

Abstract
We have determined the crystal structure of a phosphatase with a unique substrate binding domain from Thermotoga maritima, TM0651 (gi 4981173), at 2.2 Å resolution by selenomethionine single-wavelength anomalous diffraction (SAD) techniques. TM0651 is a member of the haloacid dehalogenase (HAD) superfamily, with sequence homology to trehalose-6-phosphate phosphatase and sucrose-6F-phosphate phosphohydrolase. Selenomethionine labeled TM0651 crystallized in space group C2 with three monomers per asymmetric unit. Each monomer has approximate dimensions of 65 × 40 × 35 Å3, and contains two domains: a domain of known hydrolase fold characteristic of the HAD family, and a domain with a new tertiary fold consisting of a six-stranded β-sheet surrounded by four α-helices. There is one disulfide bond between residues Cys35 and Cys265 in each monomer. One magnesium ion and one sulfate ion are bound in the active site. The superposition of active site residues with other HAD family members indicates that TM0651 is very likely a phosphatase that acts through the formation of a phosphoaspartate intermediate, which is supported by both NMR titration data and a biochemical assay. Structural and functional database searches and the presence of many aromatic residues in the interface of the two domains suggest the substrate of TM0651 is a carbohydrate molecule. From the crystal structure and NMR data, the protein likely undergoes a conformational change upon substrate binding.
Keywords: X-ray crystallography, structural proteomics, phosphatase, HAD family, new fold, gi 4981173
A large family of magnesium-dependent phosphatase/phosphotransferase enzymes is present in both prokaryotic and eukaryotic organisms (Collet et al. 1998; Selengut and Levine 2000; Wang et al. 2001). These enzymes belong to the L-2-haloacid dehalogenase (HAD) superfamily characterized by three conserved sequence motifs and a common catalytic mechanism (Fig. 1▶). Motif I is DXXX(T/V), in which the first aspartate forms a phosphoaspartate intermediate with the substrate (Collet et al. 1998). Motif II contains a conserved serine or threonine, which has been proposed to hydrogen-bond to a phosphoryl oxygen (Wang et al. 2001). Motif III, K-(X)18–30–(G/S)(D/S)XXX(D/N), also forms part of the active site and helps coordinate the magnesium ion required for activity (Morais et al. 2000; Wang et al. 2001). The structures of several members of the HAD superfamily have been determined (Welch et al. 1998; Ridder et al. 1999; Toyoshima et al. 2000; Wang et al. 2001, 2002; Galburt et al. 2002; Lahiri et al. 2002; Parsons et al. 2002; Rinaldo-Matthis et al. 2002). Most of them are multidomain proteins that share a phosphatase domain with an α/β-hydrolase fold. Characteristic protein phosphoaspartate intermediates in this family function in signal transduction in two-component response regulator proteins (Welch et al. 1998), phosphotransfer reactions in phosphatases and mutases (Wang et al. 2002), and the conversion of chemical energy to ion gradients in P-type ATPases (Toyoshima et al. 2000).
Figure 1.

Sequence comparison between TM0651 and its homologs. Several homologs of TM0651, all with defined substrate specifity, were selected for a sequence comparison. Abbreviations are as follows: (S6P) Sucrose-6F-phosphate phosphohydrolase from Synechocystis sp. PCC 6803 (Lunn et al. 2000), (SPPA) sucrose-phosphate phosphatase (sucrose-6-phosphate hydrolase) from Nostoc sp. PCC 7120 (gi:14594809; http://www.ncbi.nlm.nih.gov), (T6P) trehalose-6-phosphate phosphatase from E. coli (Giaever et al. 1988), (IDW) % whole sequence identity with TM0651, (Homo) % whole sequence homology with TM0651, (IDCA, IDCB, IDCC, IDCD) relative % cap domain sequence identities versus TM0651, S6P, SPPA, and T6P, respectively. The sequence alignment was done based on sequence comparison and the secondary structure of TM0651. The secondary structure derived from TM0651 is shown above the sequence. The blue character H represents a sequence belonging to an α-helix, a green G for 310-helix, a pink β for β-strand, and "I" for the pi-helix. The sequence identical to TM0651 is colored red. The three motifs are represented as a light yellow box with essential active site residues in dark green characters. The cap domain (residues from 82 to 189) is boxed with scarlet. The "—"s represent gaps and the residue numbers refer to those of TM0651.
An examination of the sequence lengths of the regions between motifs I and II and between motifs II and III shows that there are structural subgroups within the HAD superfamily (Selengut and Levine 2000). One group, consisting of phosphoserine phosphatase (PSP), β-phosphoglucomutase, 2-deoxyglucose-6-phosphatase, phosphoglycolate phosphatase, P-type ATPase, phosphonatase, and haloacid dehalogenase (Selengut and Levine 2000), has a large domain between motifs I and II that is believed to be critical for substrate binding or catalysis revealed by several crystal structures (Selengut and Levine 2000). A second group, consisting of phosphomannomutase, sucrose phosphatase, and trehalose phosphatase, contains a large domain between motifs II and III (Selengut and Levine 2000). No crystal structures of the enzymes in this group have been available until now. The third group, consisting of histidinol phosphatase, the DEM-1 DNA kinase/phosphatase, and MDP-1, has neither of these large domains (Selengut and Levine 2000).
An open reading frame of Thermotoga maritima codes for a hypothetical protein, TM0651, of 27.9 kD molecular mass (Nelson et al. 1999). A PSI-BLAST search of this sequence revealed around 180 proteins with full-length sequence identity ranging from 23% to 34% and an E-value below 4E-10. Most of the homologous sequences are annotated as putative hydrolases of the HAD superfamily. In the Pfam databases (Bateman et al. 2000), TM0651 matches with HAD-like hydrolase (Pfam00702) and trehalose phosphatase (Pfam02358). In Escherichia coli K12, the closest hypothetical protein to TM0651 is ybhA (Blattner et al. 1997). In Mycoplasma pneumoniae, the closest homolog MPN383 is an essential gene as assayed by global transposon mutagenesis (Hutchison et al. 1999).
We have determined the three-dimensional structure of TM0651, which belongs to the second group of the HAD superfamily, by X-ray crystallography and discuss structural characteristics of the protein.
Results and Discussion
Quality of the model
The final model includes all residues except the last, Glu268. Most of the residues are well defined by electron density in the refined models of TM0651 (Fig. 2A▶). The final model has been refined at 2.2 Å resolution to a crystallographic R-factor of 21.7%. The root-mean-square (RMS) deviations from ideal stereochemistry are 0.012 Å for bond lengths, 1.4° for bond angles, and 0.74° for improper angles. The averaged B-factors for main chain atoms and side chain atoms are 31.1 Å2 and 39.5 Å2, respectively. In the TM0651 model, the residues showing higher B-factors are located around loops (Arg35 and Glu155) and C-termini (Leu266 and Asp267). Table 1A▶ summarizes the refinement statistics as well as model quality parameters. The mean positional error in atomic coordinates for the refined model is estimated to be 0.30 Å by a Luzzati plot (Luzzati 1952). All residues lie in the allowed region of the Ramachandran plot produced with PROCHECK (Laskowski et al. 1993).
Figure 2.
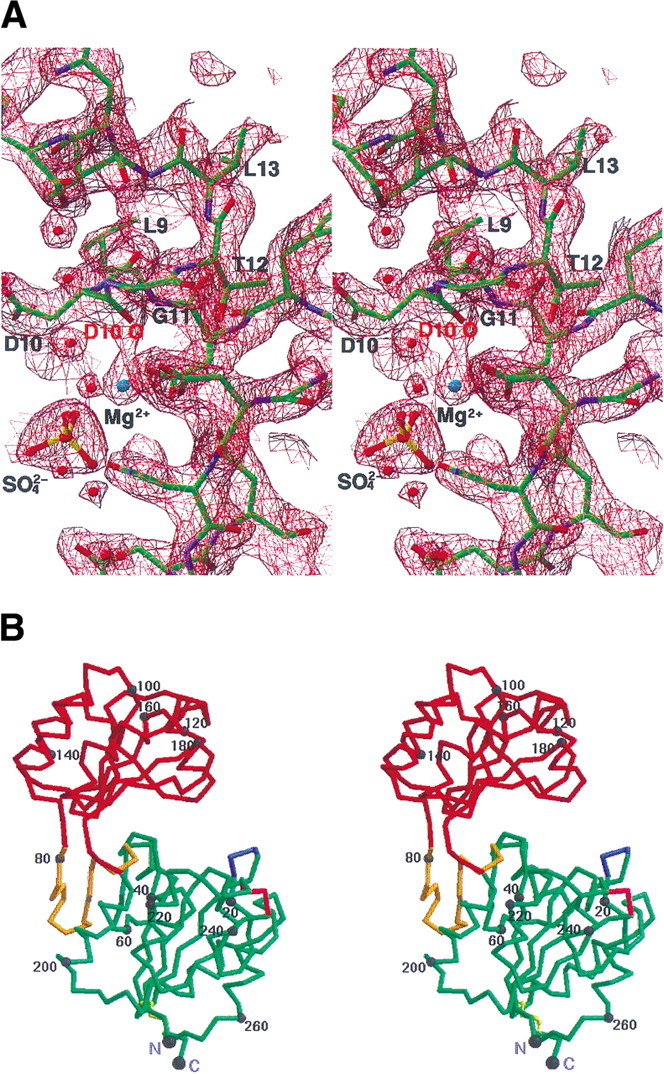

(A) A stereo view of a refined electron-density map around pi-helix countered at 1.8σ. The 2fo–fc map from the finally refined phase was calculated using all reflection data between 20 and 2.2 Å. The figure was generated using the program RIBBONS (Carson 1991). The dark red net represents the electron-density map. The residues around the pi-helix are represented by a ball-and-stick model. The residues belonging to the pi-helix are labeled. Blue represents nitrogen atoms, red for oxygen, yellow for sulfur, green for carbon, and light blue for the magnesium ion. The backbone oxygen that coordinates the magnesium ion is labeled. (B) A stereo drawing of a Cα trace of TM0651. The core domain (the minimal common structural domain in the HAD superfamily colored green plus the extra domain colored gold) and the cap domain (red) are represented by a thick line. The blue and the pink represent where the insertion occurs in other HAD superfamily structures. One disulfide bridge between Cys35 and Cys265 is drawn by a yellow ball-and-stick model. Every 20th residue is numbered and represented by a dot. The N (residue Met1) and C termini (residue Asp267) are labeled. The figure was generated by MOLSCRIPT (Kraulis 1991). (C) A topology diagram of TM0651. For the simple topologic representation, only α-helices and β-strands are represented. α-Helices are represented by cylinders and β-strands by thick arrows. Secondary structure elements are labeled. The light gray box represents the minimal common structural domain in the HAD superfamily.
Table 1A.
Statistics of X-ray diffraction data and structure refinement
| Crystal parameters and refinement statistics | |
|---|---|
| Space group | C2 |
| Cell dimensions | A = 158.41Å b = 93.88Å c = 98.42Å β = 127.91° |
| Volume fraction of protein | 58.6% |
| Vm (Å3/Dalton) | 3.09 |
| Total number of residues | 801 |
| Total non-H atoms | 6591 |
| Number of water molecules | 494 |
| Number of sulfate ions | 6 |
| Number of magnesium ions | 3 |
| Average temperature factors | |
| Protein | 35.5 Å2 |
| Solvent | 44.2 Å2 |
| Magnesium ions | 39.1 Å2 |
| Sulfate ions | 34.3 Å2 |
| Resolution range of reflections used | 20.0–2.2 Å |
| Amplitude cutoff | 0.0 σ |
| R-factor | 21.7% |
| Free R-factor | 26.3% |
| Stereochemical ideality: bond | 0.012 Å |
| angle | 1.36° |
| improper | 0.74° |
| dihedral | 23.22° |
Overall structure
The Cα trace of the atomic model of TM0651 is shown in Figure 2B▶. The protein has approximate dimensions of 65 × 40 × 35 Å3 with the core and the cap domains connected by two hinge loops. Both domains belong to the three-layer (αβ)-sandwich, an architecture in a mixed α-β class, according to the CATH classification (Fig. 2C▶; Orengo et al. 1997). The core domain is composed of an eight stranded β-sheet surrounded by six α-helices. There is one disulfide bridge between Cys35 and Cys265 that may increase thermostability of TM0651 by decreasing the flexibility of the N- and C- terminal structure (Fig. 2B▶). The cap domain consists of a six stranded β-sheet surrounded by four α-helices (Fig. 2B,C▶).
We searched for TM0651's structural homologs in the Protein Data Bank (PDB) with the program DALI (Holm and Sander 1997). As individual domain searches gave better matches compared to the whole protein search, we summarize the results of search for each domain. As expected from the sequence analysis, the DALI search revealed five HAD superfamily members that show a z-score above 10.0 for the core domain. The RMS deviations of these five with TM0651 are 2.3 Å (for 143 pairs of aligned Cα atoms for YrbI from Haemophilus influenzae [HI1679]; 1j8d-A; z = 18.0), 2.3 Å (for 133 Cα atoms for phosphoserine phosphatase; 1f5s-A; z = 14.4), 2.9 Å (for 135 Cα atoms for phosphonoacetaldehyde hydrolase; 1fez-A; z = 12.2), 2.6 Å (for 137 Cα atoms for calcium-transporting ATPase; 1eul; z = 10.9), and 2.7 Å (135 Cα atoms for l-2-haloacid dehalogenase; 1qq5-A; z = 10.6). An aspartic acid is believed to form a covalent intermediate with the substrate in all these proteins.
The cap domain did not show any high matches with other known structures (88 hits show a z-score between 2.0 and 4.3). The best hit was with a ribosome recycling factor with RMS deviation of 3.0 Å (for 63 Cα atoms; 1ek8; z = 4.3). However, the overall fold and topology of the two structures are different. Therefore, the cap domain has a unique structural topology.
In summary, a structural homology search indicated that TM0651 consists of one domain of known fold, and one domain of novel fold (Fig. 2B,C▶).
Core domain and active site structure comparison
Comparison of the five best structural homologs of the core domain of TM0651 using the DALI search revealed a common structural motif in the HAD superfamily (Fig. 2B,C▶). Five β-stands surrounded by six α-helices were common in all structures of the core domain, although the last α-helix was not well conserved in the L-2-haloacid dehalogenase structure. All the active site residues were compared with those of PSP according to the detailed studies by Wang et al. (2001). All members showed that the highly conserved active site residues were located in almost the same position as in PSP. The positions of functional residues in TM0651, Asp10 (Asp 13 in PSP structure) known to act as a general base, Asp8 (Asp11 in PSP) acting as the nucleophile, K191 (K144 in PSP) stabilizing the transition state, and Ser41 (Ser99 in PSP) interacting with a bound phosphate, are well conserved in the structure (Fig. 3A▶).
Figure 3.



(A) Comparison of active site residues. The residues around the active sites are labeled and represented by a ball-and-stick model. Gray represents TM0651, and dark gray, PSP. (B) The electrostatic surface potential of TM0651. Molecular surfaces were created by the program GRASP (red, negative; blue, positive; white, uncharged; Nicholls et al. 1991). The magnesium and the sulfate ions were not included for the surface charge calculation. The right side figure was drawn after 180° rotation of the left figure around the y-axis. The surface of the active site is highly negatively charged, as shown in the figure on the right. (C) A stereo ribbon diagram of TM0651 with aromatic residues around the inter domain gap. The figure was generated using the program RIBBONS (Carson 1991). TM0651 is drawn as a ribbon model. The aromatic residues are labeled and represented as red ball-and-stick models. The sulfate and magnesium ion are represented by light gray ball-and-stick models.
An interesting feature of the active site is the presence of a pi-helix (4.416-helix) from Leu9 to Leu13, detected by the automatic algorithm STRIDE (Frishman and Argos 1995; Fig. 2A▶). A search for the occurrence of the rare pi-helix was performed and revealed that most pi-helices have been directly linked to the formation or stabilization of a specific binding site within proteins (Weaver 2000). The pi-helix is more loosely constructed than the α-helix via the hydrogen-bonding pattern between carbonyl oxygen of ith residue and nitrogen backbone atom of (i + 5)th residue. The position of backbone oxygen of Asp10 is unique in the TM0651 structure (Fig. 2A▶). Unlike the normal pi-helix, the backbone oxygen is coordinated to a magnesium ion rather than a backbone nitrogen atom (Fig. 2A▶). Comparing all five structural homologs with TM0651, this pi-helix region is structurally similar, including the Asp10 backbone oxygen position. However, except in PSP, all the others are structurally defined as type II β-turn or type IV β-turn structures instead of pi-helices by STRIDE (Frishman and Argos 1995).
In the active site of TM0651, there are one magnesium ion and one sulfate ion. The magnesium ion that was coordinated by Asp11, Asp167, and the phosphate ion in PSP interacts with Asp8, Asp214, and a sulfate ion in TM0651. The location of the sulfate ion is almost the same as that of the phosphate ion in the PSP structure (Fig. 3A▶). The positions of four amide groups (Asp13, Ser14, Gly100, and Asn170) important for activity of PSP (Wang et al. 2001) are also similar in the TM0651 structure (Asp10, Gly11, Gly42, and Asn217, respectively). The reaction intermediate of TM0651 may be stabilized through interactions with Mg2+ as in PSP. The electrostatic surface potential of TM0651 indicates a strong negative charge distribution around the active site as shown in the other HAD family structures (Fig. 3B▶).
When the core domain structure of TM0651 is compared with that of CheY, the central regulator of bacterial chemotaxis, there is structural homology with RMS deviation of 3.4 Å (for 85 pairs of aligned Cα atoms for CheY from Escherichia coli; 3chy; z = 3.6), as expected from their analogous phosphatase activity (Volz and Matsumura 1991). However, although the CheY structure represents a minimal domain of phosphoaspartase activity, there are structural differences with HAD family. First, CheY does not have a pi-helix motif. Second, as its topology is not the same as the HAD family, its active site is overlapped only if the secondary structure of CheY is permutated as discussed by Wang et al. (2002). Therefore, if they are aligned without permutation, their active sites have approximate twofold rotational relationship.
Characteristics of the cap domain and possible substrates
One of the interesting structural features of TM0651 is the distribution of aromatic residues. Four tyrosines and two phenylalanines are located in the gap between the two domains (Fig. 3C▶). Of these, Tyr105, Tyr122, Phe178, and Tyr181 compose a cluster of aromatic rings. Aromatic residues are known to play an important role in binding carbohydrates in some proteins (Vyas 1991). In the maltodextrin/maltose-binding protein, the carbohydrate binding region has four aromatic residues (Tyr341, Trp340, Tyr155, and Trp230) that are involved in stacking interactions (Duan et al. 2001). It is also well known that galactose binding is almost always accompanied by a stacking interaction with an aromatic residue against the B face of the sugar (Pratap et al. 2002).
A Blast and an iterative PSI-Blast search using the TM0651 sequence gave hits with several phosphatases including trehalose-6-phosphate phosphatase, sucrose-phosphate phosphatase, and sucrose-6F-phosphate phosphohydrolase. A sequence alignment based on the TM0651 structure with some phosphatases of known function shows that these proteins have full-length sequence homology to TM0651 including the active site residues, except around the H5 helix (Fig. 1▶), which is located far from the active site (Fig. 2B▶). The combined information of the sequence comparison and the TM0651 structure support a carbohydrate molecule as a possible substrate for TM0651. The gap between the core and cap domains of TM0651 has dimensions of about 8 × 20 × 25 Å3 (Fig. 3C▶). As the size of phosphorylated disaccharides is around 5 × 8 × 13 Å3, the gap has plenty of room to accommodate phosphorylated carbohydrate molecules such as trehalose-6-phosphate or sucrose-6F-phosphate.
To identify another possible active site in TM0651, two structural databases containing known structures and functions were queried for similar residue constellations as in TM0651. Two of the active site templates in the PROCAT database (Wallace et al. 1997) of functional groups in enzyme active sites match residues in the TM0651 structure. Glu231 and Glu288 of the Glucan endo-1,3-β-d-glucosidase template (PDB ID: 1ghs) have an RMS deviation of 1.23 Å with Glu86 and Glu93 of TM0651. Glu160 and Glu375 of the 6-phospho-β-galactosidase template (1pbg) match with Glu117 and Glu118 of TM0651 with an RMS deviation of 1.52 Å.
A database search for the presence of a known protein motif by RIGOR (Kleywegt 1999) gave 44 motifs that are found in the TM0651 structure. Eleven of them match the clusters of hydrophobic residues in known protein structures. Other motifs located around the active site of TM0651 are related to binding substrates such as N-acetyl-d-glucosamine or inositol-1,4– bisphosphate. Finally, there are sites related to metal ion binding; a magnesium ion with an RMS deviation of 0.94 Å (1bpy: Asp190, Asp192, Asp256–Asp8, Asp218, Asp214: TM0651), and two calcium ion sites with RMS deviations of 0.96 Å (1bn8: Asp184, Asp223, Asp227–Asp8, Asp10, Asp214: TM0651) and 0.50 Å (2por: Asp93, Asp95, Asp100, Asp101–Asp218, Asp8, Asn215, Asp214: TM0651).
Thus, the metal binding site of TM0651 was well confirmed by comparison with other protein structures from databases. In addition, possible carbohydrate binding motifs were found, although no electron density was detected for those molecules in our structure. However, this is not unexpected, as reaction products would not be expected to be tightly bound. No other prominent active site was detected.
NMR experiment
It has been known that the phosphatase reaction in HAD family members proceeds through the formation of a phosphoaspartate bond (Yan et al. 1999; Cho et al. 2000, 2001; Lahiri et al. 2002). Evidence of an equivalent intermediate for TM0651 is provided by NMR data. In both the response regulators NtrC and CheY, and the HAD member PSP, the addition of beryllofluoride (BeF3−) has been shown to form a mimic of the phosphorylated protein. Crystal structures show BeF3− binds stably in the active site and interacts with the active aspartate in a geometry similar to the phosphoryl group (Yan et al. 1999; Cho et al. 2000, 2001). To test whether BeF3− binds to TM0651, BeF3− was titrated into a 15N -labeled protein sample. A heteronuclear single quantum correlation (HSQC) spectrum was collected after each addition (Mori et al. 1995). As BeF3− is added to TM0651, many amide peaks move, appearing at new chemical shifts (Fig. 4▶). There is no noticeable broadening of the peaks, indicating a slow off-rate and the formation of a stable BeF3−–TM0651 complex. The magnitude and nature of the shifts appear to be intermediate between those seen for PSP, in which the shift of most amide peaks indicated a large domain movement, and receiver domains, in which the shifts were concentrated around the active site residues, and complexation resulted in a subtle structural change (Yan et al. 1999; Cho et al. 2000, 2001).
Figure 4.
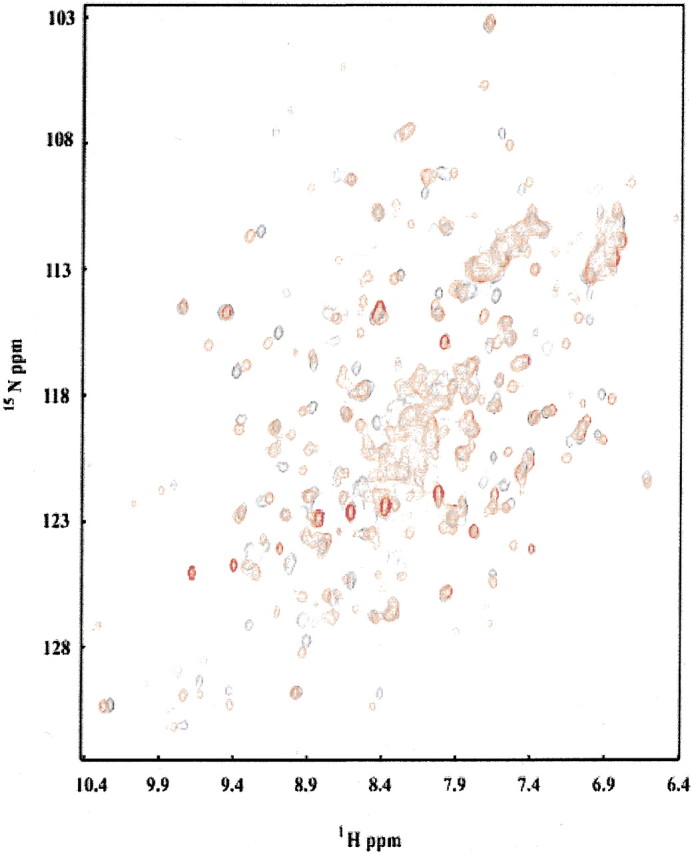
Overlay of HSQC spectra of TM0651 with (red) and without (black) BeF3−. Protein concentration was ∼0.5 mM and the NMR buffer contained 12 mM Mg2+ and 0.5 mM BeF3−.
The two domains of PSP appear to be in a "closed configuration" in the BeF3− • PSP complex (Cho et al. 2001). The widespread changes in the 1H-15N HSQC spectrum of PSP on addition of BeF3− were suggested to arise from a transition of its two domains from a more open to the closed configuration, in which phosphate would be hydrolyzed (Cho et al. 2001). The NMR results from the BeF3−–TM0651 complex suggest that a similar structural change occurs between the open and closed conformations during substrate binding for TM0651. The presence of two hinge loops connecting the core and the cap could facilitate such a conformational change (Fig. 3C▶). However, structures of the ligated state are needed to determine the details of structural changes that may occur upon formation of the BeF3−–TM0651 complex.
Phosphatase assay
To test the proposed phosphatase activity of TM0651, which was supported by NMR data and structural analysis, a general phosphatase assay was performed using p-nitrophenyl phosphate as a substrate. The Km and kcat values determined were 16 ± 3 mM and 0.1 ± 0.02 sec−1. These values reflect typical activities for nonspecific substrates (Parsons et al. 2002), but verify that TM0651 has phosphatase activity.
In summary, the structure of TM0651 provides the first view of a HAD family member containing a large domain between motifs II and III. Because the topology of the three-layer (αβ)-sandwich cap domain is unique, we cannot identify the substrate for this enzyme by structural homology. However, sequence alignment, features of the structure, NMR data, and a nonspecific phosphatase assay strongly suggest that TM0651 acts as phosphatase with a phosphorylated carbohydrate molecule as a possible substrate. Even though a specific molecular substrate for the protein is not immediately evident, the structure provides a framework to deduce and assay substrates based on clustered conserved residues and general fold characteristics.
Materials and methods
Cloning of TM0651
Cloning primers (Invitrogen Corp.) for TM0651 gene amplification from genomic DNA by PCR contained an NdeI restriction site in the forward primer (5′- CATATGTACAGGGTGTTCGTTTTT GACCT-3′) and a BamHI site in the reverse primer (5′-GGATC CTTACTCATCAAGACAATCTGTGGAAATGC-3′). PCR was performed using Deep Vent Polymerase (New England Biolabs, Inc.) and Thermotoga maritima genomic DNA. The PCR product was cloned into the pCR-BluntII-TOPO vector (Invitrogen), and the TM0651 gene insert was confirmed by DNA sequencing. The amplified TOPO vector was cut with NdeI and BamHI and the gene insert was purified by agarose gel electrophoresis. This insert was ligated into pET21a (Novagen, Inc.), which had been digested with NdeI and BamHI and then was transformed into DH5α cell. A plasmid containing the gene insert was transformed into BL21(DE3)pSJS1244 for protein expression (Kim et al. 1998).
Protein expression, purification, and crystallization
A selenomethionine derivative of the protein was expressed in a methionine auxotroph, E. coli strain B834(DE3)/pSJS1244 (Leahy et al. 1992; Kim et al. 1998), grown in M9 medium supplied with selenomethionine. In the purification process, the cell lysate was subjected to heating (80°C for 30 min) to precipitate most of E. coli proteins. After heating, the supernatant was fractionated using anion exchange chromatography on a HiTrap-Q column (Amersham Biosciences Corp.). The protein eluted in 50 mM Tris-HCl, pH 6.8, and 100 mM NaCl. SDS-PAGE showed one band around 30 kD, corresponding to the molecular weight of TM0651. Dynamic light scattering confirmed this, showing a monodisperse peak of the monomer size. The initial crystallization conditions were screened by the sparse matrix method using the Hampton Research Kits (Jancarik and Kim 1991) at room temperature. In the optimized crystallization conditions, 1 μL of the protein (108 mg/mL) in 50 mM Tris-HCl, pH 6.8, 220 mM NaCl, was mixed with 1 μL of 5 mM DTT, 1.0 M ammonium sulfate, and 0.1 M citric acid at pH 5.5. The hanging drop was equilibrated with 0.5 mL of 1.0 M ammonium sulfate, and 0.1 M citric acid at pH 5.5. Thick pyramidal shaped crystals grew in a month to approximate dimensions of 0.1 × 0.1 × 0.05 mm3.
Data collection and reduction
The selenomethionine incorporated crystals were soaked in a drop of mother liquor with 20% glycerol (about 10 μL) for about 1 min before being flashed-cooled in liquid nitrogen and used for X-ray data collection. X-ray diffraction data to 3.0 Å were collected at a single wavelength (0.97938 Å; Table 1B▶) at the Macromolecular Crystallography Facility beamline 5.0.2 at the Advanced Light Source at Lawrence Berkeley National Laboratory using an Area Detector System Co. Quantum 4 CCD detector placed 140 mm from the sample. The oscillation range per image was 1.0° with no overlap between two contiguous images. X-ray diffraction data were processed and scaled using DENZO and SCALEPACK from the HKL program suite (Otwinowski and Minor 1997). The native X-ray diffraction data set was collected to 2.2 Å. Data statistics are summarized in Table 1B▶.
Table 1B.
Statistics of the peak wavelength SAD data set and native data set
| Data set | Peak | Native |
|---|---|---|
| Wavelength (Å) | 0.97938 | 1.00000 |
| Resolution (Å) | 83.3–3.0 | 83.3–2.2 |
| Redundancy | 3.4 (3.1)a | 3.7 (3.5)a |
| Unique reflections | 51235 (2600) | 57785 (2866) |
| Completeness (%) | 99.6 (99.7) | 99.8 (99.7) |
| I/σ | 8.3 (1.5) | 15.3 (3.2) |
| Rsymb (%) | 14.9 (74.4) | 9.6 (35.3) |
a Numbers in parentheses refer to the highest resolution shell, which is 3.05–3.00 Å for all peak wavelength data set and 2.24–2.20 Å for native data set.
bRsym = Σhkl Σi|Ihkl,i − ≤I>hkl|/Σ|≤I>hkl|.
Structure determination and refinement
The program SOLVE (Terwilliger and Berendzen 1999) was used to locate the selenium sites in the crystal and found a best interpretable map from 20.0 to 3.5 Å resolution data. The initial single-wavelength anomalous dispersion phases were further improved by solvent flattening and histogram matching with the DM program in the CCP4 package (CCP4 1994). The map calculated by using the improved phases was not good enough to trace the backbone structure of the protein. However, the presence of a twofold noncrystallographic symmetry (NCS) was recognized among selenium sites, and twofold NCS density averaging was carried out using DM (CCP4 1994). The improved density map revealed that there were three subunits in the asymmetric unit, and the three partial models were built using the O program (Jones et al. 1991). Based on these, three NCS matrices were found and these matrices were refined using software in the RAVE package of real-space averaging and density-manipulation programs (Kleywegt and Jones 1999). A model containing 267 residues was derived from progressive improvement of the electron density map using DMMULTI (CCP4 1994).
The preliminary model was then refined against a native data set collected later using the program CNS (Brunger et al. 1998). The reflections in this data set between 20.0 Å and 2.2 Å were included throughout the refinement calculations. Ten percent of the data were randomly chosen for free R-factor crossvalidation. The refinement statistics are shown in Table 1A▶. Isotropic B-factors for individual atoms were initially fixed to 20 Å2, and were refined in the last stages. The 2Fo − Fc and Fo − Fc maps were used for the manual rebuilding between refinement cycles and for the location of solvent molecules. When the refined B-factor of a solvent molecule exceeded 70 Å2, it was removed. Atomic coordinates have been deposited in the Protein Data Bank (PDB) with the access code of 1NF2.
NMR sample preparation
Uniformly labeled 15N TM0651 was extracted from cells grown in M9 minimal media with 15N ammonium chloride as the sole nitrogen source. The labeled protein was purified according to the unlabeled protocol. Purified protein was concentrated to ∼0.5 mM in 20 mM HEPES pH 7.22, 50 mM NaCl, 8 mM DTT, 10 μM PMSF, 0.02% NaN3, and 12 mM MgCl2. A concentrated stock solution of BeF3− was made by adding a seven molar excess of NaF to BeCl2. This stock solution was used for the titration. 1H-15N HSQC spectra were recorded at 315 K on a DRX 600 spectrometer (Bruker Instruments).
Phosphatase assay
para-Nitrophenyl phosphate (Sigma-Aldrich) was dissolved in a solution of 50 mM Tris pH 8.0, 5 mM DTT, and 20 mM MgCl2. Serial dilutions of this stock solution were either incubated with TM0651 (total concentration 300 nM as determined by UV absorbance at 280 nm) or with an equivalent volume of buffer as a control. All reactions were carried out at 70°C, according to the thermophilic origin of the protein. Aliquots of the reaction solutions were taken at different timepoints and diluted into 100 mM NaOH to stop the reaction. Progress of the reaction was monitored at 410 nm, and the amount of p-nitrophenol released was calculated by taking the difference in absorbance between the controls and corresponding protein-containing reaction solutions using a molar absorptivity coefficient of 18,400 M−1cm−1 (Parsons et al. 2002). Kinetic constants were determined from the initial rates of hydrolysis calculated at substrate concentrations of 50, 40, 30, 20, 15, 10, 7.5, and 5 mM.
Acknowledgments
We thank Dr. David King for mass spectrometric analysis of the protein and Dr. Gerry McDermott (Advanced Light Source, Lawrence Berkeley National Laboratory) for assistance during data collection. The work described here was supported by the NIH GM 62412.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0302703.
References
- Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Howe, K.L., and Sonnhammer, E.L. 2000. The Pfam protein families database. Nucleic Acids Res. 30 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner, F.R., Plunkett III, G., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277 1453–1474. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Carson, M. 1991. RIBBONS 2.0. J. Appl. Crystallogr. 24 958–961. [Google Scholar]
- Cho, H.S., Lee, S.-Y., Yan, D., Pan, X., Parkinson, J.S., Kustu, S., Wemmer, D.E., and Pelton, J.G. 2000. NMR structure of activated CheY. J. Mol. Biol. 297 543–551. [DOI] [PubMed] [Google Scholar]
- Cho, H.S., Wang, W., Kim, R., Yokota, H., Damo, S., Kim, S.-H., Wemmer, D.E., Kustu, S., and Yan, D. 2001. BeF(3)(−) acts as a phosphate analog in proteins phosphorylated on aspartate: Structure of a BeF(3)(−) complex with phosphoserine phosphatase. Proc. Natl. Acad. Sci. 98 8525–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project Number 4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Collet, J.F., Stroobant, V., Pirard, M., Delpierre, G., and Van Schaftingen, E. 1998. A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif. J. Biol. Chem. 273 14107–14112. [DOI] [PubMed] [Google Scholar]
- Duan, X., Hall, J.A., Nikaido, H., and Quiocho, F.A. 2001. Crystal structures of the maltodextrin/maltose-binding protein complexed with reduced oligosaccharides: Flexibility of tertiary structure and ligand binding. J. Mol. Biol. 306 1115–1126. [DOI] [PubMed] [Google Scholar]
- Frishman, D. and Argos, P. 1995. Knowledge-based protein secondary structure assignment. Proteins 23 566–579. [DOI] [PubMed] [Google Scholar]
- Galburt, E.A., Pelletier, J., Wilson, G., and Stoddard, B.L. 2002. Structure of a tRNA Repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure 10 1249–1260. [DOI] [PubMed] [Google Scholar]
- Giaever, H.M., Styrvold, O.B., Kaasen, I., and Strom, A.R. 1988. Biochemical and genetic characterization of osmoregulatory trehalose synthesis in Escherichia coli. J. Bacteriol. 170 2841–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1997. Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 25 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison, C.A., Peterson, S.N., Gill, S.R., Cline, R.T., White, O., Fraser, C.M., Smith, H.O., and Venter, J.C. 1999. Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286 2165–2169. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. and Kim, S.-H. 1991. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Crystallogr. 24 409–411. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kim, R., Sandler, S.J., Goldman, S., Yokota, H., Clark, A.J., and Kim, S.-H. 1998. Overexpression of archaeal proteins in Escherichia coli. Biotech. Lett. 20 207–210. [Google Scholar]
- Kleywegt, G.J. 1999. Recognition of spatial motifs in protein structures. J. Mol. Biol. 285 1887–1897. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. and Jones, T.A. 1999. Software for handling macromolecular envelopes. Acta Crystallogr. D 55 941–944. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lahiri, S.D., Zhang, G., Dunaway-Mariano, D., and Allen, K.N. 2002. Caught in the act: The structure of phosphorylated β-phosphoglucomutase from Lactococcus lactis. Biochemistry 41 8351–8359. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Leahy, D.J., Hendrickson, W.A., Aukhil, I., and Erickson, H.P. 1992. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science 258 987–991. [DOI] [PubMed] [Google Scholar]
- Lunn, J.E, Ashton, A.R., Hatch, M.D., and Heldt, H.W. 2000. Purification, molecular cloning, and sequence analysis of sucrose-6F-phosphate phosphohydrolase from plants. Proc. Natl. Acad. Sci. 97 12914–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzati, V. 1952. Traitement statistique des erreurs dans la determinationn des structures cristallines. Acta Crystallogr. 5 802–810. [Google Scholar]
- Morais, M.C., Zhang, W., Baker, A.S., Zhang, G., Dunaway-Mariano, D., and Allen, K.N. 2000. The crystal structure of Bacillus cereus phosphonoacetaldehyde hydrolase: Insight into catalysis of phosphorus bond cleavage and catalytic diversification within the HAD enzyme superfamily. Biochemistry 39 10385–10396. [DOI] [PubMed] [Google Scholar]
- Mori, S., Abeygunawardana, C., Johnson, M.O., and Vanzijl, P.C.M. 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108 94–98. [DOI] [PubMed] [Google Scholar]
- Nelson, K.E., Clayton, R.A., Gill, S.R., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Nelson, W.C., Ketchum, K.A., et al. 1999. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature 399 323–329. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., and Thornton, J.M. 1997. CATH—A hierarchic classification of protein domain structures. Struct. Fold Des. 5 1093–1108. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Parsons, J.F., Lim, K., Tempczyk, A., Krajewski, W., Eisenstein, E., and Herzberg, O. 2002. From structure to function: YrbI from Haemophilus influenzae (HI1679) is a phosphatase. Proteins 46 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap, J.V., Jeyaprakash, A.A., Rani, P.G., Sekar, K., Surolia, A., and Vijayan, M. 2002. Crystal structures of Artocarpin, a moraceae lectin with mannose specificity, and its complex with methyl-α-d-mannose: Implications to the generation of carbohydrate specificity. J. Mol. Biol. 317 237–247. [DOI] [PubMed] [Google Scholar]
- Ridder, I.S., Rozeboom, H.J., Kalk, K.H., and Dijkstra, B.W. 1999. Crystal structures of intermediates in the dehalogenation of haloalkanoates by L-2-haloacid dehalogenase. J. Biol. Chem. 274 30672–30678. [DOI] [PubMed] [Google Scholar]
- Rinaldo-Matthis, A., Rampazzo, C., Reichard, P., Bianchi, V., and Nordlund, P. 2002. Crystal structure of a human mitochondrial deoxyribonucleotidase. Nat. Struct. Biol. 9 779–787. [DOI] [PubMed] [Google Scholar]
- Selengut, J.D. and Levine, R.L. 2000. MDP-1: A novel eukaryotic magnesium dependent phosphatase. Biochemistry 39 8315–8324. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405 647–655. [DOI] [PubMed] [Google Scholar]
- Volz, K. and Matsumura, P. 1991. Crystal structure of Escherichia coli CheY refined at 1.7 Å resolution. J. Biol. Chem. 266 15511–15519. [DOI] [PubMed] [Google Scholar]
- Vyas, N.K. 1991. Atomic features of protein–carbohydrate interactions. Curr. Opin. Struct. Biol. 1 732–740. [Google Scholar]
- Wallace, A.C., Borkakoti, N., and Thornton, J.M. 1997. TESS: A geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases. Application to enzyme active sites. Protein Sci. 6 2308–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W., Kim, R., Jancarik, J., Yokota, H., and Kim, S.-H. 2001. Crystal structure of phosphoserine phosphatase from Methanococcus jannaschii, a hyperthermophile, at 1.8 Å resolution. Structure 9 65–71. [DOI] [PubMed] [Google Scholar]
- Wang, W., Cho, H.S., Kim, R., Jancarik, J., Yokota, H., Nguyen, H.H., Grigoriev, I.V., Wemmer, D.E., and Kim, S.-H. 2002. Structural characterization of the reaction pathway in phosphoserine phosphatase: Crystallographic "snapshots" of intermediate states. J. Mol. Biol. 319 421–431. [DOI] [PubMed] [Google Scholar]
- Weaver, T.M. 2000. The pi-helix translates structure into function. Protein Sci. 9 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, M., Chinardet, N., Mourey, L., Birck, C., and Samama, J.P. 1998. Structure of the CheY-binding domain of histidine kinase CheA in complex with CheY. Nat. Struct. Biol. 5 25–29. [DOI] [PubMed] [Google Scholar]
- Yan, D., Cho, H.S., Hastings, C.A., Igo, M.M., Lee, S.-Y., Pelton, J.G., Stewart, V., Wemmer, D.E., and Kustu, S. 1999. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. 96 14789–14794. [DOI] [PMC free article] [PubMed] [Google Scholar]