

Abstract
Standard methods for measuring free energy of protein unfolding by chemical denaturation require complete folding at low concentrations of denaturant so that a native baseline can be observed. Alternatively, proteins that are completely unfolded in the absence of denaturant can be folded by addition of the osmolyte trimethylamine N-oxide (TMAO), and the unfolding free energy can then be calculated through analysis of the refolding transition. However, neither chemical denaturation nor osmolyte-induced refolding alone is sufficient to yield accurate thermodynamic unfolding parameters for partly folded proteins, because neither method produces both native and denatured baselines in a single transition. Here we combine urea denaturation and TMAO stabilization as a means to bring about baseline-resolved structural transitions in partly folded proteins. For Barnase and the Notch ankyrin domain, which both show two-state equilibrium unfolding, we found that ΔG° for unfolding depends linearly on TMAO concentration, and that the sensitivity of ΔG° to urea (the m-value) is TMAO independent. This second observation confirms that urea and TMAO exert independent effects on stability over the range of cosolvent concentrations required to bring about baseline-resolved structural transitions. Thermodynamic parameters calculated using a global fit that assumes additive, linear dependence of ΔG° on each cosolvent are similar to those obtained by standard urea-induced unfolding in the absence of TMAO. Finally, we demonstrate the applicability of this method to measurement of the free energy of unfolding of a partly folded protein, a fragment of the full-length Notch ankyrin domain.
Keywords: Protein stability, protein folding, Notch ankyrin domain, Barnase, osmolytes
Trimethylamine N-oxide (TMAO) is a naturally occurring osmolyte that is found in several marine organisms containing elevated intracellular urea concentrations (Robertson 1966, 1975; Griffith et al. 1974). Numerous studies have investigated the effect of TMAO on proteins and described its stabilizing effects (Yancey and Somero 1979; Lin and Timasheff 1994; Jaravine et al. 2000). Yancey et al. (1982) showed, by gel filtration chromatography, that TMAO promotes folding of proteins into more compact forms. They further showed, by recovery of enzymatic activity, that TMAO promotes folding to specific, biologically relevant native states (Yancey et al. 1982). Wang and Bolen (1997) provided an explanation for the ability of TMAO to promote specific refolding to the native structure, in which unfavorable thermodynamic interactions between TMAO and the peptide backbone destabilize the denatured state, shifting equilibrium toward the native state. Preferential interaction data from Lin and Timasheff (1994) are consistent with this interpretation, showing the region of solvent near the denatured state of the protein to be rarified in TMAO.
The functional dependence of protein stability on TMAO has been analyzed in several systems and has led to different interpretations. Some studies suggest a linear dependence of free energy of unfolding on TMAO concentration, analogous to the dependence of unfolding free energy on chemical denaturants such as urea and guanidine-HCl. For example, Zou et al. (2002) showed that transfer free energies of cyclic dipeptides increase linearly with increasing molar TMAO concentrations. In addition, Jaravine et al. (2000) showed that free energies calculated from hydrogen exchange measurements on cold-shock protein A vary linearly with TMAO concentration. However, Lin and Timasheff (1994) reported a linear dependence on the thermal unfolding midpoint (Tm) of RNase T1 with TMAO molarity. Because linearity of both Tm and of free energy of unfolding with a cosolvent can only be simultaneously achieved under special circumstances (see Appendix), this observation seems at odds with the linear dependencies of free energy on TMAO described above. Lin and Timasheff (1994) reported free energies of unfolding of RNase T1 that are not linear with TMAO concentration. A similar finding was described by Anjum et al. (2000), who reported a linear dependence of Tm on osmolyte concentration (glycine, proline, sarcosine, and glycine-betaine) for unfolding of lysozyme, ribonuclease A, cytochrome c, and myoglobin, but a free energy of unfolding that is independent of osmolyte concentration.
The combined effects of TMAO and denaturants on peptides and proteins have been shown to be additive in several systems. Transfer free energies of amino acid side chains from water into a mixture of 2 M urea and 1 M TMAO are approximately equal to the algebraic sums of the individual transfer free energies from water into 2 M urea alone and water into 1 M TMAO alone (Wang and Bolen 1997). Experimentally determined preferential interaction parameters of urea with protein are independent of TMAO at 0.5, 1.0, and 2.0 M urea (Lin and Timasheff 1994).
Stabilizing agents such as TMAO can be used to estimate the stability of unstable proteins. For fully unfolded proteins, TMAO can be titrated in to generate a refolding curve, and the same linear relationship used to analyze urea and guanidine denaturation curves can be used to estimate stability in the absence of TMAO (Baskakov and Bolen 1998). For partly folded proteins, titration with TMAO yields a native baseline at high concentrations; however, the denatured state is not sufficiently populated to determine stability from the partial refolding curve. Stabilities of partly folded proteins have been estimated by combining urea or guanidine denaturation with signal estimates for the native state using high concentrations of stabilizing agents, such as ammonium sulfate and glycerol (Shortle et al. 1990). However, this method requires that the native-state signal be independent of the concentrations of both the stabilizing and destabilizing agents.
Here we used a mixed solvent system of urea and TMAO to determine the stability of partly folded proteins by simultaneously adjusting the concentration of both solvents and by treating the effects of both solvents on the observed conformational transitions using global analysis. We investigated the dependence of stability of two well-folded proteins, Barnase and Notch ankyrin domain, on mixtures of the two solvents. The dependence of the stabilities of these two proteins on urea and TMAO mixtures justifies the use of a simple model for the combined effects of these two cosolvents on protein stability. Finally, we show that this model can be used to quantify the free energy of unfolding of a partly folded polypeptide from structural transitions in the mixed-solvent system.
Results and Discussion
The partial unfolding transition of a four-repeat fragment of the Notch ankyrin domain
To estimate unfolding free energies from solvent-induced denaturation, a complete unfolding transition must be observed between two well defined baseline regions. For Nank4–7*, urea-induced unfolding produces only a partial unfolding transition that lacks a native baseline (leftmost transition in Fig. 1A▶). Thus, Nank4–7* is only partly folded even in the absence of urea. Consistent with this, the far-UV CD signal of Nank4–7* is significantly lower (less negative) than for the full-length construct (cf. leftmost transitions in Figs. 1A and 2A▶▶, respectively).
Figure 1.

Structural transitions of Nank4–7*, a partly folded protein, in a mixed urea/TMAO solvent system monitored by circular dichroism at 222 nm. (A) Urea-induced denaturation in increasing TMAO concentrations (from left to right at 0, 0.125, 0.25, 0.375, 0.5, 0.75 M TMAO). Solid lines are the result of fitting Equation 4 to the data. (B) CD signal at 222 nm in various TMAO concentrations in the absence of urea; the solid line is meant to guide the eye.
Figure 2.

Structural transitions of two fully folded proteins in a mixed urea/TMAO solvent system. (A) Urea denaturation curves (CD at 222 nm) of Nank1–7* in increasing TMAO concentrations (from left to right: 0, 0.125, 0.25, 0.375, and 0.5 M TMAO). (B) Urea denaturation curves of Barnase (CD at 230 nm) in increasing TMAO concentrations (from left to right: 0, 0.2, 0.4, 0.6, 0.8, and 1.0 M TMAO). Solid lines are the result of fitting Equation 2 to each urea-induced unfolding transition separately.
For two completely unfolded proteins, RCAM-T1 and a mutant of staphylococcal nuclease (T62P), the addition of TMAO induces complete refolding (Baskakov and Bolen 1998). As expected, the addition of TMAO to Nank4–7* appears to result in complete refolding, as evidenced by a progressive increase in CD signal in the absence of urea to a plateau value that is similar in magnitude to that seen for the full-length construct (Fig. 1B▶). However, owing to the fact that Nank4–7* is partly folded in the absence of TMAO, this induced refolding transition lacks a denatured baseline. Thus, unlike the fully unfolded proteins described above, this partial TMAO-induced refolding transition can not reliably provide estimates of stability, as is also the case with the partial urea-induced unfolding transition of Nank4–7*.
One way to accurately determine the stability of partly folded proteins would be to combine denatured baseline information from urea-induced unfolding with native baseline information from TMAO-induced refolding. For Nank4–7*, this strategy can produce conformational transitions that include both native and denatured baselines, as can be seen in the urea-induced unfolding transition in 0.75 M TMAO (Fig. 1▶, rightmost curve). This denaturation can be fitted to obtain ΔG°urea=0 in 0.75 M TMAO. However, to obtain a value for the free energy of unfolding in the absence of TMAO, the dependence of ΔG° on TMAO and its sensitivity to urea must be determined and accounted for.
The effect of TMAO on unfolding free energy
To determine the effect of TMAO on the free energy of unfolding, we measured urea-induced unfolding of Barnase and Nank1–7* in increasing TMAO concentrations. These proteins show full, baseline-resolved unfolding transitions upon urea denaturation in the absence of TMAO (see Fig. 2A,B▶, leftmost curves); thus they can be used to assess the effects of TMAO on stability over a range of cosolvent concentrations. Increasing TMAO concentrations shifted the urea-induced unfolding transitions of these proteins to higher urea concentrations, without affecting the apparent steepness of the transitions.
Free energy of unfolding in the absence of urea was estimated using the linear extrapolation method (Equation 2) at each TMAO concentration. Free energy values show a roughly linear dependence on molar TMAO concentration for both Barnase and Nank1–7* (Fig. 3▶, closed symbols). The sensitivity of unfolding free energy of Nank1–7* is significantly larger than that of Barnase (Fig. 3▶, Table 1▶), which is consistent with the larger size of Nank1–7*. The observed linear dependence of unfolding free energy on TMAO is consistent with the linear dependence seen for cspA (Jaravine et al. 2000). However, the linear dependence seen here is at odds with the nonlinear TMAO dependence of the unfolding free energy of RNase T1 reported by Lin and Timasheff (1994). Although resolution of this apparent discrepancy will require additional stability measurements, we note that in the same study, the free energy of unfolding also shows a nonlinear dependence on urea (Lin and Timasheff 1994), which is inconsistent with a study of RNase T1 stability by Pace and coworkers (Thomson et al. 1989).
Figure 3.

Dependence of unfolding free energy (closed symbols) and urea m-values (open symbols) on TMAO concentration for Nank1–7* (circles) and Barnase (triangles). Parameters are obtained by fitting urea-induced unfolding curves in different TMAO concentrations using Equation 2. Straight lines are the result of linear regression to each parameter. Correlation coefficients from linear fits to unfolding free energies are 0.98 for Nank1–7* and 0.99 for Barnase.
Table 1.
Thermodynamic parameters calculated from linear extrapolation of urea denaturation and from global analysis of structural transitions in mixtures of urea and TMAO
| Linear fitb |
Global fitc |
|||||
|---|---|---|---|---|---|---|
| nresa | ΔG°H2O (kcal • mole−1) | murea (kcal • mole−1 • M−1) | ΔG°H2O (kcal • mole−1) | murea (kcal • mole−1 • M−1) | mTMAO (kcal • mole−1 • M−1) | |
| Nank1-7* | 256 | 6.55 ± 0.13 (n = 4) | 2.76 ± 0.09 | 6.31 | 2.75 | −6.52 |
| Barnase | 110 | 7.58 ± 0.11 (n = 4) | 1.84 ± 0.02 | 7.95 | 1.55 | −1.66 |
a nres is the number of residues. For the Notch ankyrin domain, only 207 of these residues are likely to participate in the unfolding transition, because the first sequence repeat appears to be disordered (Bradley and Barrick 2002; Zweifel and Barrick).
b The linear fit is from urea-induced unfolding transitions in the absence of TMAO. Errors are standard deviations on parameters from n independent melts (given in parentheses).
c In the global fit, Eq. 4 was fit to the unfolding transitions in Fig. 2▶.
The effect of TMAO on murea (and of urea on mTMAO)
At concentrations required to bring about structural transitions of proteins, urea and TMAO both make up a significant fraction of the solution (Schellman 2002). Thus, mixtures of these two cosolvents may be expected to significantly alter each other’s effects on protein stability. For partly folded proteins, where full unfolding transitions can only be obtained at high concentrations of both cosolvents (i.e., neither cosolvent brings about a complete structural transition in the absence of the other), extrapolation to water requires accounting for potential interaction between cosolvents.
We have used fully folded proteins (Barnase and Nank1–7*) to evaluate how urea and TMAO influence each other’s effect on protein stability. For both Barnase and Nank1–7*, the sensitivity of stability to urea denaturation, murea in Equation 2, is essentially independent of TMAO concentration (Fig. 3▶, open symbols). Based on the definition of murea, this observation establishes the following equality:
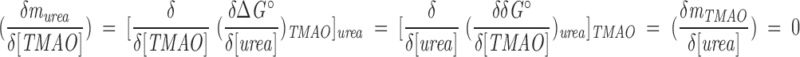 |
(1) |
The middle relationship results from the independence of second partial derivatives on the order of differentiation (this independence is restricted to functions with continuous second derivatives, as is expected of the cosolvent dependence of the free energy of unfolding). In words, Equation 1 states that because TMAO has no influence on the effect of urea on protein stability (Fig. 3▶), urea has no influence on the effect of TMAO on protein stability. Thus, over the concentration range explored here (0–1 M TMAO, 0–5 M urea), these two cosolvents are independent. The independent effects of urea and TMAO on the stabilities of Barnase and Nank1–7* seen here are consistent with the additivity of amino acid solubilities in a 2 M urea/1 M TMAO mixture (Wang and Bolen 1997) and are also consistent with the observation that the preferential interaction coefficient of urea with RNase T1 is independent of TMAO over the range 0–1 M TMAO/0–2 M urea (Lin and Timasheff 1994). Other examples of additivity of cosolvents and cosolutes on stability of macromolecular structures include the additive effects of a variety of pairs of neutral salts on the Tm of collagen (Bello et al. 1956) and the destabilizing effects of methanol and guanidinium chloride on free energy of ubiquitin unfolding (Jourdan and Searle 2001).
Recognition that the cross derivatives in Equation 1 are zero justifies the use of a model in which unfolding free energy is linear in both urea and TMAO molarity (Equation 4). The structural transitions of Nank1–7* and Barnase in the mixed solvent system are well fitted using Equation 4 (not shown; root mean square deviation is 0.2×103deg • cm2 • dmole res−1 for Nank1–7* and 0.04×103deg • cm2 • dmole res−1 for Barnase). Thermodynamic parameters obtained from global fitting (ΔG°H2O and murea) are in good agreement with those obtained by fitting urea-induced unfolding transitions in the absence of TMAO with Equation 2 (leftmost curves, Fig. 2▶ and Table 1▶). Using sensitivity analysis, we find that the thermodynamic parameters are determined to within fairly tight confidence intervals (data not shown), which may be partly attributable to the high coverage of cosolvent space for the two fully folded proteins.
Based on the quality of the fit of the global model to the unfolding of Barnase and Nank1–7*, we used the global model to estimate the stability of Nank4–7*, which is partly folded in the absence of urea (see above). The global model (Equation 4) fits well to the series of Nank4–7* urea unfolding curves at different TMAO concentrations (Fig. 1A▶, solid lines). The value of ΔG°H2O determined from the global fit for Nank4–7* is −0.09 kcal/mole, consistent with the observation that the CD signal in the absence of both cosolvents is midway between the native (TMAO) and denatured (urea) baselines. The use of mixed TMAO/urea cosolvent systems should provide a means to quantify the stability of partly folded proteins without requiring assumptions about native baseline signals.
Materials and methods
Protein expression and purification
Nank1–7* contains seven tandem ankyrin repeat sequences (Zweifel and Barrick 2001a). The construct investigated here contains an N-terminal His6 tag, and has the two internal cysteines substituted by serines. A shorter construct containing the four C-terminal sequence repeats (Nank4–7*) was subcloned using PCR, and contains a C-terminal His6 tag. Recombinant DNA methods used to generate these constructs were essentially as in (Zweifel and Barrick 2001a).
Notch ankyrin polypeptides were expressed in E. coli strain BL21(DE3) as in (Zweifel and Barrick 2001a). Cells were lysed using a French Press (SLM-Aminco) and were clarified by centrifugation at 31,000g. Whereas most of the Nank1–7* polypeptide remained in the supernatant following lysis, Nank4–7* partitioned into the pellet and was purified by solubilization in urea; antiprotease cocktail (Sigma, P8465) was included, per the manufacturer’s instructions, to prevent degradation of this partly folded polypeptide. Notch ankyrin polypeptides were purified as in (Zweifel and Barrick 2001a), with the following modifications: (1) The lysis buffer consisted of 50 mM Tris•HCl and 300 mM NaCl, pH 8.0, and (2) the final dialysis buffer was 25 mM Tris•HCl, pH 8.0.
Barnase was expressed from pMT1022, a construct provided by Dr. Robert W. Hartley (National Institutes of Health, Bethesda, MD). pMT1022 is a derivative of pMT441 (Okorokov et al. 1994), differing by three point mutations: Two of these mutations disrupt the HindIII sites in the lambda repressor gene, and a third mutation results in a Pro to Gly substitution in the phoA signal sequence (at position −10 from the Barnase Ala1); this Pro to Gly substitution yields uniformly processed Barnase lacking the phoA signal sequence. To purify Barnase, cells were resuspended in 50 mM sodium acetate, pH 4.5, (30 mL per L of culture) in the presence of protease inhibitors and were then lysed using a French press. The cleared lysate was diluted with an equal volume of the lysis buffer and then loaded onto an SP Sepharose Fast-Flow column (Amersham Biosciences AB) equilibrated in the same buffer. Barnase was eluted with a linear gradient to 1.5 M ammonium acetate, pH 8.0. Fractions containing Barnase were pooled, concentrated, and chromatographed on a Sephacryl S300 gel filtration column (Amersham Biosciences AB) in 25 mM Tris•HCl and 150 mM NaCl, pH 8.0. Fractions containing Barnase (∼99% pure) were pooled, concentrated, and stored at −80°C. Proper signal-sequence processing was confirmed by mass spectrometry.
Equilibrium denaturations
Urea-induced denaturations were performed in an Aviv 62A DS Spectropolarimeter (Aviv Associates) as described (Zweifel and Barrick 2001b). All denaturations were performed in 25 mM Tris•HCl, pH 8.0. For the Notch ankyrin polypeptides, unfolding transitions were measured at 15°C and monitored by circular dichroism (CD) at 222 nm, a wavelength at which α-helical structure can be quantified. For Barnase, unfolding transitions were measured at 25°C and monitored by CD at 230 nm. The CD signal for Barnase at 230 nm has contributions from both secondary structure and from the large number of aromatic side chains in this protein (Vuilleumier et al. 1993). At higher TMAO concentrations, equilibration times were increased (from 500 to 900 sec) to compensate for increased relaxation times. Ultrapure urea (Amresco) was treated with mixed-bed resin (AG501-X8, Biorad Laboratories) for 1 h and subsequently filtered. Tris•HCl was added to 25 mM, the pH was adjusted to 8.0, and the final urea concentration was determined by refractometry (Pace 1986). TMAO (ICN Biomedicals) was dissolved directly into buffer, and the pH was adjusted to 8.0 with HCl.
Data analysis
For Barnase and Nank1–7*, free energies of unfolding in the absence of urea (ΔG°(urea=0)) and sensitivities of unfolding free energies to urea (murea) were estimated from urea-induced unfolding transitions at fixed TMAO concentrations using the linear extrapolation method (Pace 1986; Santoro and Bolen 1988). In this model, free energy of unfolding is assumed to vary linearly with urea molarity:
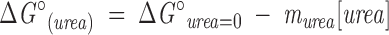 |
(2) |
KU is the unfolding equilibrium constant ([D]/[N]) and is related to the free energy of unfolding as ΔG° = −RTlnKU. The dependence of the unfolding equilibrium constant on urea is obtained by substituting Equation 2 into an exponential rearrangement of Equation 3. The observed CD signal, Yobs, is a population-weighted average of the signal of each state, YD and YN
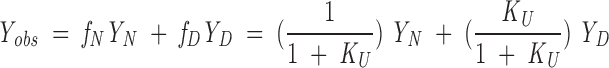 |
(3) |
where fN and fD are the fraction of protein in the native and denatured states, respectively. YN and YD are treated as linear functions of urea concentration. Equation 3 was fitted to urea unfolding transitions using nonlinear least-squares optimization with the program Kaleidagraph 3.0 (Synergy Software) to determine ΔG°urea=0 and murea, along with the four baseline parameters.
The combined effects of urea and TMAO on unfolding free energies were modeled as being linear in both cosolvents.
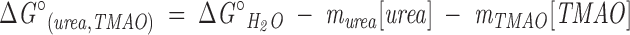 |
(4) |
The linear dependence of free energy of unfolding on TMAO molarity is justified in experiments presented below. Equation 4 was globally fitted to unfolding transitions in mixtures of urea and TMAO (using Nonlin for Macintosh; Brenstein 1989) to yield the free energy of unfolding in the absence of both cosolvents.
Acknowledgments
This work was supported by NIH grant GM60001. We thank Jayant Udgaonkar, Utpal Nath, and Robert W. Hartley for providing Barnase expression constructs.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
TMAO, trimethylamine N-oxide
Nank1-7*, ankyrin domain of Drosophila melanogaster Notch receptor containing seven ankyrin repeats
Nank4-7*, fragment of the ankyrin domain of D. melanogaster Notch receptor containing the four C-terminal ankyrin repeats
Tris•HCl, Tris[hydroxymethyl]aminomethane
Appendix
The relationship between the influence of a cosolvent on Tm and on the free energy of unfolding
Both Tm and ΔG° for protein unfolding reactions are measures of protein stability, and thus the sensitivity of these two parameters to the addition of a cosolvent represents the difference in the thermodynamic interaction of the cosolvent with the native and denatured ensembles. However, due to the relationship between Tm and ΔG°, and to the complicated temperature dependence of protein stability (Privalov and Khechinashvili 1974; Privalov 1979; Becktel and Schellman 1987; Schellman 2002), the particular functional dependence of Tm on cosolvent puts restrictions on the functional dependence of ΔG° on cosolvent, and vice versa. In this appendix, that relationship is examined from the perspective of the effect of an assumed linear relation between Tm and cosolvent concentration on the cosolvent dependence of ΔG° (Lin and Timasheff 1994). Approaching the problem from the other side, that is, examining the effects of an assumed linear relationship between ΔG° and cosolvent concentration on the cosolvent dependence of Tm leads to the same conclusion.
We will represent the assumed linear relation between Tm and cosolvent molarity [x] (urea, guanidine, TMAO, or glycine, for example) as
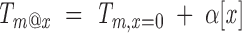 |
A1 |
where Tm,x=0 is the value of Tm in the absence of cosolvent, and α is a constant, independent of both [x] and T. What are the consequences of this relationship on the cosolvent dependence of the free energy of unfolding? At each denaturant concentration, there will be a temperature midpoint (referred to as Tm@x, to emphasize that this is the midpoint temperature obtained at a particular cosolvent concentration; this point in [x], T space is equivalent to the cosolvent concentration midpoint obtained at a given temperature), and at this paired [x], T value,
 |
(A2) |
where the Tm@x subscript indicates that energies and entropies are evaluated at the Tm value that is obtained at a given [x]. The (x) term serves to indicate that each thermodynamic function likely has a dependence on the denaturant concentration, [x]. It should be recognized that because of the effects of cosolvent on stability, Equation A2 is equal to zero not at a single Tm, but at a range of temperatures and cosolvent concentrations at which the native and denatured ensembles have equal populations. These values map to a line (a straight line, if Equation A1 applies) in the [x], T plane at which ΔG°=0.
The assumed linear dependence of Tm on denaturant concentration (Equation A1) can be rearranged and substituted into Equation A2, leading to
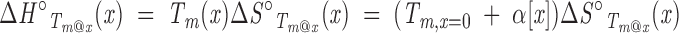 |
(A3) |
This equation puts restrictions on the cosolvent dependencies of ΔH°Tm@x and ΔS°Tm@x. For instance, if ΔS°Tm@x is linear in [x], ΔH°Tm@x will show a second-order dependence.
The consequences of Equation A3 on the free energy of unfolding at temperatures other than Tm can be evaluated by considering the explicit temperature dependence of ΔG°, ΔH°, and ΔS°. In the simplest case dependence, ΔH° and ΔS° are independent of temperature (ΔCp=0; thus, for a given value of [x], ΔH°Tm@x(x) and ΔS°Tm@x(x) apply at temperatures other than Tm)
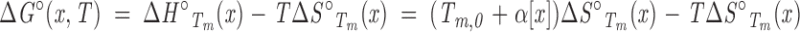 |
(A4) |
Equation A4 shows an explicit linear dependence of ΔG° on cosolvent molarity as long as ΔS°Tm@x(x) is independent of cosolvent. In other words, linear cosolvent dependencies of Tm and ΔG° can both be accommodated as long as ΔH°Tm@x is linear in [x] (with slope and intercept equal to αΔS°Tm@x and Tm,0, respectively), and ΔS°Tm@x is independent of [x] (as is required by Equation A3, if ΔH°Tm@x is to be linear).
Consideration of the effect of [x] on ΔG° in Equation A4 is useful because the relationship is simple, but the restriction that ΔCp=0 limits its application to a narrow range of temperature (at best). In a more appropriate expression, ΔCp is treated as a non-zero value that is independent of temperature (Privalov and Khechinashvili 1974; Privalov 1979; Becktel and Schellman 1987; Schellman 2002):
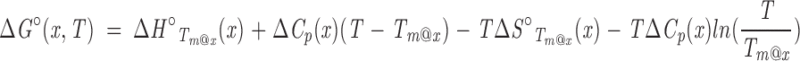 |
(A5) |
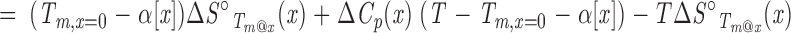 |
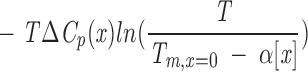 |
Note that Equation A5 is complicated by the fact that Tm@x, which serves as a reference temperature in integration of the Gibbs-Helmholtz equation to yield Equation A5, varies implicitly with denaturant concentration, as do the temperatures at which the constants of integration (ΔH°Tm@x and ΔS°Tm@x) are evaluated. In contrast to Equation A4, the free energy of unfolding in Equation A5 is nonlinear in cosolvent concentration as a result of the logarithmic term, even in the absence of explicit cosolvent dependencies of ΔH°Tm@x, ΔS°Tm@x, and ΔCp. If, as before, ΔH°Tm@x depends linearly on cosolvent molarity (so that ΔS°Tm@x is independent of cosolvent by Equation A3)3 and ΔCp is independent of cosolvent, ΔG° will have a linear-log dependence on [x] (first and fourth terms, respectively, in Equation A5). If ΔCp is also treated as linear in [x], ΔG° will have a quadratic-log dependence on [x] (second and fourth terms, respectively). The same functional dependence results from a linear variation of ΔS° on [x]. Together, consideration of Equations A4 and A5 shows that Tm and ΔG° can be simultaneously linear in [x] only if ΔCp is zero and ΔS°Tm@x is independent of cosolvent, thus imparting a linear dependence of ΔH°Tm@x on cosolvent (Equation A3). Because ΔCp for denaturation of globular proteins is non-zero, this condition cannot be formally met; that is, ΔG° and Tm cannot simultaneously show linear dependences in cosolvent. However, over narrow ranges of temperature, ΔG° may appear to be roughly linear in T (i.e., showing a dependence approximated by Equation A4), especially for proteins where ΔCp is small (typically small proteins; Myers et al. 1995). Under such conditions, ΔG° and Tm both appear to be linear in [x], so long as ΔH°Tm@x and ΔS°Tm@x are linear in and independent of denaturant, respectively.
Several studies have examined the effects of cosolvents on ΔH°, ΔS°, and ΔCp for protein unfolding (Pfeil and Privalov 1976; Makhatadaze and Privalov 1992; Agashe and Udgaonkar 1995; DeKoster and Robertson 1997; Zweifel and Barrick 2002). Makhatadaze and Privalov (1992)) examined the thermodynamics of interaction of urea and guanidine with native and denatured proteins and found significant effects of these cosolvents on both enthalpy and entropy of interaction. These observations are qualitatively consistent with calorimetric studies of the interaction of diketopiperazines with urea (Zou et al. 1998): Urea shows a favorable enthalpy of interaction, and an unfavorable entropy of interaction with the peptide unit. Analogous studies of the interaction between diketopiperazines and TMAO show that both enthalpies and entropies of interaction are significant (Zou et al. 2002). In a study of the effects of temperature and urea on the unfolding of HPr, Nicholson and Scholtz (1996) estimated the entropy and enthalpy of unfolding to decrease in a roughly linear manner with increasing urea concentrations. As the dependence of ΔG° of unfolding of HPr on denaturant was found to be linear over a wide range of temperature and urea, these denaturant dependencies would indicate a nonlinear dependence of Tm on urea, as can be seen in Table 2 of the Nicholson and Scholtz (1996) report. Similar results were found in studies of Barstar (Agashe and Udgaonkar 1995). Recent studies on the urea dependence of the stability of the Notch ankyrin domain show a nonlinear dependence of ΔCp on denaturant concentration, which would also argue against a linear relationship between Tm and urea (Zweifel and Barrick 2002). Thus, both model compound and stability studies indicate that, at least for denaturants such as urea, either Tm or ΔG° should show a nonlinear dependence on denaturant concentration. The widely observed linear dependence of ΔG° on denaturants such as urea and guanidine (Greene and Pace 1974; Thomson et al. 1989; Santoro and Bolen 1992; Agashe and Udgaonkar 1995; Nicholson and Scholtz 1996) argues against a linear dependence of Tm on denaturants. The linear dependence of ΔG° on TMAO observed for proteins in the present study suggests that Tm should show a nonlinear dependence on TMAO concentration, although a more detailed study of the effects of TMAO on thermal denaturation of these proteins would help clarify the issue.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0372903.
Footnotes
Because in Equation A5, ΔCp is assumed to be non-zero, the value of ΔH°Tm@x is expected to vary with [x], simply because the temperature at which it is evaluated (Tm@x) varies with [x] as assumed in Equation A1. Even if ΔH° does not depend on [x] at a fixed temperature, ΔHTm@x will show an [x] dependence because of its temperature dependence through ΔCp.
References
- Agashe, V.R. and Udgaonkar, J.B. 1995. Thermodynamics of denaturation of barstar: Evidence for cold denaturation and evaluation of the interaction with guanidine hydrochloride. Biochemistry 34 3286–3299. [DOI] [PubMed] [Google Scholar]
- Anjum, F., Rishi, V., and Ahmad, F. 2000. Compatibility of osmolytes with Gibbs energy of stabilization of proteins. Biochim. Biophys. Acta 1476 75–84. [DOI] [PubMed] [Google Scholar]
- Baskakov, I. and Bolen, D.W. 1998. Forcing thermodynamically unfolded proteins to fold. J. Biol. Chem. 273 4831–4834. [DOI] [PubMed] [Google Scholar]
- Becktel, W.J. and Schellman, J.A. 1987. Protein stability curves. Biopolymers 26 1859–1877. [DOI] [PubMed] [Google Scholar]
- Bello, J., Riese, H.C.A., and Vinograd, J.R. 1956. Mechanism of gelation of gelatin. Influence of certain electrolytes on the melting points of gels of gelatin and chemically modified gelatins. J. Phys. Chem. 60 1290–1306. [Google Scholar]
- Bradley, C.M. and Barrick, D. 2002. Limits of cooperativity in a structurally modular protein: Response of the Notch ankyrin domain to analogous alanine substitutions in each repeat. J. Mol. Biol. 324 373–386. [DOI] [PubMed] [Google Scholar]
- Brenstein, R.J. 1989. Nonlin for Macintosh 0.9.8b4. Robelko Software, Carbondale, IL.
- DeKoster, G.T. and Robertson, A.D. 1997. Calorimetrically derived parameters for protein interactions with urea and guanidine-HCl are not consistent with denaturant m values. Biophys. Chem. 64 56–98. [DOI] [PubMed] [Google Scholar]
- Greene, R.F. and Pace, C.N. 1974. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, α-chymotrypsin, and β-lactoglobulin. J. Biol. Chem. 249 5388–5393. [PubMed] [Google Scholar]
- Griffith, R.W., Umminger, B.L., Grant, B.F., Pang, P.K.T., and Pickford, G.E. 1974. Composition of bladder urine of the coelacanth Latimeria chalumnae. J. Exp. Zool. 196 371–380. [DOI] [PubMed] [Google Scholar]
- Jaravine, V.A., Rathgeb-Szabo, K., and Alexandrescu, A.T. 2000. Microscopic stability of cold shock protein A examined by NMR native state hydrogen exchange as a function of urea and trimethylamine N-oxide. Protein Sci. 9 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan, M. and Searle, M.S. 2001. Insights into the stability of native and partially folded states of ubiquitin: Effects of cosolvents and denaturants on the thermodynamics of protein folding. Biochemistry 40 10317–10325. [DOI] [PubMed] [Google Scholar]
- Lin, T.-Y. and Timasheff, S.N. 1994. Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein. Biochemistry 33 12695–12701. [DOI] [PubMed] [Google Scholar]
- Makhatadaze, G.I. and Privalov, P.L. 1992. Protein interactions with urea and guanidinium chloride. A calorimetric study. J. Mol. Biol. 226 491–505. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, E.M. and Scholtz, J.M. 1996. Conformational stability of the Escherichia coli HPr protein: Test of the linear extrapolation method and a thermodynamic characterization of cold denaturation. Biochemistry 35 11369–11378. [DOI] [PubMed] [Google Scholar]
- Okorokov, A.L., Hartley, R.W., and Panov, K.I. 1994. An improved system for ribonuclease Ba expression. Protein Expr. Purif. 5 547–552. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Pfeil, W. and Privalov, P.L. 1976. Thermodynamic investigations of proteins ii. Calorimetric study of lysozyme denaturation by guanidine hydrochloride. Biophys. Chem. 4 33–40. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. 1979. Stability of proteins: Small globular proteins. Adv. Protein Chem. 33 167–241. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. and Khechinashvili, N.N. 1974. A thermodynamic approach to the problem of stabilization of globular protein structure: A calorimetric study. J. Mol. Biol. 86 665–684. [DOI] [PubMed] [Google Scholar]
- Robertson, J.D. 1966. Osmotic constituents of the blood plasma and parietal muscle of Myxine glutinosa L. In Some contemporary studies in marine science (ed. H. Barnes), pp. 631–644. Allen and Unwin, London.
- ———. 1975. Osmotic constituents of the blood plasma and parietal muscle of Squalous acanthias l. Biol. Bull. 148 303–319. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method 1. Unfolding of phenymethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- ———. 1992. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry 31 4901–4907. [DOI] [PubMed] [Google Scholar]
- Schellman, J.A. 2002. Fifty years of solvent denaturation. Biophys. Chem. 96 91–101. [DOI] [PubMed] [Google Scholar]
- Shortle, D., Stites, W.E., and Meeker, A.K. 1990. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry 29 8033–8041. [DOI] [PubMed] [Google Scholar]
- Thomson, J.A., Shirley, B.A., Grimsley, G.R., and Pace, C.N. 1989. Conformational stability and mechanism of folding of ribonuclease-T1. J. Biol. Chem. 264 11614–11620. [PubMed] [Google Scholar]
- Vuilleumier, S., Sancho, J., Loewenthal, R., and Fersht, A.R. 1993. Circular dichroism studies of barnase and its mutants: Characterization of the contribution of aromatic side chains. Biochemistry 32 10303–10313. [DOI] [PubMed] [Google Scholar]
- Wang, A. and Bolen, D.W. 1997. A naturally occurring protective system in urea-rich cells: Mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry 36 9101–9108. [DOI] [PubMed] [Google Scholar]
- Yancey, P.H. and Somero, G.N. 1979. Counteraction of urea destabilization of protein structure by methylamine osmoregulatory compounds of elasmobranch fishes. Biochem. J. 183 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey, P.H., Clark, M.E., Hand, S.C., Bowlus, R.D., and Somero, G.N. 1982. Living with water stress: Evolution of osmolyte systems. Science 217 1214–1222. [DOI] [PubMed] [Google Scholar]
- Zou, Q., Habbermann-Rottinghaus, S.M., and Murphy, K.P. 1998. Urea effects on protein stability: Hydrogen bonding and the hydrophobic effect. Proteins 31 107–115. [PubMed] [Google Scholar]
- Zou, Q., Bennion, B.J., Daggett, V., and Murphy, K.P. 2002. The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea. J. Am. Chem. Soc. 124 1192–1202. [DOI] [PubMed] [Google Scholar]
- Zweifel, M.E. and Barrick, D. 2001a. Studies of the ankyrin repeats of the Drosophila melanogaster Notch receptor. 1. Solution conformational and hydrodynamic properties. Biochemistry 40 14344–14356. [DOI] [PubMed] [Google Scholar]
- Zweifel, M.E. and Barrick, D. 2001b. Studies of the ankyrin repeats of the Drosophila melanogaster Notch receptor. 2. Solution stability and cooperativity of unfolding. Biochemistry 40 14357–14367. [DOI] [PubMed] [Google Scholar]
- ———. Relationships between the temperature dependence of solvent denaturation and the denaturant dependence of protein stability curves. Biophys. Chem. 101–102 221–237. [DOI] [PubMed]
- ———. 2002. Relationships between the temperature dependence of solvent denaturation and the denaturant dependence of protein stability curves. Biophys. Chem. 101–102 221–237. [DOI] [PubMed] [Google Scholar]