

Abstract
Metallo-β-lactamases are zinc enzymes able to hydrolyze the four-membered ring of β-lactam antibiotics, representing one of the latest generations of β-lactamases. These enzymes belong to the zinc metallo-hydrolase family of the β-lactamase fold. Enzymes belonging to this family have a bimetallic active site whose structure varies among different members by point substitutions of the metal ligands. In this work, we have grafted new metal ligands into the metal binding site of BcII from Bacillus cereus that mimic the ligands present in other members of this superfamily. We have characterized spectroscopically and modeled the structure of the redesigned sites, which differ substantially from the wild-type enzyme. Despite the changes introduced in the active site, the mutant enzymes retain almost full activity. These results shed some light on the possible evolutionary origin of these metalloenzymes.
Keywords: Metallo-β-lactamase, metal site engineering, zinc enzymes, metal substitution, spectroscopy
Metallo-β-lactamases (class B β-lactamases, EC 3.5.2.6) are zinc-dependent enzymes able to hydrolyze the amide bond of β-lactam antibiotics (Bush et al. 1995; Frere 1995). These metalloenzymes initially received little attention, but the recent plasmid-mediated spread of metallo-β-lactamase genes among pathogenic strains has raised the concerns of the biomedical community. This problem is empowered by the broad hydrolytic spectrum of metallo-β-lactamases and the inefficacy of clinically approved inhibitors toward them (Rasmussen and Bush 1997; Cornaglia et al. 1999; Cricco and Vila 1999).
Sequence and structural information on enzymes from different strains reveals that all known metallo-β-lactamases are evolutionarily related, and possess a similar protein fold (Concha et al. 1996; Rasmussen and Bush 1997; Fabiane et al. 1998; Ullah et al. 1998; Cricco et al. 1999; Concha et al. 2000). All of them are able to bind up to two Zn(II) equivalents in their active sites through a largely conserved metal ligand set (Fig. 1A▶). One of the metal ions is bound to three histidine residues (Zn1), while the coordination environment of the other (Zn2) differs between metallo-β-lactamases isolated from different sources. In subclass B1 metallo-β-lactamases, Zn2 is coordinated by an aspartate, a cysteine, and a histidine residue, while in the subclass B3 metallo-β-lactamase from Stenotrophomonas maltophilia, the cysteine ligand is replaced by a histidine located in a different position of the protein sequence (Figs. 1B, 2▶▶). Metallo-β-lactamases are active either as mono-Zn(II) or bi-Zn(II) (Orellano et al. 1998; Paul-Soto et al. 1999; Rasia and Vila 2002; Wommer et al. 2002). In the binuclear forms, the hydroxide moiety bound to Zn1 is believed to be the catalytic nucleophile while Zn2 behaves as a cocatalytic site (Wang et al. 1999). An exception to this general trend is the metallo-β-lactamase CphA from Aeromonas hydrophila, which is active with only one Zn(II). Uptake of a second metal equivalent inhibits this enzyme (Hernandez-Valladares et al. 1997). Bacillus cereus metallo-β-lactamase (BcII hereafter) is active with one Zn(II), while binding of a second Zn(II) equivalent further activates the enzyme. This enzyme thus represents a unique protein template to probe the function of mono- and binuclear states in metallo-β-lactamases.
Figure 1.

Structure of the metal binding sites of enzymes belonging to the zinc metallo-hydrolase family of the β-lactamase fold. (A) Binuclear Bacillus cereus β-lactamase (PDB code 1bc2; Fabiane et al. 1998); (B) Stenotrohomonas maltophilia β-lactamase L1 (PDB code 1sml; Ullah et al. 1998); (C) human glyoxalase II (PDB code 1qh3; Cameron et al. 1999); (D) Desulfovibrio gigas ROO (PDB code 1e5d; Frazao et al. 2000). These figures were drawn using MOLMOL (Koradi et al. 1996). Residues are labeled according to the consensus numbering scheme for β-lactamases in all cases (Galleni et al. 2001).
Figure 2.

Sequence alignment of representative proteins of the zinc metallo-hydrolase family of the β-lactamase fold. (Top line), Consensus residues. Metal ligands are indicated in bold characters. Metallo-β-lactamases: BCII, B. cereus; CCRA, Bacteroides fragilis; IMP-1, Pseudomas aeruginosa; CPHA, Aeromonas hydrophila; L1, Stenotrophomonas maltophilia. Other proteins: HGLYII, Homo sapiens glyoxalase II; ROO, Desulfovibrio gigas rubredoxin:oxygen oxidoreductase; ZIPD, Escherichia coli phosphodiesterase; SCYC, Streptomyces arenae cyclase; BCPSF, Bos taurus cleavage and polyadenylation specificity factor; MDNAR, Mus musculus DNA cross-link repair gene; PMPH, Plesiomonas sp. methyl parathion hydrolase (Daiyasu et al. 2001).
Accumulation of sequence and structure data led recently to the identification of many proteins homologous to metallo-β-lactamases. The crystal structures of human glyoxalase II (Fig. 1C▶; Cameron et al. 1999) and Desulfovibrio gigas rubredoxin:oxygen oxydoreductase (ROO, Fig. 1D▶; Frazao et al. 2000) showed that these enzymes have a domain sharing the metallo-β-lactamase fold and a bimetallic active site in the same location within the protein scaffold. This group of enzymes is known as the zinc metallo-hydrolase family of the β-lactamase fold (Daiyasu et al. 2001). Both ROO and glyoxalase II differ from metallo-β-lactamases in the coordination environment of the two metal ions. A structurally based sequence alignment shows that the putative ligands of the two metal ions differ within this family of proteins (Fig. 2▶; Daiyasu et al. 2001). Specifically, the coordination environment of the second metal ion is the most variable structural feature within the active site of the zinc metallo-hydrolase family of the β-lactamase fold. In particular, a histidine ligand in position 121 seems to be conserved within the family, except in most metallo-β-lactamases. Knowledge of the factors that shape the Zn2 structure may contribute to the understanding of the role of Zn2 in catalysis, which has not yet been elucidated. Therefore we decided to explore the malleability of the Zn2 site in BcII by grafting potential new ligands in this position. Here, we report that engineering a histidine, as is found in most proteins of the superfamily, or an isosteric glutamate ligand in the Arg121 position of B. cereus metallo-β-lactamase results in a substantial structural modification of the second metal binding site. Surprisingly, these structural changes do not abolish activity of the chimeric enzymes.
Results
Activity and zinc content in the mutant enzymes
The R121H and R121E mutants of BcII were obtained by oligonucleotide-directed mutagenesis, as described in Materials and Methods. Both mutants were active toward benzylpenicillin and nitrocefin, and their activity was abolished by incubation of the enzymes with EDTA.
After extensive dialysis against metal-free buffer, all three enzymes (wild type, R121H, and R121E βcII) contained more than one equivalent of bound Zn(II) per mole of enzyme (Table 1▶). In the presence of 5 μM Zn(II) during the dialysis steps, both mutants showed a higher metal content than the wild-type enzyme, indicating a higher affinity for the formation of a binuclear site (Table 1▶). Moreover, the metal:enzyme ratio is close to two, suggesting that in these conditions, at 5 μM added Zn(II) both mutant enzymes are in their binuclear forms.
Table 1.
Zinc content of the wild type and mutant enzymes
| Zinc/enzyme ratio |
|||
|---|---|---|---|
| Added Zn(II) during dialysis (μM) | Wild type | R121H | R121E |
| 0 | 1.4 | 1.5 | 1.2 |
| 5 | 1.5 | 1.8 | 1.9 |
Protein samples (1 mL 60–80 μM) were dialyzed against 150 mL of a buffer containing 15 mM Hepes at pH 7.5 (1) treated with chelex and (2) with 5 μM added ZnSO4. Five buffer changes were performed. The values correspond to the average of two independent determinations. Standard deviations were below 10%.
The mutant enzymes are active when assayed in metal-free reaction medium. Their reactivity toward benzylpenicillin is enhanced in the presence of increasing concentrations of added Zn(II), reaching its maximum value at >2 μM added Zn(II) (Table 2▶). Analysis of the activity data at the light of the Zn(II) content determination suggests that kinetic measurements performed at >2 μM Zn(II) can be taken as representative of the catalytic activity of the binuclear forms of the enzymes. The activity of the binuclear form of the R121E mutant is comparable to that of the wild-type enzyme, while the R121H mutant shows a fivefold decrease. The relative catalytic performance of both mutants toward nitrocefin is also maintained (Table 3▶).
Table 2.
Zinc dependence of the catalytic efficiency of the wild type and mutant enzymes
| kcat/KM (M−1s−1) |
|||
|---|---|---|---|
| Zn(II) (μM) | Wild type | R121H | R121E |
| 1 | 1.2 · 106 | 2.2 · 105 | 1.0 · 106 |
| 2 | 1.5 · 106 | 2.3 · 105 | 1.2 · 106 |
| 5 | 1.7 · 106 | 2.5 · 105 | 1.2 · 106 |
| 10 | 1.6 · 106 | 2.5 · 105 | 1.1 · 106 |
Kinetic measurements were performed in 15 mM Hepes buffer at pH 7.5, using benzylpenicillin as substrate. The values correspond to the average of at least three independent determinations. Standard deviations were below 5%.
Table 3.
Activity of the wild type and mutant enzymes against benzylpenicillin and nitrocefin
| Wild type | R121H | R121E | |
|---|---|---|---|
| Benzylpenicillin, pH 7.5 | |||
| KM (μM) | 310 | 300 | 430 |
| kcat (s−1) | 510 | 75 | 500 |
| kcat/KM (M−1s−1) | 1.7 · 106 | 0.25 · 106 | 1.2 · 106 |
| Nitrocefin, pH 6.0 | |||
| KM (μM) | 11.5 | 38 ± 7 | 18.0 ± 4.1 |
| kcat (s−1) | 11.6 | 7.1 ± 1.0 | 25.7 ± 4.2 |
| kcat/KM (M−1s−1) | (1.0 ± 0.1) · 106 | (0.19 ± 0.06) · 106 | (1.4 ± 0.5) · 106 |
Kinetic measurements were performed in (1) 15 mM Hepes buffer pH 7.5 and (2) TACS buffer pH 6.0, in the presence of 10 μM Zn(II) in the reaction medium. Reported values are the average of at least three independent determinations. Standard deviations were below 5% except when indicated.
We determined the catalytic efficiencies of the Co(II)-adducts in conditions in which the enzymes should be present mostly as mononuclear [10 μM Co(II)] or binuclear [100 μM Co(II)] species, according to the spectrophotometric titration of the apoenzymes (see below). In both cases, the Co(II)-substituted wild-type enzyme is noticeably more active than the native Zn(II) enzyme (Table 4▶). For all three enzymes, the activity of the binuclear form is higher than that of the mononuclear. The activity enhancement produced by uptake of a second Co(II) equivalent is larger in the mutant enzymes than in the wild-type enzyme, indicating that the mononuclear Co(II)-forms of the mutant enzymes are relatively less active. This difference in relative activities might be because of different metal coordination geometries or to different relative occupancies of the two metal binding sites of the mono Co(II)-adducts, as inferred from the intensity of the ligand field bands in the visible spectrum of Co(II) R121H (see below).
Table 4.
Activity of the Co(II)-substituted enzymes
| Wild type |
R121E |
R121H |
||||
|---|---|---|---|---|---|---|
| 10 μM Co(II) | 100 μM Co(II) | 10 μM Co(II) | 100 μM Co(II) | 10 μM Co(II) | 100 μM Co(II) | |
| KM (μM) | 6 | 12 | 20 | 26 | 52 | 46 |
| kcat (s−1) | 30 | 70 | 8 | 50 | 9 | 37 |
| kcat/KM (M−1 s−1) | 5 · 106 | 6.2 · 106 | 4 · 105 | 2 · 106 | 1.7 · 105 | 8.1 · 105 |
Enzyme activity against benzylpenicillin was determined in 15 mM Hepes buffer pH 7.5 containing the indicated amount of CoCl2. Apoenzyme samples were first incubated in the presence of the indicated amount of Co(II) and then added to the reaction medium containing the same concentration of Co(II) to start the reaction.
UV-visible spectra of metal derivatives
Substitution of Zn(II) by Co(II) is a common strategy for the structural study of Zn(II) sites in proteins (Bertini and Luchinat 1983). The electronic spectra of Co(II)-substituted Zn(II) enzymes yield valuable information on the coordination geometry of the metal ion. In Co(II)-substituted metallo-β-lactamases, the absorption bands observed above 450 nm correspond to electronic transitions within metal d orbitals (ligand field transitions), whose intensity can be correlated to the metal coordination number. An intense absorption band at 300–350 nm is attributed to a Cys-Co(II) ligand to metal charge transfer transition (Orellano et al. 1998; de Seny et al. 2001).
Addition of Co(II) to the apoenzymes in buffer at pH 7.5 allows the incorporation of two Co(II) ions to the active site of the enzyme. When one equivalent of Co(II) is added, a partial occupation of the Co1 and the Co2 sites is observed (de Seny et al. 2001). Whereas the ligand field bands of mono-Co(II) R121E are identical to those of mono-Co(II) wild-type βcII, the mono-Co(II) R121H shows a different pattern (Fig. 3A▶). We interpret this as being due to the formation of an adduct in which Co(II) is incorporated in a position that do not correspond to any of the canonical metal binding sites, as this intense band is not present neither in the Co1Co2 nor in the Zn1Co2 adducts (see below). Further addition of Co(II) gives rise to the Co1Co2 derivative. In this binuclear Co(II) adduct, the four band pattern characteristic of the tetrahedrally coordinated Co1 is present in all three enzymes, suggesting that the mutations do not lead to a modification of the geometry of the Co1 site (Fig. 3B▶). The R121H spectrum still shows a contribution from the mono-Co(II) adduct under these conditions. The charge transfer bands of both the mono- and bi-Co(II) adducts are again blue shifted, and are split in both mutants. The observed changes in the absorption spectra with Co(II) concentration suggest that the apparent affinities for the formation of mono- and binuclear sites are not altered despite the introduced mutations and the change in the coordination geometry of Co2.
Figure 3.
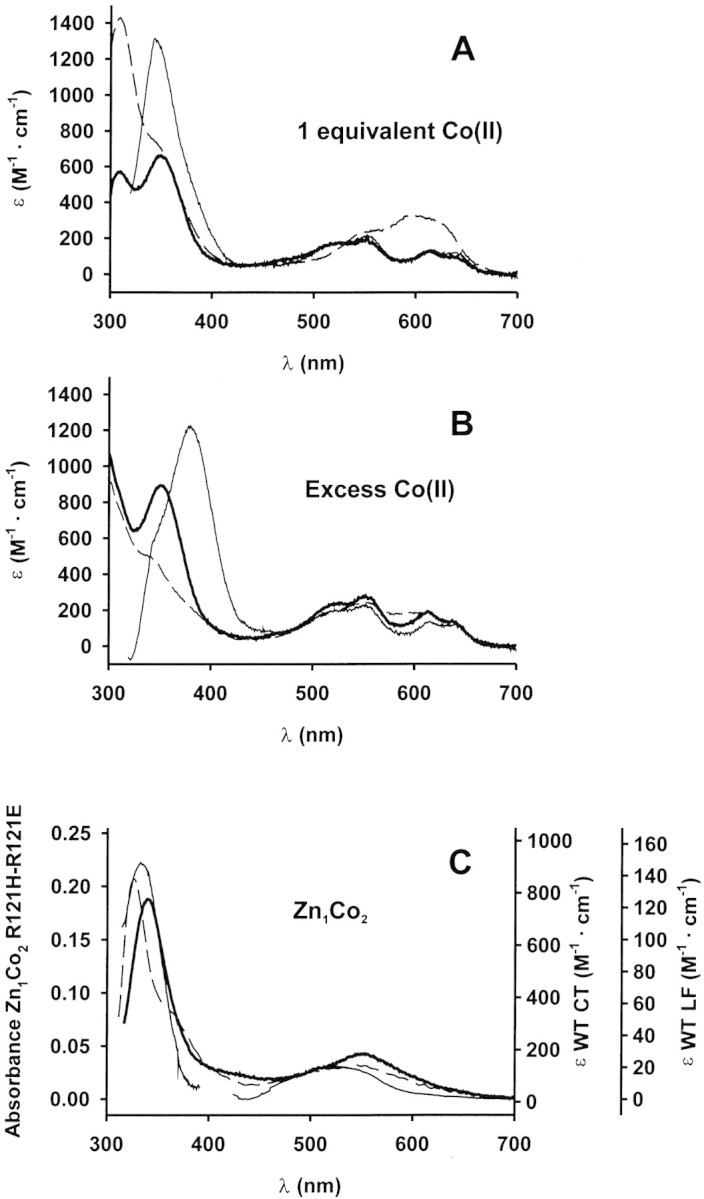
Electronic spectra of metal derivatives. (A, B) UV-visible spectra of the Co(II) adducts in Hepes 15 mM pH 7.0. Enzyme concentrations were 120 μM (wild type, thin line), 91 μM (R121H, dashed line), and 77 μM (R121E, bold line). (A) 1 equivalent of Co(II) added. (B) Excess Co(II) added: 500 μM Co(II) for wild type and R121H BcII and 1 mM Co(II) for R121E BcII. (C) UV-visible spectra of Zn1Co2 derivatives. Left vertical axis (absorbance) corresponds to the mutant enzymes spectra, and right axes (molar absorptivities) correspond to the wild-type enzyme spectrum. The 300–400 nm region of the wild-type BcII Zn1Co2 derivative spectrum (thin line) corresponds to 0.15 mM enzyme reconstituted with 20 mM Co(II). The visible region of the same spectrum was recorded with 8 mM enzyme with 9 mM added Co(II). Spectra of the mutant enzymes were recorded in samples containing 1.7 mM enzyme and 10 mM Co(II) (R121H dashed line), and 3.5 mM enzyme and 10 mM Co(II) (R121E bold line).
To selectively probe the second metal binding site, we obtained the hybrid Zn1Co2 derivatives by addition of Co(II) to enzyme samples extensively dialyzed against 20 mM sodium succinate, 1M NaCl, 20 μM ZnSO4 pH 6.0. This treatment results in the selective incorporation of Co(II) to the second metal binding site (Orellano et al. 1998). Co(II)-substitution in the second metal binding site might not be complete because part of the enzyme may be present in the Zn1Zn2 form. Therefore, molar absorptivities cannot be accurately determined, and the spectral absorbances are reported. Both Zn1Co2 derivatives lack the ligand field bands observed in the Co1Co2 species. This confirms that Co(II) does not replace Zn(II) from the first metal binding site (Fig. 3C▶). The spectra of these hybrid metal derivatives show altered Cys→Co(II) charge transfer bands compared to the wild-type βcII, indicating that the metal binding features of Cys221 are modified. The ligand field absorption features of this site (that were obscured in both the mono and bi-Co(II) derivatives due to the more intense absorption of the Co1 site) are visibly altered respect to wild-type βcII, confirming that coordination geometry of the Co2 site is modified.
Altogether, the results obtained from the electronic spectra of the Co(II) derivatives indicate that the geometry of the first metal binding site is preserved in the mutants, and that alterations are confined to the coordination sphere of the second metal binding site.
XANES spectroscopy
We recorded XANES spectra of the mono-Zn(II) forms of the wild-type and mutant enzymes to directly probe the coordination environment of the zinc sites. Spectra of the three mono-Zn(II) enzymes show close similarity with that of human carbonic anhydrase II (HCAII), indicating that the Zn(II) ion is four-coordinated, with three His and one water ligand (Fig. 4▶). In the hybrid Zn1Co2-derivatives, X-ray absorption allows us to probe separately each metal ion, by working at the corresponding absorption edge. Co(II) uptake in the second metal binding site takes place without inducing noticeable distortions in the first metal binding site, as revealed by the similar XANES spectrum of Zn1Co2-R121H (Fig. 4▶). Unfortunately, XANES experiments to selectively probe Co(II) in the second metal binding site could not be performed due to the excess Co(II) present in samples of the Zn1Co2 derivatives that obscures the XAS signals coming from the second metal binding site of the enzyme.
Figure 4.

XANES of wild type and mutant BcII recorded at the Zn K-edge. Samples were lyophilized and measured as powder. Spectra were recorded at room temperature. The intensity of all the spectra was normalized to unity at 9800 eV.
NMR spectroscopy
1H NMR spectroscopy of Co(II) substituted metalloproteins enables the detection of resonances belonging to proton nuclei near the paramagnetic Co(II) ion, that are shifted outside the diamagnetic envelope (Bertini and Luchinat 1996). We decided to apply this technique to complement XANES data, and thus elucidate the coordination sphere of Co(II) in the hybrid Zn1Co2 derivatives. The 1H NMR spectrum of Zn1Co2 wild-type BcII shows two broad signals (a,b) at 210 ppm already assigned to the Cys221 β-CH2. Four downfield shifted signals (c-f) were assigned to protons from Asp120 and His263 (Fig. 5A▶). Signal c is solvent-exchangeable, and was assigned to the Nɛ proton of His263 (Orellano et al. 1998).
Figure 5.

1H NMR spectra of Zn1Co2 derivatives. (A) Wild-type BcII, (B) R121E BcII, and (C) R121H BcII. Spectra for wild type and R121H BcII were recorded in H2O at 200 MHz and 298 K in 20 mM sodium succinate pH 6.0, 1 M NaCl. Resonances indicated with an asterisk are absent in spectra recorded in D2O. Enzyme concentrations are 5 mM (wild type), 9 mM (R121H), and 6 mM (R121E), respectively. Samples were prepared by adding one Co(II) equivalent to the enzymes dialyzed extensively against the same buffer containing 20 μM Zn(II).
The 1H NMR spectrum of Zn1Co2 R121H and R121E are also reported in Figure 5▶. Broad, downfield shifted signals, characteristic of β-CH2 Cys moieties are found at 230 ppm (R121E, Fig. 5B▶) and 160 ppm (R121H, Fig. 5C▶). This further confirms that Cys221 is bound to Co(II) in the second metal binding site. Histidine ligands can be readily identified on the basis of deuterium exchange of the imidazole NH resonances. The spectrum of Zn1Co2 R121H possesses only one signal absent in D2O (corresponding to an exchangeable proton), whereas no exchangeable signals are evident in the spectrum of Zn1Co2 R121E. The observed shift for the exchangeable resonance in the NMR spectra of Zn1Co2 R121H presents a value somehow smaller than those typically observed for Co(II) bound His. However, no other reasonable candidate for this signal can be found in the ligand sphere of the Co2 site. These data suggest that one His residue is coordinated to the second metal binding site in Zn1Co2 R121H, while no His ligands are present in the second metal binding site in Zn1Co2 R121E. The presence of several resonances of fractional intensity in Zn1Co2 R121H suggests that additional Co(II) ions may be bound under these conditions.
Discussion
Analysis of the spectroscopic data
In the binuclear metallo-β-lactamase L1 from S. maltophilia, His121 binds Zn2 (Fig. 1B▶; Ullah et al. 1998). Thus, the mutated residues in R121H and R121E are potential ligands of the second metal binding site. We studied spectroscopically different Co(II) derivatives to explore the coordination sphere of the two metal binding sites in the mutant enzymes.
The ligand field bands observed in the visible spectra of Co1Co2 R121E and R121H BcII correspond to the first metal binding site. These patterns closely resemble the spectrum of Co1Co2 wild-type BcII (Orellano et al. 1998), and the high pH form of Co(II) substituted HCA II (Bertini et al. 1980). This metallo-hydrolase possesses one metal binding site with a coordination environment identical to that of the first metal binding site of BcII (His3 water). The band intensities clearly correspond to a tetrahedral Co(II) site (Bertini and Luchinat 1983). XANES spectra recorded at the Zn-edge for the mono-Zn(II) derivatives of wild type, R121H and R121E BcII, and HCAII are similar; and this spectral pattern is not modified in the Zn1Co2 R121H derivative. These data indicate that the basic features of the first metal binding site are preserved in the mutant enzymes.
Spectroscopic data suggest that structural changes are confined to the second metal binding site in both mutants, or to the mono-Co(II) derivative of R121H. The Cys→Co(II) charge transfer bands are shifted in the mono-Co(II), Co1Co2 and Zn1Co2 derivatives. The analysis of the hybrid Zn1Co2 adduct allowed us to selectively probe the second metal binding site. The ligand field bands in the UV-visible spectra of both Zn1Co2 derivatives are noticeably different from that of wild-type BcII, evidencing an altered coordination environment respect to wild-type BcII.
The 1H NMR spectra of the Zn1Co2 adducts confirm that Cys221 is bound to Co(II) in the second metal binding site in both mutants. Instead, the number of His ligands differs. No signals corresponding to His ligands are found for the second metal binding site of R121E, whereas in Zn1Co2 R121H, one histidine binds Co(II). This suggests that His263 is not present in the coordination sphere of R121E BcII. In R121H, either His263 or His121 are good candidates for being the His ligand in the second metal binding site. Several signals of fractional intensity are present in the 1H NMR spectrum of Zn1Co2 R121H, possibly arising from Co(II) bound in a lower affinity site.
An explanation consistent with all the above data is that the engineered residue (His121 or Glu121) binds Zn2, forcing His263 to be detached from the metal ion. This hypothesis can be tested by comparing the binuclear sites of wild type BcII and L1 from S. maltophilia (Fig. 6▶). Residues His 116, His 118, Asp 120, and Zn1 were overlaid using the software Deep View (Guex and Peitsch 1997), which resulted in the superimposition of the Cα atoms of Arg121 (βcII) and His121 (L1). The Arg121 residue was mutated to His in BcII, and the side chain was fixed using the exhaustive search tool to eliminate Van der Waals contacts and maximize hydrogen bonding. Modeling the cocatalytic zinc ion of BcII in the same position as found in L1, reveals that Cys221 and His121 can bind the metal ion, whereas His263 is beyond bonding distance from Zn(II), in agreement with the spectroscopic data. His 263 is loosely bound to Zn2 in wild-type BcII, as revealed by the long Nɛ-Zn2 distances (2.4–2.6 Å) in the two molecules present in the asymmetric unit (PDB file 1bc2). In one structure of mono-Zn(II) BcII (PDB file 2bc2), His 263 points toward the bulk solvent. These evidences of the flexibility of His263 make it reasonable to assume that this residue may be detached from the metal ion in the presence of a more buried ligand. This picture holds for the R121E mutant, in which the ligand set of the second metal binding site would be Cys221, Glu121, and Asp120. The structure proposed for the second metal binding site in the mutant enzymes based on the spectroscopic characterization and modeling is clearly different from that observed in the wild-type enzyme.
Figure 6.

Modeled active site of R121H BcII (A) compared with wild-type BcII (B).
Activity and zinc content
Metal content analysis showed that the mutant enzymes bind two equivalents of Zn(II) per mole of enzyme at 5 μM Zn(II). The higher zinc/enzyme ratio present in the mutant enzymes at both Zn(II) concentrations measured suggests a higher macroscopic affinity for the formation of a binuclear site compared to the wild type enzyme. This may be because of the alteration of the second metal binding site that results in a Zn2 site being more deeply buried into the protein environment.
The mutant enzymes retain high β-lactamase activity. In the case of R121E, the catalytic efficiency is comparable to that of the wild-type enzyme, while in R121H the activity is only fivefold reduced respect to the wild type. The relative activities of the mutant enzymes are independent of the substrate and of the concentration of added Zn(II). Although the structures proposed for the second metal binding site of both mutant enzymes are identical, steric restrictions for the simultaneous coordination of Asp120 and His121 may alter the conformation of Asp120. This residue was shown to be important for the catalytic activity and it was suggested to play a key role in catalysis as a proton shuttle (Bounaga et al. 1998; de Seny et al. 2002). The same restrictions would not apply to the R121E mutant, as the glutamate side chain can adopt different possible conformations and could bind Zn2 without disturbing Asp 120.
An alternative explanation for the different behavior of the mutant enzymes would be the formation of a metal site with an altered coordination geometry in the R121H mutant at low metal concentration, that is supported by UV-Visible spectroscopy on the mono-Co(II) derivative (Fig. 3A▶).
The kinetic parameters determined at 5 μM added Zn(II) are assigned to the binuclear form of the enzymes based on the metal stoichiometry determined by AAS. In both R121H and R121E mutant enzymes, almost full β-lactamase activity is retained despite the significant rearrangement induced in the second metal binding site. These results suggest that the catalytic activity relies mainly on the structure of the first metal binding site, while the Zn2 site may adopt different conformations giving rise to active enzymes.
Evolutionary implications
The different functions found in enzymes within the metallo-β-lactamase fold are determined both by the presence of additional domains (a flavodoxin-like domain in ROO and a substrate binding helical domain in glyoxalase II) and by subtle changes in the metal coordination sphere (Figs. 1, 2▶▶). Most enzymes within this family were shown to have hydrolytic activity against a wide range of compounds, suggesting that nature might have exploited this stable fold and a flexible metal site to catalyze a similar reaction on different substrates (Daiyasu et al. 2001). Within the structurally characterized metallo-β-lactamases, the metal ligands are fully conserved with the exception of the more divergent enzyme produced by S. maltophilia (L1). In this metallo-β-lactamase, two active site mutations (R121H and C226S) lead to a different Zn2 coordination environment (Fig. 1B▶). In glyoxalase II, an aspartate residue bridges both metal ions (Fig. 1C▶). Finally, in ROO a histidine residue from the first metal site is replaced by a glutamate, and His121, present in the protein sequence, does not bind the second metal ion (Fig. 1D▶). These changes in coordination environment, although probably not sufficient, may be necessary to change the catalytic activity of these enzymes, and show the flexibility of these metal sites contained in the same protein scaffold. The fact that the R121H and R121E BcII mutants retain high β-lactamase activity despite a significant rearrangement of the Zn2 site reveals that this active site can tolerate some considerable modifications without substantial loss of activity. This tolerance should be a necessary feature for an enzyme fold to evolve the catalytic function over a wide landscape of chemical reactions.
Materials and methods
Materials
All chemicals were of the best quality available. Restriction endonucleases and other DNA modifying enzymes were purchased from Promega. Oligonucleotides were synthesized by Biosynthesis, Inc. Nitrocefin was a kind gift of Glaxo Wellcome. Metal-free buffers were prepared by adding Chelex 100 to regular buffer solutions, and stirring for half an hour. Metal-free dialysis tubing was prepared by boiling for half an hour in distilled water with 1 mM EDTA. Protein concentrations were determined spectrophotometrically using ɛ280 = 30,500 M−1 cm−1 (Paul-Soto et al. 1999).
Site-directed mutagenesis and enzyme purification
The mutant enzymes R121H and R121E BcII were generated by oligonucleotide-directed mutagenesis on single-stranded DNA (Kunkel et al. 1987). Escherichia coli CJ236 cells harboring the plasmid pKS-NH3+, which contains a fragment coding for the first 112 amino acids of BcII, were infected with the helper phage R408. Single-strand plasmid DNA was purified from phage particles. Second-strand synthesis was carried out using the Klenow fragment of DNA polymerase I from E. coli, primed with the following oligonucleotides: 5′-CCGCCAATATGATCAGCAT GCGCATGTG-3′ for R121H and 5′-CCGCCAATTTCATCAG CATGCGCATGTG-3′ for R121E. These primers introduced the desired mutations in position 121 (indicated in bold) and a translationally silent mutation that generates an Sph I restriction site (underlined). Double strand DNA synthesized in this way was used to transform E. coli JM109 cells. Transformant clones were screened for the presence of the Sph I restriction site and sequenced to verify the introduction of the mutation (DNA Sequencing Facility, University of Maine). The BcII gene was reconstructed by ligation of the mutated amino terminal fragment with the wild type carboxy terminal fragment and the pETGEX-CT overexpression vector. Expression and purification of the mutant proteins was performed as previously reported, with typical yields of 20–30 mg pure enzyme/lt. culture (Orellano et al. 1998). Purity of the enzyme preparations was checked by SDS-PAGE.
Zn(II) dependence of the enzymatic activity
Hydrolysis of benzylpenicillin and nitrocefin were followed spectrophotometrically at 235 and 485 nm, respectively. Substrate concentrations were calculated by using Δɛ235 = −800 M−1·cm−1 (benzylpenicillin) and Δɛ485 = 17,400 M−1·cm−1 (nitrocefin). Kinetic measurements were performed in the polybuffer TACS (Tris 50 mM, sodium acetate 50 mM, sodium cacodylate 50 mM, NaCl 0.5 M) at pH 6.0 or in Hepes buffer 15 mM pH 7.5. Reactions were followed in an Ultraspec II LKB spectrophotometer thermostatted at 25°C with a Lauda RC6 circulating water bath. 50 μg/mL bovine serum albumin was always added to the reaction medium.
Metal derivatives
Apoenzymes in Hepes buffer pH 7.5 were prepared as described by de Seny et al. (2001). Apoenzyme preparations retained ≤1% of the penicillinase activity shown by the holoenzymes and their activity could be restored by addition of Zn(II). The Co(II) derivatives were obtained by addition of CoCl2 to solutions of the apoenzyme. To generate the hybrid Zn1Co2 adducts, the mutant enzymes were dialyzed extensively against 20 mM sodium succinate at pH 6.0, 1 M NaCl, 20 μM ZnSO4. Addition of CoCl2 to this preparation results in the selective incorporation of Co(II) to the second metal binding site (Orellano et al. 1998). A remaining fraction of Zn1Zn2 enzyme is not expected to interfere with measurements focused on the Co(II) substituted site.
Spectroscopic measurements
Electronic spectra were recorded in a Gilford Response II spectrophotometer. X-ray absorption spectroscopy (XAS) at Zinc K-Edge was performed in the XAS line placed at the D04 dipolar magnet of the storage ring of the LNLS–National Synchrotron Light Laboratory. Spectra, collected up to ∼150 eV above the Zn K-edge (∼9650 eV), were acquired with an average electron current of 100 mA at 1.37 GeV. An energy resolution of about 0.7 eV was achieved using a Si(111) monochromator and a 1-mm entrance slit. Samples for XANES measurements were lyophilized and measured as powder in fluorescence mode (I/I0). The fluorescence signal I was acquired using one NaI(Tl) scintillator placed 90° relative to the incident beam. The intensity of the incoming X-ray beam I0 was measured using a conventional ionization chamber. The samples, located 45° relative to the incident beam, were placed in 1-mm thick Teflon sample holders supplemented with 0.1-mm thick Mylar windows. Spectra were recorded at room temperature and each one results from the average of at least 10 individual runs. The threshold absorption energy was assumed fixed for all spectra to the value determined using a metallic zinc foil as reference. The intensity of all the spectra was normalized to unity at 9800 eV.
1H NMR spectra were recorded on a Bruker ACE200 spectrometer, and processed with the WinNMR program (Bruker). Deuterated samples were obtained by solvent exchange in Centricon units. Chemical shifts were referenced to the chemical shift of water at the appropriate temperature, which in turn was calibrated against internal DSS. 1D experiments were recorded using the superWEFT pulse sequence. The low signal-to-noise ratio of the spectra precluded their assignment through NOE experiments.
Acknowledgments
This work has been supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (PICT 97–01–00190, PICT 99–01–6616 and PICT 97–0115), CONICET (PIP 0582/99), University of Rosario, and an early career grant from Fundación Antorchas to A.J.V. Partial financial support by National Synchrotron Light Laboratory (LNLS), Brazil and Centro Latinoamericano de Física (CLAF) is also acknowledged. R.M.R. is recipient of a graduate fellowship from CONICET, and A.J.V. and M.C. are staff members of CONICET. A.J.V. is a Carrillo-Oñativia fellow (Ministry of Health, Argentina) and an International Research Scholar of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BcII, metallo-β-lactamase from Bacillus cereus
L1, metallo-β-lactamase from Stenotrophomonas maltophilia
ROO, rubredoxin-oxygen oxydoreductase from Desulfovibrio gigas
HCAII, human carbonic anhydrase II
BSA, bovine serum albumin
EDTA, ethylenedinitrilotetraacetic acid
XANES, X-ray absorption near-edge structure
AAS, atomic absorption spectroscopy
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0301603.
References
- Bertini, I. and Luchinat, C. 1983. An insight on the active site of zinc enzymes through metal substitution. In Metal ions in biological systems (ed. H. Siegel), pp. 101–156. Marcel Dekker Inc., New York.
- ——1996. NMR of paramagnetic substances (ed. A.B.P. Lever), Elsevier Science, Amsterdam.
- Bertini, I., Luchinat, C., and Scozzafava, A. 1980. The acid-base equilibria of carbonic anhydrase. Inorg. Chim. Acta 46 85–89. [Google Scholar]
- Bounaga, S., Laws, A.P., Galleni, M., and Page, M.I. 1998. The mechanism of catalysis and the inhibition of the Bacillus cereus zinc-dependent β-lactamase. Biochem. J. 331 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush, K., Jacoby, G.A., and Medeiros, A.A. 1995. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39 1211–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, A.D., Ridderstrom, M., Olin, B., and Mannervik, B. 1999. Crystal structure of human glyoxalase II and its complex with a glutathione thiolester substrate analogue. Structure. Fold. Des. 7 1067–1078. [DOI] [PubMed] [Google Scholar]
- Concha, N., Rasmussen, B.A., Bush, K., and Herzberg, O. 1996. Crystal structure of the wide-spectrum binuclear zinc β-lactamase from Bacteroides fragilis. Structure 4 823–836. [DOI] [PubMed] [Google Scholar]
- Concha, N.O., Janson, C.A., Rowling, P., Pearson, S., Cheever, C.A., Clarke, B.P., Lewis, C., Galleni, M., Frere, J.M., Payne, D.J., et al. 2000. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: Binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39 4288–4298. [DOI] [PubMed] [Google Scholar]
- Cornaglia, G., Riccio, M.L., Mazzariol, A., Lauretti, L., Fontana, R., and Rossolini, G.M. 1999. Appearance of IMP-1 metallo-β-lactamase in Europe. Lancet 353 899–900. [DOI] [PubMed] [Google Scholar]
- Cricco, J.A. and Vila, A.J. 1999. Class B β-lactamases: The importance of being metallic. Curr. Pharm. Des. 5 915–927. [PubMed] [Google Scholar]
- Cricco, J.A., Rasia, R.M., Orellano, E.G., Ceccarelli, E.A., and Vila, A.J. 1999. Metallo-β-lactamases: Does it take two to tango? Coord. Chem. Rev. 190–192 519–535. [Google Scholar]
- Daiyasu, H., Osaka, K., Ishino, Y., and Toh, H. 2001. Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. FEBS Lett. 503 1–6. [DOI] [PubMed] [Google Scholar]
- de Seny, D., Heinz, U., Wommer, S., Kiefer, M., Meyer-Klaucke, W., Galleni, M., Frere, J.M., Bauer, R., and Adolph, H.W. 2001. Metal ion binding and coordination geometry for wild type and mutants of metallo-β-lactamase from Bacillus cereus 569/H/9 (BcII): A combined thermodynamic, kinetic, and spectroscopic approach. J. Biol. Chem 276 45065–45078. [DOI] [PubMed] [Google Scholar]
- de Seny, D., Prosperi-Meys, C., Bebrone, C., Rossolini, G.M., Page, M.I., Noel, P., Frere, J.M., and Galleni, M. 2002. Mutational analysis of the two zinc-binding sites of the Bacillus cereus 569/H/9 metallo-β-lactamase. Biochem J. 363 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiane, S.M., Sohi, M.K., Wan, T., Payne, D.J., Bateson, J.H., Mitchell, T., and Sutton, B.J. 1998. Crystal structure of the zinc-dependent β-lactamase from Bacillus cereus at 1.9 Å resolution: Binuclear active site with features of a mononuclear enzime. Biochemistry 37 12404–12411. [DOI] [PubMed] [Google Scholar]
- Frazao, C., Silva, G., Gomes, C.M., Matias, P., Coelho, R., Sieker, L., Macedo, S., Liu, M.Y., Oliveira, S., Teixeira, M., et al. 2000. Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nat. Struct. Biol. 7 1041–1045. [DOI] [PubMed] [Google Scholar]
- Frere, J.M. 1995. β lactamases and bacterial resistance to antibiotics. Mol. Microbiol. 16 385–395. [DOI] [PubMed] [Google Scholar]
- Galleni, M., Lamotte-Brasseur, J., Rossolini, G.M., Spencer, J., Dideberg, O., and Frere, J.M. 2001. Standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 45 660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. Swiss-model and the Swiss-Pdbviewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Hernandez-Valladares, M., Felici, A., Weber, G., Adolph, H.W., Zeppezauer, M., Rossolini, G.M., Amicosante, G., Frere, J.M., and Galleni, M. 1997. Zn(II) dependence of the Aeromonas hydrophila AE036 metallo-β-lactamase activity and stability. Biochemistry 36 11534–11541. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–32. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A., Roberts, J.D., and Zakour, R.A. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154 367–382. [DOI] [PubMed] [Google Scholar]
- Orellano, E.G., Girardini, J.E., Cricco, J.A., Ceccarelli, E.A., and Vila, A.J. 1998. Spectroscopic characterization of a binuclear metal site in Bacillus cereus β-lactamase II. Biochemistry 37 10173–10180. [DOI] [PubMed] [Google Scholar]
- Paul-Soto, R., Bauer, R., Frére, J.M., Galleni, M., Meyer-Klaucke, W., Nolting, H., Rossolini, G.M., de Seny, D., Hernández Valladares, M., Zeppezauer, M., et al. 1999. Mono- and binuclear Zn(II) β-lactamase. J. Biol. Chem. 274 13242–13249. [DOI] [PubMed] [Google Scholar]
- Rasia, R.M. and Vila, A.J. 2002. Exploring the role and the binding affinity of a second zinc equivalent in B. cereus metallo-β-lactamase. Biochemistry 41 1853–1860. [DOI] [PubMed] [Google Scholar]
- Rasmussen, B.A. and Bush, K. 1997. Carbapenem-hydrolizing β-lactamases. Antimicrob. Agents Chemother. 41 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah, J.H., Walsh, T.R., Taylor, I.A., Emery, D.C., Verma, C.S., Gamblin, S.J., and Spencer, J. 1998. The crystal strucuture of the L1 metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 Å resolution. J. Mol. Biol. 284 125–136. [DOI] [PubMed] [Google Scholar]
- Wang, Z., Fast, W., Valentine, A.M., and Benkovic, S.J. 1999. Metallo-β-lactamase: Structure and mechanism. Curr. Op. Chem. Biol. 3 614–622. [DOI] [PubMed] [Google Scholar]
- Wommer, S., Rival, S., Heinz, U., Galleni, M., Frere, J.M., Franceschini, N., Amicosante, G., Rasmussen, B., Bauer, R., and Adolph, H.W. 2002. Substrate-activated zinc binding of metallo-β-lactamases: Physiological importance of mononuclear enzymes. J. Biol. Chem. 277 24142–24147. [DOI] [PubMed] [Google Scholar]