living organisms, and it catalyses the synthesis of glutamine from glutamic acid, ATP, and ammonium. One of the recently discovered mechanisms of GS regulation involves protein-protein interactions with a small 65-residue-long protein named IF7. Here, we study the structure and stability of IF7 and its binding properties to GS, by using several biophysical techniques (fluorescence, circular dichroism, Fourier transform infrared and nuclear magnetic resonance spectroscopies, and gel filtration chromatography) which provide complementary structural information. The findings show that IF7 has a small amount of residual secondary structure, but lacks a well defined tertiary structure, and is not compact. Thus, all of the studies indicate that IF7 is a "natively unfolded" protein. The binding of IF7 to GS, its natural binding partner, occurs with an apparent dissociation constant of KD = 0.3 ± 0.1 μM, as measured by fluorescence. We discuss the implications for the GS regulation mechanisms of IF7 being unfolded.
Keywords: Inactivating factor, structure, glutamine synthetase, "natively unfolded" protein, intrinsic disorder
A fundamental question in cellular physiology is how cells are able to recognize and respond to changes in their environment. To answer that question, it is necessary to identify not only the physiological signal, but also the protein(s) that senses and induces the responses to that signal. Different signal transduction pathways operate in bacteria to regulate gene expression. This regulation occurs at the transcriptional as well as the posttranscriptional levels. In some cases, a metabolite functions as the signal that triggers and alters gene expression by interacting directly with a transcription factor. However, protein-protein interaction mechanisms occur frequently in the regulatory systems.
The response of bacteria to nitrogen limitation represents a paradigm of those regulatory systems (Merrick and Edwards 1995; Magasanik 1996). In bacteria, glutamine and 2-oxoglutarate are the most important molecules involved in nitrogen sensing. The intracellular concentration of these metabolites, and also the ratio between them, change depending on nitrogen nutritional conditions (Hu et al. 1999). Glutamine synthetase (GS), the enzyme that catalyses the synthesis of glutamine from glutamic acid, ATP, and ammonium, is then also subject to regulation depending on nitrogen availability. GS type I, the most common type found in prokaryotes, is a large protein with a molecular weight of about 600,000 Da. It consists of 12 identical subunits arranged in two superimposed hexagonal rings (Almassy et al. 1986; Yamashita et al. 1989; Liaw et al. 1993). When nitrogen is abundant, the GS activity is down-regulated; conversely, as the nitrogen level lowers, the GS activity increases. This regulation occurs by the following mechanisms: feedback inhibition of the activity, reversible covalent modification of the enzyme, and transcriptional regulation of the structural gene (Ritzer 1996). We showed that cyanobacterial GS from Synechocystis sp. PCC6803 is controlled posttranslationally by a mechanism that involves the interaction of the protein with two inhibitory proteins: a 65-residue-long protein named IF7, and a 149-residue-long protein named IF17 (García-Domínguez et al. 1999). The presence of either of the two proteins is sufficient per se for GS inactivation in vitro, but the effects of both proteins in vivo seem to be cumulative (García-Domínguez et al. 1999). IF7 and IF17 show sequence similarities to each other, and the theoretical predictions suggest the presence of an α-helical structure in both proteins. Because a protein homologous to IF7 is also present in Anabaena sp. strain PCC7120 (García-Domínguez et al. 1999), the regulation mechanism of GS by IF7 must be present in other cyanobacteria, and deserves to be investigated. Furthermore, the small size of IF7 makes it a suitable model for nuclear magnetic resonance (NMR) studies and biophysical characterization.
Here we studied the structure, conformational propensities, and stability of IF7 using several biophysical techniques, namely, fluorescence, circular dichroism (CD), Fourier transform infrared spectroscopy (FTIR), NMR spectroscopy, and gel filtration chromatography. The near-UV CD, fluorescence, and NMR experiments show that in the explored pH range (pH 3–12), IF7 lacks a significant amount of tertiary structure. In addition, the gel filtration experiments indicate that IF7 is not compact and has a well hydrated core. However, there is evidence of residual secondary structure, as concluded by the far-UV CD and FTIR experiments. Thus, IF7 shows the features of a "natively unfolded" protein. Finally, we describe the binding of IF7 to GS of cyanobacterium Synechocystis sp. PCC6803, measured by fluorescence. Binding occurs with a dissociation constant of KD = 0.3 ± 0.1 μM. The importance of IF7 being unfolded for the GS regulation is discussed.
Results
Structure and stability of IF7
We used several biophysical probes which provide complementary information about the structure of IF7.
Fluorescence experiments
Steady-state fluorescence.
We have used fluorescence spectra to map any change in the tertiary structure of the protein upon pH (Schmid 1997). IF7 has a sole tryptophan at position 42 and one tyrosine at position 57. The emission fluorescence spectrum of IF7 at pH 6.5, either by excitation at 280 nm or 295 nm, is red-shifted, with a maximum at 354 nm. This indicates that the tryptophan is solvent-exposed. The shape of the spectra and maximum wavelengths did not vary as the pH changed from 3 to 12 (data not shown).
Examination of tryptophan exposure by fluorescence quenching.
To further examine whether the tertiary structure around Trp42 and Tyr57 is absent at pH 6.5, we studied iodide quenching by excitation at 280 nm (Table 1▶). As can be observed, the Ksv parameter remained essentially constant under all conditions explored, in both the absence and the presence of any chemical denaturant, suggesting that the aromatic rings were solvent-exposed.
Table 1.
Quenching parameters in KIa
| Conditions | KSV (M−1) |
|---|---|
| pH 6.5 | 3.6 ± 0.3 |
| 6 M GdmCl | 4.0 ± 0.2 |
| 6 M urea | 4.4 ± 0.3 |
a Errors are data fit errors to Eq. 1. The constants were obtained by fitting of fluorescence intensity at 338 nm versus concentration of quenching agent (similar constants were obtained by fitting the intensities at 335, 336, and 337 nm; data not shown). Experiments were carried out at 298 K, pH 6.5, 10 mM phosphate buffer.
The presence of KCl (see Materials and Methods) did not modify the structure of IF7, as can be concluded from the following findings: (1) The positions of the maxima wavelengths in the fluorescence spectra during the quenching experiment did not change, and (2) the shape and the ellipticity of the CD spectra carried out in the KCl concentration range of 0–1 M did not change significantly (data not shown).
Chemical denaturation.
Chemical denaturation in the presence of GdmCl showed a gradual increase in the fluorescence as the concentration of denaturant increased, with small gradual changes in the maxima of the spectra (data not shown). This linear behavior is expected for solvent-exposed aromatic rings (Schmid 1997).
ANS binding.
ANS binding at pH 6.5 was used to monitor the presence of clustered hydrophobic regions in IF7 and then the existence of partially folded conformations (Semisotnov et al. 1991). The spectrum of ANS in the presence of protein showed a maximum at 480 nm, as in the spectrum of ANS alone. Changes in the fluorescence intensity of the ANS spectrum were not observed when IF7 was added (data not shown). This suggests that no exposure of any clustered hydrophobic region occurred in IF7 at pH 6.5. Similar results were obtained at other pH values between 3 and 12.
Circular dichroism experiments
Far-UV CD.
We used far-UV CD in the analysis of the unfolding of IF7 as a spectroscopic probe that is sensitive to protein secondary structure (Woody 1995; Kelly and Price 2000). The CD spectrum of IF7 at any pH at 298 K showed a minimum negative ellipticity at ∼200 nm, characteristic of random coil or denatured proteins (Fig. 1A▶; Cantor and Schimmel 1980; Kelly and Price 2000). There was a small shoulder at 222 nm, which suggests the presence of α-helix- or turn-like conformations; however, the presence of aromatic signals cannot be ruled out (Cantor and Schimmel 1980). The estimated population of α-helix- or turn-like structure from Equation 4 below is 13%. The same shape was observed at low temperature (283 K).
Figure 1.

Far-UV CD of IF7. (A) Spectrum of IF7 at pH 6.5 (dotted line) and at 6 M GdmCl, pH 6.5 (continuous line) at 298 K. (B) Change in the ellipticity at 222 nm upon GdmCl concentration changes at 298 K. (C) Changes in the spectra at 222 nm upon pH changes at 298 K. The continuous line is the fitting to Equation 6; see text for details. (D) Thermal denaturation profiles at pH 6.5 followed by the changes in the ellipticity at 222 nm. The conditions were 20–30 μM of protein; buffer concentration was 10 mM in all cases. Spectra were acquired in 0.1-cm-pathlength cells.
Chemical- and pH-denaturation.
The ellipticity at 222 nm decreased (more positive values) gradually as the concentration of GdmCl was increased (Fig. 1B▶), indicating that the denaturation was a noncooperative process. At high GdmCl concentrations, the CD spectra showed a positive band around 220 nm (Fig. 1A▶), as could be expected in completely random-coil proteins (Cantor and Schimmel 1980; Kelly and Price 2000). These results further support the previous suggestion that IF7, under native conditions, could have some residual secondary structure, although the general shape of the spectra corresponds to that of random conformations.
Urea denaturation experiments showed the same behavior as that described for GdmCl (data not shown). Both chemical denaturation processes were fully reversible.
The shape of the spectrum did not change over the pH range explored. There was, however, a sigmoidal change in the ellipticity at 222 nm as the pH increased (Fig. 1C▶), towards more negative ellipticity values. The apparent pKa of that transition at 222 nm was 6.6 ± 0.1, which is close to the titration midpoint of solvent-exposed histidine residues (Cantor and Schimmel 1980). The same pKa values were calculated by observing the ellipticity at other wavelengths (data not shown).
Near-UV.
We used near-UV CD to detect possible changes in the asymmetric environment of aromatic residues (Cantor and Schimmel 1980; Kelly and Price 2000). The near-UV of IF7 was very weak, and there were no intense bands. There are two possible explanations: First, the absence of a near-UV signal could be because of the absence of an asymmetric environment for the aromatic residues, and thus it would further support the previous suggestions of the lack of a well defined tertiary structure in IF7; alternatively, the absence of a near-UV signal could be due to the low content of aromatic residues (data not shown). We favor the first explanation because of the agreement with the results of the other biophysical probes.
Thermal denaturation experiments.
To further investigate the possible presence of residual secondary structure in IF7, we carried out thermal denaturation experiments followed at 222 nm. The ellipticity at this wavelength did not change in a sigmoidal fashion; rather, it showed a linear behavior until it reached a plateau at high temperatures (Fig. 1D▶). This suggests absence of cooperativity, as expected for a noncompact structure. The denaturation process was fully reversible.
Fourier transform infrared spectroscopy
Steady-state FTIR.
FTIR is a powerful method for investigation of secondary protein structure. The main advantage in comparison with CD and fluorescence is that FTIR is much more sensitive to the presence of β-structure or random-coil. In the case of proteins, structural information can be obtained by analyzing the amide I region of the spectrum (1700–1600 cm−1). The absorbance of this band is mainly because of the stretching vibration of the carbonyl peptide bond, whose frequency is highly sensitive to hydrogen bonding and thus to protein secondary structure (Surewicz and Mantsch 1988). We acquired the experiments at this low temperature to stabilize the possible residual secondary structure observed by CD (see above). The band is centered near 1644 cm−1, which is characteristic of nonordered conformations. Amide I band analysis (see Materials and Methods) showed maxima centered at 1695, 1683, 1672, 1657, 1644, 1628, 1615, and 1609 cm−1. The 1609 and 1615 cm−1 components correspond to side chain vibrations, and the other maxima are assigned to vibration of groups involved in different secondary structural motifs (Surewicz and Mantsch 1988); the 1628 cm−1 band is assigned to (π, 0) β-sheet structure (Byler and Susi 1986); the 1644 cm−1 band is assigned to random-coil structure (Arrondo and Goñi 1999); the 1657 cm−1 is assigned to α-helix or disordered structure (Braiman and Rothschild 1988; Denning et al. 2002); the 1672 cm−1 component is assigned to turns and loops (Byler and Susi 1986), and also to (0, π) β-sheet vibration band (Arrondo et al. 1987; Denning et al. 2002); the 1683 cm−1 band includes contributions from turns; and the origin of the band at 1695 cm−1 is uncertain, but it could be because of a turn-like conformation (Arrondo et al. 1987). The percentages of secondary structure calculated from the area of the fitted bands are in Table 2▶. The determination of the percentage of secondary structure by deconvolution of the FTIR amide I band has been used for a long time. It has proved to be a very robust method by comparing the calculated percentage of structure obtained by FTIR with that obtained by X-ray in several model proteins (Byler and Susi 1986). In all cases, the results from both techniques are in good general agreement.
Table 2.
Secondary structural analysis of IF7 as determined by FTIRa
| Wavenumber (cm−1) | Structural assignmentb | % of total secondary structure |
|---|---|---|
| 1683 | Turns | 2 |
| 1672 | Turns/loops/(0, π) β-sheet | 21 |
| 1657 | Loops/disordered/α-helixc | 17 |
| 1644 | Random-coil | 38 |
| 1628 | β-sheet MT, 01 | 22 |
a Errors in the wavenumber are estimated to be ± 2 cm−1.
b There are three more bands, which have not been indicated in the table. The first one centered at 1695 cm−1, whose assignment is uncertain (its contribution to the total area of the amide I band is less than the 1%). The other two bands, at 1609 and 1615 cm−1, are assigned to side chains; they account for 5% of the whole area of the amide I band. The percentages of secondary structure on the third column of the table do not take into account these three bands.
c The assignment of this band is uncertain (see text for details).
No band was observed in the amide II region (because of the stretching of the amide protons) 5 min after the protein was dissolved in deuterated water (hydrogen/deuterium exchange experiments). This suggests, as was shown in the NMR experiments (see below), that no stable hydrogen bonds are present in IF7.
Thermal denaturation FTIR.
The thermal denaturation of IF7 showed basically the same behavior observed in the CD experiments, but it was irreversible. This is probably because of aggregation or chemical modification (at high temperatures) of the protein because of the high concentrations used (data not shown). Aggregation problems because of the lyophilization process during FTIR sample preparation can be ruled out because the shapes of the NMR spectra of lyophilized protein and those of concentrated solutions are the same (see below).
Nuclear magnetic resonance
NMR can give information about the general fold of a polypeptide chain in solution at the residue level. The one-dimensional NMR spectrum of IF7 at 298 K showed a slight degree of chemical shift dispersion: The amide, the aromatic, and the methyl protons (Fig. 2▶) were clustered in those regions expected for random coil proteins (Wüthrich 1986), namely, between 8.0 and 8.7 ppm (for the amide signals), between 6.8 and 7.5 ppm (for the aromatic signals), and between 0.8 and 1.0 ppm (for the methyl protons). Further, the proton belonging to the indole moiety of the sole tryptophan residue appeared at 10.23 ppm, close to the value observed in disordered polypeptides (10.22 ppm; Fig. 2A▶; Wüthrich 1986). The line widths of amide region protons were not significantly broader than those of proteins with similar size, suggesting that the protein is a monomer even at 1.5 mM. A similar spectrum was observed at 278 K (data not shown).
Figure 2.

NMR spectrum of IF7. Amide (A) and methyl (B) region of IF7. Conditions were 1.5 mM protein, phosphate buffer (10 mM), pH 6.5, 298 K.
In the hydrogen/deuterium exchange experiment (data not shown), all of the amide protons disappeared within 5 min. Only the aromatic protons corresponding to Phe17, Phe41, Phe53, Trp42, Tyr57, His14, and His15 could be observed. This suggests that no stable hydrogen-bonding structure is present in IF7.
Gel filtration experiments and shape parameters
IF7 eluted at pH 6.5 at 14.18 mL, which yields a Stokes radius determined from Equation 8 of 15.6 Å (Fig. 3▶). Because the inherent precision of molecular radius determinations of globular proteins by gel partitioning methods has been shown to be ± 0.2 Å (Darlin et al. 2000), it can be concluded that the measured value is larger than that expected from a 65-residue-long globular protein (13.7 Å, obtained from the linear relationship between the molecular weight of the standard globular proteins and their Stokes radii; Fig. 3A▶; Creighton 1993). There are two possible explanations for this result: Either native IF7 is an oligomer, or it has an elongated shape relative to the globular proteins used as standards. We can rule out the first explanation because (1) the presence of oligomer populations would result in elution volumes close to those observed for the higher-molecular-weight standards, (2) cross-linking experiments of IF7 did not show the presence of dimers at the concentrations used in the gel filtration experiments (data not shown), and (3) the NMR experiments (see above) indicated that the protein is a monomer even at 1.5 mM. Thus, we can conclude that IF7 is not an oligomer, and it probably has an elongated shape.
Figure 3.

Hydrodynamic properties of IF7. (A) Determination of the Stokes radius by gel filtration chromatography on an HR Superdex G75. The elution volume of IF7 is indicated by an arrow and a square (see text for details). The numeration corresponds respectively to the elution volumes of ribonuclease A (1), chymotrypsinogen A (2), ovalbumin (3), and albumin (4). (B) Gel filtration elution peak of IF7 in phosphate buffer, pH 6.5 (10 mM) at 298 K; flow, 1mL/min.
We can further elaborate on the IF7 shape by using the gel filtration methods to calculate the frictional ratio, f/f0, a measure of the departure of the molecule from spherical shape (Siegel and Monty 1966). Globular proteins have usually frictional ratios close to 1, but elongated or nonglobular proteins have larger ratios (Siegel and Monty 1966). The molecular weight of IF7 is 7475.4 Da and 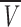 = 0.70456 cm3/g, as calculated from amino acid composition (Creighton 1993). Then, Equation 12 leads to ro = 12.7 Å. This results in a frictional coefficient f/f0 = 1.2, consistent with an extended, elongated, nonspherical conformation.
= 0.70456 cm3/g, as calculated from amino acid composition (Creighton 1993). Then, Equation 12 leads to ro = 12.7 Å. This results in a frictional coefficient f/f0 = 1.2, consistent with an extended, elongated, nonspherical conformation.
The wmax for globular proteins is usually 0.3 g of water per gram of protein or even lower; higher values indicate that the protein is more hydrated and that there is a larger number of solvent-exposed regions. The wmax of IF7, obtained from Equation 13, is 0.6 g of water per gram of IF7, suggesting that the protein is highly hydrated.
In conclusion, all of the hydrodynamic parameters obtained from the gel filtration experiments indicate that IF7 has an elongated shape and that it is very well hydrated.
Structure of GS
To characterize the binding of IF7 to GS, we first examined the spectroscopic properties of GS.
Fluorescence measurements
The maxima of the fluorescence spectra of GS was 337 nm (data not shown). This maximum moved to red-shift wavelengths (350 nm) and the fluorescence intensity decreased as the concentration of either GdmCl or urea increased. The measured denaturation midpoint, when using GdmCl as denaturant, was smaller (data not shown). As could be expected from an oligomer, the midpoint of the chemical denaturation increased as the concentration of protein increased, changing in any case with a sigmoidal behavior (data not shown).
CD measurements
The shape of the CD spectrum of GS was typical of an α-helix conformation (Cantor and Schimmel 1980; Kelly and Price 2000), as could be expected from the X-ray structure of other GS proteins (Almassy et al. 1986; Yamashita et al. 1989; Liaw et al. 1993). There were intense minima at 222 nm and 208 nm. The shape of the spectrum did not change in the concentration range explored, 5–50 μM (data not shown).
Thus, the results of both techniques indicate unambiguously that GS is well folded.
Determination of the GS to IF7 binding constant
The value of KD obtained by fluorescence (see Materials and Methods) at 1.5 μM of GS is 0.3 ± 0.1 μM. At concentrations lower than 0.5 μM of GS, the results could not be analyzed reliably because of the limitations of the technique. Binding curves obtained at GS concentrations of 1 μM yielded the same KD (data not shown). It is interesting to note here that the use of IF7 concentrations four times or more larger than the GS concentration used yielded sample precipitation during the binding experiments. These results are in agreement with functional assays, where it has been shown that the complete inactivation of GS occurs when the concentration of IF7 is four times larger than the GS concentrations (García-Domínguez et al. 1999). The complete GS inactivation could implicate the precipitation of IF7, GS, and/or the complex at high IF7 concentrations, as concluded from the absorbance measurements. Neither precipitated forms nor aggregated species were found at lower IF7 concentrations. We were not able to determine reliably the value of the number of interacting sites, since the fitting calculations with different n values yielded the same dissociation constant and showed a good statistical χ2 parameter.
Discussion
IF7 is a "natively unfolded" protein
Structural features
All of the biophysical studies in this work show that IF7 lacks a well defined tertiary structure and is mainly a disordered protein, with neither stable hydrogen bonds nor a well formed core. The lack of dispersion of the amide signals in the NMR spectrum and the absence of cooperativity during the thermal and chemical denaturation experiments (followed by CD, FTIR, and fluorescence) also support that the tertiary structure of IF7, if any, is very weak and no hydrophobic core is formed.
However, there is evidence of residual secondary structure, as concluded from the FTIR and CD experiments. Far-UV CD spectra show the presence of secondary structure as supported by the shoulder at 222 nm (Fig. 1A▶), which disappears at high denaturant concentrations (Fig. 1B▶). This shoulder could be because of either the presence of residual helical- or turn-like structure [13%, as calculated by Equation 4] or aromatic residues (Cantor and Schimmel 1980; Kelly and Price 2000). Moreover, FTIR also suggests the existence of turn-like conformations, although the percentage (∼21%) seems to be higher than that observed by CD (Table 2▶). The differences between the values reported by both techniques could be because of (1) the deconvolution procedures (in the case of FTIR), (2) the presence of aromatic residues (CD), or (3) the empirical character of Equation 5 (CD). Because theoretical predictions suggest that IF7 has a long α-helix comprising residues 3–45 (García-Domínguez et al. 1999), part of this helix could be populated, and its presence could be reported by FTIR and CD measurements. It is worth noting that FTIR also predicts 22% β-sheet (Table 2▶), which could not be confirmed by CD, because of its lower sensitivity to this type of secondary structure.
The described structural features of IF7 are not unusual, rather they are common to a class of proteins known as "natively unfolded" proteins. In brief, those features include (1) absence of tertiary structure, and most of the secondary structure, (2) lack of a tightly packed core (i.e., globularity), and (3) a high degree of flexibility (Dunker et al. 2002; Dyson and Wright 2002; Uversky 2002). Here, we show that IF7 is nonglobular and devoid of tertiary structure, thus fulfilling the first criterion for this class of proteins. To address the compactness of IF7, we must use the gel filtration results. The experimentally determined Stokes radius for IF7 is 15.6 Å, suggesting that IF7 is larger than expected for a protein of its size. The hydration results indicate that IF7 has a well hydrated core.
To address the issue of the flexibility of IF7, we used two different approaches. First we used the Vihinen scale (Vihinen et al. 1994), which reflects the protein flexibility by considering a flexibility scale for each amino acid. According to this scale, the most rigid amino acids are W, C, F, I, V, Y, L, and M. In the IF7 sequence (Fig. 4▶), these amino acids account for only 25% of the total sequence; conversely, highly flexible amino acids according to the Vihinen scale (G, Q, S, N, E, K, D, and P) are overrepresented in IF7 (48%). Secondly, we used sensitivity to proteolytic digestion. IF7 was sensitive to low concentrations of proteinase K (M. Muro-Pastor, J. Reyes, and F. Barrera, unpubl.) and trypsin (F. Barrera and J. Neira, unpubl.), thus suggesting a quite flexible structure as is the case with other "natively unfolded" proteins (Denning et al. 2002).
Figure 4.

Sequence of IF7. The amino acid sequence of IF7 is given in the one-letter code. Residues which are underrepresented in "natively unfolded" proteins and those overrepresented, according to the findings of Dunker et al. (Li et al. 1999, 2000; Romero et al. 2001; Dunker et al. 2002) are in blue and in red, respectively. The rest of the residues are in black.
Then, based on structural features alone, we can conclude that IF7 behaves as a "natively unfolded" protein; that is, IF7 is essentially disordered in solution, lacking a well fixed tertiary structure, and containing weak and flickering secondary structure.
Amino acid sequence features
"Natively unfolded" proteins also share sequence peculiarities, such as (1) numerous uncompensated charged groups, which results in a large net charge at neutral pH; (2) a low content of hydrophobic amino acid residues; and (3) a large number of the E, K, R, G, Q, S, P, and A amino acids and a low number of I, L, V, W, F, Y, C, and N, as shown by Dunker and coworkers (Li et al. 1999, 2000; Romero et al. 2001) and others (Tompa 2002). It is interesting to note here that the set of residues that is underrepresented in "natively unfolded" proteins, according to the Dunker criteria, is essentially the same (except for the M and N) as the set of rigid residues found in the Vihinen scale (Vihinen et al. 1994). The accumulation of both sets of amino acids in "natively unfolded" proteins accounts for the general low hydrophobicity and the high net charge observed by Uversky and coworkers in their experimental survey of those proteins (Uversky 2002); their conclusions indicate that hydrophobicity and net charge are responsible for the compactness of any polypeptide chain.
IF7 also shows all of the sequence features of "natively unfolded" proteins. Its theoretical isoelectric point is 10.9, which corresponds to a net mean charge of 4 at neutral pH. Its content of hydrophobic residues is very low, yielding an average of hydropathicity of −0.8 (Kyte and Doolittle 1982). In addition, residues I, L, V, W, F, Y, C, and N account for 21% of the total number of amino acids, and E, K, R, G, Q, S, P, and A account for 58% of the total number of residues (A, Q, and S are the most frequently appearing residues of the protein—11%, 12%, and 12%, respectively; Fig. 4▶).
In conclusion, considering the conformational propensities and sequence characteristics of IF7, it can be classified unambiguously as a "natively unfolded" protein.
Binding of IF7 to GS
The finding that GS is reactivated (i.e., IF7 is no longer bound) by increasing the pH up to 9 (Mérida et al. 1991; García-Domínguez et al. 1999) underlines that the electrostatic factor is responsible for the binding. The electrostatic factor seems, with the low hydrophobicity, responsible for the "natively unfolded" state of unbound IF7 (see above). An excessive accumulation of negative charges in GS and IF7 at high pHs does probably make IF7 detach. We do not know whether the structure of bound IF7, if any, changes with pH, but the ellipticity of unbound (and "natively unfolded") IF7 does change (Fig. 1C▶). However, it remains to be established whether a change in the environment of an aromatic residue or a proper conformational change is responsible for the ellipticity changes in unbound IF7 (Fig. 1C▶). Even if a conformational change is occurring, we cannot conclude, based exclusively on these data, that stabilization of residual structure in unbound IF7 promotes unzippering of the complex in light of the shift in equilibrium conditions.
The binding between GS and IF7 occurs with an apparent dissociation constant of 0.3 ± 0.1 μM, which corresponds to a ΔG of −8.9 ± 0.3 kcal mole−1 at 298 K. Because we have shown by gel shift experiments that IF17 has a higher affinity for GS than does IF7 (García-Domínguez et al. 1999), the measured KD value sets a limit for the affinity of IF17. As no affinity constant was determined from the activity measurements (García-Domínguez et al. 1999), it has not been possible to compare the value of the KD measured here. Despite our attempts, we have not been able to determine exactly the stoichiometry of the GS-IF7 complex. Nor could we determine unambiguously whether IF7 folds upon binding to GS, although CD experiments suggest a change in the conformational propensities of one, if not both, molecules upon binding (data not shown).
Finally, we may speculate over the advantages of a disordered IF7 relative to a folded protein. It has been suggested that if the protein is unfolded there are two clear advantages (Dunker et al. 2002). First, the free energy arising from the protein-ligand contacts would be reduced by the free energy needed to fold the intrinsic disorder. And secondly, if a protein is disordered, a particular molecule could bind to different partners with only small structural accommodations. In vivo, the effects of IF7 and IF17 are cumulative, with different inactivation kinetics, but the presence of either of the two proteins is sufficient per se for GS inactivation in vitro (García-Domínguez et al. 1999). If IF7 were unfolded, it would have enough flexibility to form a loose overall assembly with either the isolated GS or the formed GS-IF17 complex. In this latter case, IF7 could undergo different rounds of disorder–order transitions to strengthen the overall complex. Experiments are underway in our laboratories to elucidate the importance of IF17 in the binding of IF7 and GS.
Materials and methods
Materials
Ultra-pure GdmCl and urea were from ICN Biochemicals. Exact concentrations of GdmCl and urea were calculated from the refractive index of the solution (Pace 1986). Standard suppliers were used for all other chemicals. Water was deionized and purified on a Millipore system.
Protein purification
Synechocystis GS type I was expressed and purified in E. coli as described elsewhere (García-Domínguez et al. 1999). IF7 was expressed in E. coli as described previously (García-Domínguez et al. 1999) and purified from the soluble fraction by the following procedure. After induction of IF7 expression, cells were collected and resuspended in buffer A (10 mM Hepes, pH 7.0, 1 M ammonium sulfate) with 1 mM phenylmethylsulfonyl fluoride and 2 μg/mL leupeptine added. Cells were disrupted by sonication, the lysate was centrifuged at 15,000 g for 15 min, and the resulting cell-free extract was loaded onto a phenyl-Sepharose column equilibrated with buffer A. Elution was performed with a linear gradient (1–0 M ammonium sulfate) in 60 mL of 5 mM Hepes, pH 6.5 (buffer B). Fractions with high GS inactivation activity were combined, diluted fivefold, and applied to a CM-Sephadex C25 column equilibrated with buffer B. After washing the column with the same buffer containing 0.2 M NaCl, the elution was carried out with a linear gradient (0.2–0.6 M NaCl) in 60 mL of buffer B. Fractions that showed GS inactivation activity were pooled, concentrated using a stirred ultrafiltration cell with a YM3 membrane (AMICON), and subjected to gel filtration chromatography using a HiLoad 16/60 Superdex 75 gel filtration column (Amersham Biosciences) running on an AKTA FPLC system. Active fractions were concentrated using the ultrafiltration system described above, and their purity was analyzed by SDS/PAGE. The protein concentration was calculated from the absorbance of stock solutions measured at 280 nm, using the extinction coefficients of model compounds (Pace and Scholtz 1997).
Fluorescence measurements
Fluorescence spectra for IF7 were collected either on an Aminco-Bowman SLM 8000 spectrofluorometer (Spectronics Instruments) interfaced with a Haake water bath, or in a Cary Eclipse spectrofluorometer (Varian) interfaced with a Peltier cell. Sample concentrations were in the range of 2–6 μM, and the final concentration of the buffer was, in all cases, 10 mM. A 0.5-cm-path-length quartz cell (Hellma) was used for measurements in the SLM spectrofluorometer, and a 1-cm-path-length quartz cell (Hellma) was used in the Varian spectrofluorometer.
Steady-state fluorescence measurements
Samples of IF7 were excited at 280 nm and at 295 nm at 298 K in the pH range 3–12 to characterize putative different behaviors of either tryptophan or tyrosine residues (Pace and Scholtz 1997). No differences, except in the spectra intensity, were observed either in the band shape or maximum wavelength, and then excitation at 280 nm was used for all of the experiments. The slit width was typically equal to 4 nm for the excitation light and 8 nm for the emission light. The fluorescence experiments were recorded between 300 and 400 nm. The signal was acquired for 1 sec, and the wavelength increment was 1 nm. Blank corrections were made in all spectra. The salts and acids used in buffer preparation were: pH 2.0–3.0, phosphoric acid; pH 3.0–4.0, formic acid; pH 4.0–5.5, acetic acid; pH 6.0–7.0, NaH2PO4; pH 7.5–9.0, Tris acid; pH 9.5–11.0, Na2CO3; pH 11.5–13.0, Na3PO4. The pH was measured with an ultrathin Aldrich electrode in a Radiometer (Copenhagen) pH-meter.
Fluorescence experiments with GS were carried out using the same parameters as those described for IF7 experiments. Here, the range of concentrations used was 5–20 μM of protomer.
Fluorescence quenching experiments
Quenching of intrinsic tryptophan and tyrosine fluorescence by iodide (Lakowicz 1999) was examined at different solution conditions. Excitation was at 280 nm; emission was measured from 300 to 400 nm. In the absence of GdmCl, ionic strength was kept constant by addition of KCl; also, Na2S2O3 was added to a final concentration of 0.1 M to avoid formation of I3−. The slit width was set at 8 nm for both excitation and emission. The data were fitted to the following equation (Lakowicz 1999),
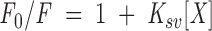 |
(1) |
where Ksv is the Stern-Volmer constant for collisional quenching, F0 is the fluorescence in the absence of KI, and F is the measured fluorescence at any KI concentration. The range of KI concentrations explored was 0–0.7 M. Experiments were carried out at pH 6.5, 10 mM phosphate buffer at 298 K.
ANS binding
ANS binding was detected by collecting fluorescence spectra at pH 6.5, 10 mM phosphate buffer at 298 K. Excitation wavelength was 380 nm, and emission was measured from 400 to 600 nm. Slit widths were 4 nm for excitation, and 8 nm for emission. Stock solutions of ANS were prepared in water and diluted into the samples to yield a final 50 μM dye concentration. Dye concentrations were determined using an extinction coefficient of 8000 M−1 cm−1 at 372 nm. In all cases, blank solutions were subtracted from the corresponding spectra.
Binding experiments
Increasing amounts of IF7, in the range 0.5–4 μM, were added to a solution of a fixed concentration of GS (1.5 μM of protomer) in 10 mM phosphate buffer, pH 6.5, and the fluorescence was measured after a 2-h incubation at 298 K. Experiments were carried out in a Varian Cary Eclipse spectrofluorimeter with excitation at 280 nm, and emission fluorescence was collected between 300–400 nm. The excitation and emission slits were 5 nm, and the data pitch interval was 1 nm. The fluorescence values of a blank solution containing only IF7 were subtracted for each point. The fluorescence of IF7 changed linearly in the explored concentration range. The dissociation constant of the complex was calculated by fitting the plot of the observed fluorescence change of GS versus added IF7 to the following equation,
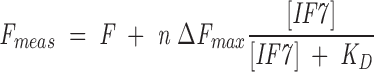 |
(2) |
where Fmeas is the measured fluorescence after subtraction of the blank, ΔFmax is the change in the fluorescence measured when all of IF7 is forming the complex, F is the fluorescence intensity when no IF7 has been added, n is the number of identical noninteracting binding sites for IF7, and KD is the dissociation constant. The dissociation constant was determined by following the fluorescence at 315 nm, but it could also be obtained by following the change at other wavelengths (data not shown). At all concentrations used, the absorbance of IF7 was kept lower than 0.2 units to avoid inner filter effects, during fluorescence excitation (Birdsall et al. 1983).
The presence of aggregated forms at any IF7 concentrations during the titration was followed by absorbance experiments collected in the 400–600-nm range. If aggregated forms occur, light scattering would occur and a broad intense band should appear. Absorbance experiments were carried out in a Shidmadzu UV-1601 ultraviolet spectrophotometer, using a 1-cm-path-length cell (Hellma).
Because the [IF7] used in the simplified Equation 2 is in terms of concentration of the free IF7, we also used the complete equation, which gives the fluorescence as a function of the total (i.e., the GS-bound and free IF7) concentration of IF7 as
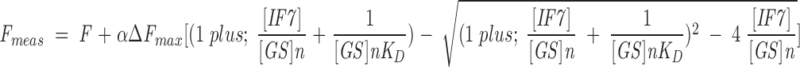 |
(3) |
where [IF7] and [GS] represent here the total concentrations of IF7 and GS, respectively, and α is a constant. Fit of the experimental data to this equation yielded larger associated errors for the n, KD, and ΔFmax compared to Equation 2, but the fitted values were similar to those obtained with Equation 2.
We did not measure the binding constant by CD titration experiments because of the large amounts of protein required.
Circular dichroism measurements
Circular dichroism spectra of IF7 were collected on a Jasco J810 spectropolarimeter fitted with a thermostated cell holder and interfaced with a Neslab RTE-111 water bath. The instrument was periodically calibrated with (+) 10-camphorsulphonic acid.
Steady-state measurements
Isothermal wavelength spectra at different pHs were acquired at a scan speed of 50 nm/min with a response time of 2 sec and averaged over four scans at 298 K. Far-UV measurements were performed using 20–40 μM of protein in 10 mM buffer, using 0.1- or 0.2-cm-pathlength quartz cells (Hellma). Near-UV spectra were acquired using 30–40 μM of protein in a 0.5-cm-pathlength cell. All spectra were corrected by subtracting the proper baseline. The mean residue ellipticity, [Θ], was calculated according to
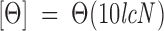 |
(4) |
where Θ is the measured ellipticity, l is the pathlength cell, c is the protein concentration, and N is the number of amino acids. The helical content of IF7 at any pH was approximated from its mean residue ellipticity at 222 nm (Zurdo et al. 1997),
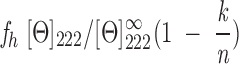 |
(5) |
where fh is the helical fraction of the protein, [Θ222] is the observed mean residue ellipticity, [Θ∞222] is the mean ellipticity for an infinite α-helix at 222 nm (−34,500 deg cm2 dmole−1), k is a wavelength-dependent constant (2.57 at 222 nm), and n is the number of peptide bonds (64 for IF7).
For the near-UV CD experiments, the pathlength cell was 0.5 cm, with a protein concentration of 40–50 μM.
CD experiments with GS were carried out using the same parameter set described in the IF7 experiments. The range of concentrations used was 5–50 μM of protomer.
Thermal denaturation
Thermal denaturation experiments with IF7 were performed at constant heating rates of 60 K/h and a response time of 8 sec. Thermal scans were collected in the far-UV region by following the ellipticity at 222 nm from 298 K (or 278 K) to 363 K (or 368 K) in 0.1-cm-pathlength quartz cells (Hellma) with a total protein concentration of 20–40 μM. The reversibility of thermal transitions was tested by recording a new scan after cooling down to 278 K the thermally denatured samples, and comparing it with the spectra obtained in the first scan. In all cases, both spectra were identical (data not shown). The possibility of drifting of the CD spectropolarimeter was tested by running two samples containing only buffer, before and after the thermal experiments. No difference was observed between the scans. Every thermal denaturation experiment was repeated at least twice with fresh new samples. In all cases, after the reheating experiment, the samples were transparent and no precipitation was observed.
Chemical- and pH-denaturation experiments
In the GdmCl-denaturation experiments with IF7, far-UV CD spectra were acquired at a scan speed of 50 nm/min, and four scans were recorded and averaged at 298 K. The response time was 2 sec. The cell pathlength was 0.1 cm, with a protein concentration of 20–40 μM in the far-UV CD experiments. Spectra were corrected by subtracting the baseline in all cases. The chemical denaturation reaction is fully reversible, as demonstrated by the sigmoidal curves obtained by starting from diluted 7 M GdmCl samples at different pHs (data not shown). Every chemical denaturation experiment was repeated at least three times with fresh new samples.
In the pH-induced unfolding experiments with IF7, the pH was measured after completion of the experiments, and essentially no differences were observed with those pHs calculated from the buffer stock solutions. The pH range explored was 3–12. The proper blank solutions were subtracted in all cases. Buffer concentration was 10 mM in all cases, and the buffers were the same as those used in fluorescence measurements.
Analysis of the pH denaturation curves
The pH-denaturation experiments were analyzed assuming that both species, protonated and deprotonated, contribute to the CD spectrum at 222 nm,
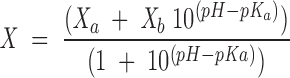 |
(6) |
where X is the ellipticity, Xa is the ellipticity at low pHs (acid form), Xb is the ellipticity at high pHs (basic form), and pKa is the apparent pK of the titrating group. The apparent pKa reported was obtained from three different measurements, carried out with fresh new samples.
Fitting by nonlinear least-squares analysis to Equations 2, 3, and 6 was carried out by using the general curve fit option of Kaleidagraph (Abelbeck software) on a personal computer.
Fourier transform infrared spectroscopy
The protein was lyophilized and dissolved in deuterated buffer Tris 10 mM and 0.1 M NaCl (pH 6.5). No pH corrections were done for the isotope effects. Samples of IF7 at a final concentration of 10 mg/mL were placed between a pair of CaF2 windows separated by a 50-μm-thick spacer in a Harrick demountable cell. Spectra were acquired on a Nicolet 520 instrument equipped with a deuterated triglycine sulphate detector and thermostated with a Braun water bath at 283 K. The cell container was continuously filled with dry air.
Steady-state measurements
Here, 600 scans per sample were taken, averaged, apodized with a Happ-Genzel function, and Fourier transformed to give a final resolution of 2 cm−1. The signal/noise ratio of the spectra was better than 1000:1. Buffer contributions were subtracted, and the resulting spectra were used for analysis after smoothing. The spectra smoothing was carried out applying the maximum entropy method (Echabe et al. 1997). In order to quantify the different secondary structure components, the amide I band was decomposed into its constituents by curve-fitting (based on a combination of Gaussian and Lorentzian functions), using the number and position of bands obtained from the deconvolved (using a Lorentzian bandwidth of 18 cm−1 and a resolution enhancement factor of 2) and the Fourier derivative (using a power of 3 and a breakpoint of 0.3) spectra (Moffat and Mantsch 1992; Jackson and Mantsch 1995; Arrondo and Goñi 1999).
Thermal denaturation measurements
Thermal denaturation experiments followed by FTIR were carried out with a scanning rate of 50 K/h, and acquired every 3 K. Fifty scans per temperature were averaged. The heating experiments were not reversible under these conditions.
Nuclear magnetic resonance spectroscopy
1H NMR experiments were carried out in a Bruker AMX-600 spectrometer at 298 K and 278 K with 32,000 data points and using presaturation to eliminate the water signal. Typically, 1024 scans were acquired, and the spectral width was 6000 Hz in all cases. Exchange samples were prepared by dissolving a lyophilized aliquot in 500 μL of precooled exchange buffer (10 mM, phosphate buffer, pH 6.5). To discard any artifact caused by unfolded protein from freeze-drying, the sample in water was diluted 10 times in deuterated water, and the spectrum was compared with that from the concentrated solution obtained from lyophilization. Both spectra were identical (data not shown). In addition, the spectra at the two temperatures used were identical. Spectra were acquired at different concentrations ranging from 0.1 to 1.5 mM at 298 K. No changes in chemical shifts or line broadening were observed in any case. The spectra were processed using BRUKER-UXNMR software working on an SGI work station. An exponential window function and polynomial baseline corrections were applied. The final one-dimensional data contained 64,000 data points. 1H chemical shifts were quoted relative to external TSP.
Gel filtration chromatography
Analytical gel filtration experiments were carried out by using an analytical gel filtration Superdex 75 HR 16/60 column (Amersham Biosciences) running on an AKTA FPLC system at 298 K. Flow rates of 1 mL/min were used, and aliquots of 100 μL were loaded into the column after equilibration. The column was equilibrated with four column volumes of elution buffer. The elution buffers for the pH experiments were those described above, containing 150 mM NaCl added to avoid nonspecific interactions with the column. To check for the presence of aggregated species at any pH, protein concentrations ranging from 20–60 μM were used. No differences in the elution volumes were observed among the different concentrations used. The column was calibrated using the gel filtration low-molecular-weight calibration kit (Amersham Biosciences). The standards used and their corresponding Stokes radii were: ribonuclease A (16.4 Å), chymotrypsinogen (20.9 Å), ovoalbumin (30.5 Å), and bovine serum albumin (35.5 Å; Hinkle et al. 1999). Each experiment at the different pHs was repeated three times with fresh new samples. Protein elution was monitored by following absorbance at 280 nm.
The elution of a macromolecule in gel filtration experiments is usually given by the partition coefficient, which is defined as the fraction of solvent volume within the gel matrix accessible to the macromolecule (Ackers 1967). The weight average partition coefficients (σ) of protein standards and IF7 were calculated by
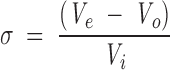 |
(7) |
where Ve is the elution volume of the protein, and Vo and Vi are the void and internal volumes of the column, with values of 8.13 ± 0.06 mL and 28.43 ± 0.03 mL, respectively. The Vo and Vi volumes were respectively determined using Blue dextran (5 mg/mL, in 10 mM phosphate buffer containing 150 mM NaCl) and L-tryptophan (0.5 mg/mL, in the above buffer) by averaging four measurements for each agent.
The partition coefficients were determined for the molecular size standards, and they were transformed by using the inverse error function complement of σ, (erfc−1[σ]), yielding a linear relationship with the molecular Stokes radius, Rs (Ackers 1967; Darlin et al. 2000)
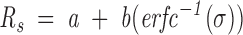 |
(8) |
where a and b are the calibration constants for the column. The error function complement is, by the mathematical definition (Spiegel 1985)
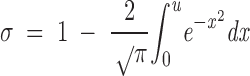 |
(9) |
and the function under the integral is the gaussian function of probability (the error function). The value of this integral under a specified area (the upper limit of the integral given by u) is found in probability tables of mathematical handbooks (Spiegel 1985). Then, once the partition coefficient is calculated from the experimental values, the difference 1 − σ must be calculated. The result of this difference is the area under the function of probability (i.e., the value of
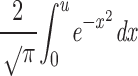 |
and it must be looked for in the table containing the values of such function. The value of the upper limit of the integral (and thus the value of the inverse complementary error function) is obtained by looking at the corresponding line and column of such table.
Fitting of the calculated erfc−1(σ) to Equation 8 by linear least-squares analysis was carried out on Kaleidagraph (Abelbeck software) on a PC. Once the calibration parameters are obtained, the Stokes radius of any macromolecule can be determined by using Equation 8.
The frictional coefficient of a solvated molecule, f/f0, can be obtained as follows. According to the Stokes law for a solvated molecule, the translational friction coefficient, f, is given by
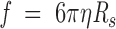 |
(10) |
where η is the solvent viscosity. The f for an ideal unsolvated spherical molecule, f0, with the same mass and partial specific volume, is given by
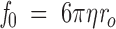 |
(11) |
where ro is the radius of the spherical anhydrous macromolecule; then f/f0 = Rs/r0. For a protein, ro can be calculated ab initio considering that the anhydrous molecular volume (M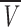 N) equals the volume of a sphere (Creighton 1993)
N) equals the volume of a sphere (Creighton 1993)
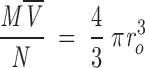 |
, which yields,
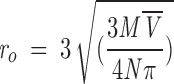 |
(12) |
where M is the molecular weight of the protein, 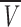 is the partial specific volume of the protein, and N is the Avogadro’s number.
is the partial specific volume of the protein, and N is the Avogadro’s number.
If it is assumed that all deviations from unity in the frictional coefficient are because of the hydration effects (either because of an elongated shape or to open, solvent-exposed conformation), an upper limit, wmax, for the hydration in grams of water bound per gram of protein is given by (Darlin et al. 2000)
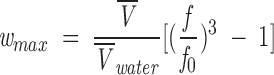 |
(13) |
where Vwater is the partial specific volume of water (1 cm3/g).
Acknowledgments
We thank the two anonymous referees for their insightful and critical remarks. This work was supported by Projects GV-00-024-5 (J.L.N.), CTIDIB/2002/6 (J.L.N.) from Generalitat Valenciana and Project BIO2000-1081, from the Spanish Ministerio de Ciencia y Tecnología (J.L.N.), and Project BMC2001-0635 (F.J.F.) from the Spanish Ministerio de Ciencia y Tecnología. M.I.M.-P. is recipient of a Ramón y Cajal contract from the Spanish Ministerio de Ciencia y Tecnología. F.N.B. is recipient of a pre-doctoral fellowship from Generalitat Valenciana. We thank Prof. Manuel Rico for the use of the NMR spectrometer in Madrid, Dr. Marika Lindahl for careful reading of the manuscript and for further suggestions, and May García, María del Carmen Fuster, María T. Garzón, and Javier Casanova for excellent technical assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ANS, 8-anilinonapthalene-1-sulfonic acid
CD, circular dichroism
FTIR, Fourier transform infrared spectroscopy
GdmCl, guanidinium hydrochloride
GS, glutamine synthetase
IF7, the 65-residue-long inactivating factor of GS
NMR, nuclear magnetic resonance spectroscopy
UV, ultraviolet
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0303203.
References
- Ackers, G.K. 1967. Molecular sieve studies of interacting protein systems. I. Equations for transport of associating systems. J. Biol. Chem. 242 3026–3034. [PubMed] [Google Scholar]
- Almassy, R.J., Janson, C.A., Hamlin, R., Xuong, N.H., and Eisenberg, D.E. 1986. Novel subunit–subunit interactions in the structure of glutamine synthetase. Nature 323 304–309. [DOI] [PubMed] [Google Scholar]
- Arrondo, J.L.R. and Goñi, F.M. 1999. Structure and dynamics of membrane proteins as studied by infrared spectroscopy. Prog. Biophys. Mol. Biol. 72 367–405. [DOI] [PubMed] [Google Scholar]
- Arrondo, J.L.R., Mantsch, H.H., Mullner, N., Pikula, S., and Martonosi, A. 1987. Infrared spectroscopic characterization of the structural changes connected with the E1–E2 transition in the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 262 9037–9043. [PubMed] [Google Scholar]
- Birdsall, B., King, R.W., Wheeler, M.R., Lewis Jr., C.A., Goode, S., Dunlap, R.B., and Roberts, G.C.K. 1983. Correction for light absorption in fluorescence studies of protein-ligand interactions. Anal. Biochem. 132 353–361. [DOI] [PubMed] [Google Scholar]
- Braiman, M.S. and Rothschild, K.J. 1988. Fourier transform infrared techniques for probing membrane protein structure. Annu. Rev. Biophys. Biophys. Chem. 17 541–570. [DOI] [PubMed] [Google Scholar]
- Byler, D.M. and Susi, H. 1986. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 25 469–487. [DOI] [PubMed] [Google Scholar]
- Cantor, C.R. and Schimmel, P.R. 1980. Biophysical chemistry. W.H. Freeman, New York.
- Creighton, T.E. 1993. Proteins. Structures and macromolecular properties, 2nd ed. W.H. Freeman, New York.
- Darlin, P.J., Holt, J.M., and Ackers, G.K. 2000. Coupled energetics of λ cro repressor self-assembly and site-specific DNA operator binding I: Analysis of cro dimerization from nanomolar to micromolar concentrations. Biochemistry 39 11500–11507. [DOI] [PubMed] [Google Scholar]
- Denning, D.P., Uversky, V., Patel, S.S., Fink, A.L., and Rexach, M. 2002. The Saccharomyces cerevisiae nucleoporin Nup2p is a natively unfolded protein. J. Biol. Chem. 277 33447–33455. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002. Intrinsic disorder and protein function. Biochemistry 41 6573–6580. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., and Wright, P.E. 2002. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12 54–60. [DOI] [PubMed] [Google Scholar]
- Echabe, I., Encinar, J.A., and Arrondo, J.L.R. 1997. Effects of noise on the obtention of protein structure by band decomposition of the infrared spectrum. Biospectroscopy 3 469–475. [Google Scholar]
- García-Domínguez, M., Reyes, J.C., and Florencio, F.J. 1999. Glutamine synthetase inactivation by protein–protein interaction. Proc. Natl. Acad. Sci. 96 7161–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle, A., Goranson, A., Butters, C.A., and Tobacman, L.S. 1999. Roles for the troponin tail domain in thin filament assembly and regulation. A deletional study of cardiac troponin T. J. Biol. Chem. 274 7157–7164. [DOI] [PubMed] [Google Scholar]
- Hu, P., Leighton, T., Ishkhanova, G., and Kustu, S. 1999. Sensing of nitrogen limitation by Bacillus subtilis: Comparison to enteric bacteria. J. Bacteriol. 181 5042–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M. and Mantsch, H.H. 1995. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30 95–120. [DOI] [PubMed] [Google Scholar]
- Kelly, S.M. and Price, N.C. 2000. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 1 349–384. [DOI] [PubMed] [Google Scholar]
- Kyte, J. and Doolittle, R.F. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157 105–132. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Plenum Press, New York.
- Li, X., Romero, P., Rani, M., Dunker, A.K., and Obradovic, Z. 1999. Predicting protein disorder for N-, C-, and internal regions. Genome Informatics 10 30–40. [PubMed] [Google Scholar]
- Li, X., Obradovic, Z., Brown, C.L., Garner, E., and Dunker, A.K. 2000. Comparing predictors of disordered protein. Genome Informatics 11 172–184. [PubMed] [Google Scholar]
- Liaw, S.H., Pan, C., and Eisenberg, D.E. 1993. Feedback inhibition of fully unadenylated glutamine synthetase from Salmonella typhimurium by glycine, alanine, and serine. Proc. Natl. Acad. Sci. 90 4996–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magasanik, B. 1996. Regulation of nitrogen utilization in Escherichia coli and Salmonella: Cellular and molecular biology. (eds. F.C. Neidhart et al.), pp. 1344–1356. Am. Soc. Microbiol. Press, Washington, DC.
- Mérida, A., Candau, P., and Florencio, F.J. 1991. In vitro reactivation of in vivo ammonium-inactivated glutamine synthetase from Synechocystis sp. PCC 6803. Biochem. Biophys. Res. Commum. 181 780–786. [DOI] [PubMed] [Google Scholar]
- Merrick, M.J. and Edwards, R.A. 1995. Nitrogen control in bacteria. Microbiol. Rev. 59 604–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat, D.J. and Mantsch, H.H. 1992. Fourier resolution enhancement of infrared spectral data. Methods Enzymol. 210 192–200. [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure (ed. T.E. Creighton), 2nd ed., pp. 253–259. Oxford University Press, Oxford, UK.
- Ritzer, L.J. 1996. Ammonia assimilation and the biosynthesis of glutamine, glutamate, aspartate, asparagine, L-alanine, and D-alanine in Escherichia coli and Salmonella: Cellular and molecular biology. (eds. F.C. Neidhart et al.), pp. 391–407. Am. Soc. Microbiol. Press, Washington, D.C.
- Romero, P., Obradovic, Z., Li, X., Garner, E.C., Brown, C.J., and Dunker, A.K. 2001. Sequence complexity of disordered protein. Proteins 42 38–48. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1997. Optical spectroscopy to characterize protein conformation and conformational changes. In Protein structure (ed. T.E. Creighton), 2nd ed., pp. 261–297. Oxford University Press, Oxford, UK.
- Semisotnov, G.V., Rodionova, N.A., Razgulyaev, O.I., Uversky, V.N., Gripas, A.F., and Gilmanshin, R.I. 1991. Study of the "molten globule" intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31 119–128. [DOI] [PubMed] [Google Scholar]
- Siegel, L.M. and Monty, K.J. 1966. Kinetic properties of the TPNH-specific sulfite and hydroxylamine reductase of Salmonella typhimurium. Biochim. Biophys. Acta 112 346–362. [DOI] [PubMed] [Google Scholar]
- Spiegel, M.R. 1985. Probability and statistics. McGraw Hill, New York.
- Surewicz, W.K. and Mantsch, H.H. 1988. New insight into protein secondary structure from resolution-enhanced infrared spectra. Biochim. Biophys. Acta 952 115–130. [DOI] [PubMed] [Google Scholar]
- Tompa, P. 2002. Intrinsically unstructured proteins. Trends Biochem. Sci. 27 527–533. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. 2002. What does it mean to be natively unfolded? Eur. J. Biochem. 269 2–12. [DOI] [PubMed] [Google Scholar]
- Vihinen, M., Torkkila, E., and Riikonen, P. 1994. Accuracy of protein flexibility predictions. Proteins 19 141–149. [DOI] [PubMed] [Google Scholar]
- Woody, R.W. 1995. Circular dichroism. Methods Enzymol. 246 34–71. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. Wiley, New York.
- Yamashita, M.M., Almassy, R.J., Janson, C.A., Cascio, D., and Eisenberg, D.E. 1989. Refined atomic model of glutamine synthetase at 3.5 Å resolution. J. Biol. Chem. 264 17681–17690. [DOI] [PubMed] [Google Scholar]
- Zurdo, J., Sanz, J.M., Gonzalez, C., Rico, M., and Ballesta, J.P.G. 1997. The exchangeable yeast ribosomal acidic protein YP2β shows characteristics of a partly folded state under physiological conditions. Biochemistry 36 9625–9635. [DOI] [PubMed] [Google Scholar]