

Abstract
Phage display enables the presentation of a large number of peptides on the surface of phage particles. Such libraries can be tested for binding to target molecules of interest by means of affinity selection. Here we present SiteLight, a novel computational tool for binding site prediction using phage display libraries. SiteLight is an algorithm that maps the 1D peptide library onto a three-dimensional (3D) protein surface. It is applicable to complexes made up of a protein Template and any type of molecule termed Target. Given the three-dimensional structure of a Template and a collection of sequences derived from biopanning against the Target, the Template interaction site with the Target is predicted. We have created a large diverse data set for assessing the ability of SiteLight to correctly predict binding sites. SiteLight predictive mapping enables discrimination between the binding and nonbinding parts of the surface. This prediction can be used to effectively reduce the surface by 75% without excluding the binding site. In 63% of the cases we have tested, there is at least one binding site prediction that overlaps the interface by at least 50%. These results suggest the applicability of phage display libraries for automated binding site prediction on three-dimensional structures. For most effective binding site prediction we propose using a random phage display library twice, to scan both binding partners of a given complex. The derived peptides are mapped to the other binding partner (now used as a Template). Here, the surface of each partner is reduced by 75%, focusing their relative positions with respect to each other significantly. Such information can be utilized to improve docking algorithms and scoring functions.
Keywords: Binding-site, phage display library, artificial evolution, active site prediction, graph algorithms
Protein binding sites are modules interacting with proteins, other macromolecules, and small ligands. These interactions are responsible for protein complex formation as well as governing diverse biologic pathways. Predicted binding sites are a promising starting point for pharmacologic target identification, drug design studies, and protein engineering. In addition, these sites can assist in identifying protein function, guide docking, and establish networks of protein–protein interactions.
Several computational methods exist for predicting protein interaction sites. These attempt to predict binding sites at different resolutions: entire domains, a sequence window, or at the single amino acid level. The majority of these methods are sequence-based. The “proline-brackets” method takes advantage of the high frequency of prolines near interaction sites (Kini and Evans 1995). Other approaches are based on correlated mutations (Pazos et al. 1997) or coevolution of proteins with their interaction partners (Lichtarge et al. 1996a,b, 1997; Goh et al. 2000; Sowa et al. 2001). According to the correlated mutations approach, residues close to an interaction site are expected to mutate simultaneously during evolution. On the other hand, the coevolution approach looks for simultaneous mutations in two interacting partners rather than in a single protein.
Some of the sequence-based methods also rely on the genomic context (Dandekar et al. 1998). Gene order conservation is considered a fingerprint for interacting proteins. A different type of conservation, primary structure conservation, was also utilized for binding site prediction methods. Several sorts of conservation were taken into consideration in different methods: residues that are conserved within a subfamily of proteins but that differ between subfamilies, residues that are conserved in a few subfamilies (Fariselli et al. 2002), and domain homologies (Marcotte et al. 1999). Domain homologies were used for site prediction on genome sequences (Marcotte et al. 1999). Interaction between two proteins is suggested if in another organism homologous domains are assembled as a single protein. Interaction sites involving hydrophobic residues are proposed to be predicted using the mean α-helical hydrophobic moment (Xavier et al. 2000). In contrast to the above-mentioned methods for interaction site prediction based on primary structure, which uses additional information, a support vector training machine uses solely the physicochemical properties of the sequence (Bock and Gough 2001).
Some methods use both sequence and structural data for the prediction. Sequence profile, together with solvent accessible surface area and neighboring residues, were used for binding site prediction using neural networks (Huan-Xiang and Yibing 2001). Finally, structure-based methods are also used for binding site prediction. Superposition requires a homologous protein with a known binding site, whereas docking (reviewed in Halperin et al. 2002) requires structures (or models) of the two binding molecules. Some of the existing computational methods for binding site prediction cannot be applied to all proteins. Domain shuffling and hydrophobic residue involvement in the interaction are examples of assumptions that do not hold for all proteins, and are a fundamental requirement for computational methods for binding site prediction. Nevertheless, protein binding sites are generally hydrophobic (Tsai et al. 1997), with large, though variable, extent of nonpolar surface areas. Further, information about active site residues is sometimes available from site-directed mutagenesis, chemical cross-linking, and phylogenetic data (Gabb et al. 1997; Blizynuk and Gready 1999). Here, even in the absence of experimental data, it is sometimes possible to predict the correct binding site (Aloy et al. 2001). Functional regions in proteins have also been identified by surface mapping of phylogentic information (Armon et al. 2001). Potential hydrogen bonding groups, enzyme clefts, and charged sites on a protein surface, have all been used for binding site prediction (Gilson and Honig 1987; Laskowski 1995; Laskowski et al. 1996; Frommel et al. 1996; Bliznyuk and Gready 1999; Pettit and Bowie 1999). Because binding sites are at least partially flexible, searches for part-flexible part-rigid sites have also produced encouraging results (Todd et al. 1998; Freire 1999; Todd and Freire 1999; Luque and Freire 2000). Algorithms that predict the location of hinges and modes of motions (e.g., Hayward et al. 1997), or those that carry out structural comparisons of a protein family, in particular, if they allow hinge-bending movements (Shatsky et al. 2000, 2002) should be useful as well. Binding sites may, in principle, be predicted through residue hot spots (Bogan and Thorn 1998; DeLano 2000, 2002; Hu et al. 2000).
Several experimental strategies can be used for the analysis of the spatial organization of protein complexes. These include chemical cross-linking, two-hybrid systems, hydrogen–deuterium exchange, protein microarrays, random mutagenesis, inhibition assays, alanine scanning, protection from chemical alteration, or proteolytic digestion and phage display, to name a few. These methods can provide four types of data: (1) constraints (i.e., proximity of specific residues from opposing partners in a complex); (2) binding site location (i.e., assignment of the binding site to a specific fragment); (3) hot spots determination (i.e., identification of those residues that contribute dominantly to the binding energy); and (4) binding site characterization (i.e., characterize a set of sequences that bind a target molecule or the consensus properties required for binding). Some of these methods can be applied not only in a case-specific manner, but also provide a generic tool. One fruitful method for mapping interactions of protein complexes is screening phage display libraries for peptide ligands (Geysen et al. 1986; Ferrer and Harrison 1999; Li et al. 2001; Wu et al. 1999, 2000; reviewed by Sidhu et al. 2003).
A critical aspect of phage display is the construction of combinatorial peptide libraries. Synthetic oligonucleotides, fixed in length but with unspecified codons, can be cloned as fusions to capsid genes of a filamentous phage (Enshell-Seijffers et al. 2001). These libraries, often referred to as random peptide libraries, can then be tested for binding to target molecules of interest. This is often done using a form of affinity selection known as Biopanning (Kay et al. 1996). Once a combinatorial library is built, it can be applied to a wide array of macromolecules, proteinaceous and nonproteinaceous, those that are known to interact with small peptides and those that had previously undefined specificity for peptides. Only a modest amount of time, effort, and resources are needed for biopanning a library displaying up to 1013 different peptides.
The potential of phage display for computational binding site prediction has been shown recently (Tong et al. 2002). Phage display and a large-scale two-hybrid system were combined for computational prediction of interaction sites. Consensus sequences based on phage display peptides were used to search genomic sequences for potential ligands. The intersection between the phage display prediction and the two-hybrid system results is expected to yield biologically relevant sites. This strategy was applied successfully to SH3 binding proteins in yeast. The SH3 binding motifs are sequential rather than "truly" 3D, that is, they are not discontinuous order-independent residues on the binding site surface. Nevertheless, although this strategy was applied only to chain-contiguous epitopes and was not yet examined on a diverse data set, the correlation between the phage display binding site mapping, the two-hybrid system, and previous biochemical data in the examined proteins is encouraging.
Here we present SiteLight, a novel computational tool for prediction of a binding site on a 3D structure using phage display libraries. SiteLight is applicable to complexes made up of a protein termed Template, and any type of molecule, termed Target. Given the 3D structure of a Template and a collection of sequences derived from biopanning against the Target, SiteLight predicts the interaction site of the Template with the Target. The algorithm can be divided into three main stages: (1) a combinatorial division of the Template surface to overlapping patches; (2) a one-dimensional (1D) to 3D alignment of peptide sequences with surface patches; (3) scoring the derived matches and assessing the results. SiteLight was implemented in C++, and runs on the order of a minute (on Red-Hat Linux 7.1, 1 processor, Pentium 4 1.80 GHz, 256 KB cache machine).
To assess the ability of SiteLight to correctly predict binding sites, we have created a data set that includes experimental results from 25 complexes and 39 phage display libraries. A variety of complex types are represented in the data set. SiteLight was tested from three different aspects. First, algorithm validity: SiteLight was run on peptides derived computationally from a known binding site. SiteLight’s ability to select the binding site out of the entire protein surface was examined. This simple experiment confirms the correctness of the method. Second, phage display libraries verification: From each phage display library one peptide that yields the best results was selected. The purpose of this presentation is to show that in each library there is at least one peptide that can be mapped to the binding site. This supports the applicability of phage display libraries to 3D binding site mapping. Third, assessing SiteLight’s performance: SiteLight was run with all the library peptides (in contrast to using only one peptide that yields the best results) and without prior knowledge of the binding site location. Because the correct binding site is known from the crystal structure of the complex, we should be able to confirm or refute the binding site predicted by SiteLight. To the best of our knowledge, this is the first study that attempts to validate the applicability of phage display libraries for automated binding site prediction on 3D structures.
Results
The data set
The newly released Artificially Selected Proteins/Peptides Database (ASPD; Valuev et al. 2002), was searched for complexes that fulfilled two criteria: (1) One of the macromolecules was used as a target for biopanning a phage display library. This molecule is named Target. (2) The second macromolecule is a protein. This molecule is termed Template. Nineteen complexes and 30 libraries were obtained from this procedure. The ASPD is available at: wwwmgs.bionet.nsc.ru/mgs/gnw/aspd/. To further augment and supplement the data set, we have manually searched the literature. This yielded six additional complexes and eight libraries. The data set is listed in Table 1▶. As the table shows, a variety of types of complexes are represented in the data set: proteinous antigen–antibody, hapten–antibody, dimers, domain–domain, receptor–hormone, enzyme–inhibitor/substrate, and other protein–protein complexes. The structures have been taken from the PDB (Berman et al. 2000). A detailed description of the methods used to establish the data set is given in Materials and Methods.
Table 1.
The data set used to assess SiteLight’s ability to predict sites
| No. | Target mol. | Template protein | Complex | Targ. chain | Temp. chain | ASPD entry | Library size | Seq. leng. | Library type |
|---|---|---|---|---|---|---|---|---|---|
| Protein–Protein | |||||||||
| 1A | Bovine Hsc70 | Bag chaperone regulator1 | 1HX1 | A | B | PH1PO059 | 8 | 15 | I |
| 1B | Bovine Hsc70 | Bag chaperone regulator1 | 1HX1 | A | B | H1PO059 | 3 | 6 | I |
| 2 | subtilisin | eglinC | 1CSE | E | I | PH1BS037 | 18 | 5 | I |
| 3 | Factor VIIa | soluble tissue factor | 1DAN | LH | TU | PH1BS102 | 34 | 11 | V |
| 4 | Fyn SH3 | V1 Nef | 1AVZ | C | AB | PH1BS005 | 18 | 11 | I |
| 5 | Nef | Hck-SH3 | 1AVZ | AB | C | PH1PO057 | 19 | 6 | II |
| 6A | Kallikrein | Hirustasin | 1HIA | AB | I | PH1PO062 | 18 | 6 | IV |
| 6B | Kallikrein | Hirustasin | 1HIA | AB | I | PH1PO063 | 11 | 6 | IV |
| 7 | plasmin | streptokinase | 1BML | A | C | PH1NE002 | 32 | 7 | V |
| 8 | Human α thrombin | Haemadin | 1E0F | AD | I | PH1PO064 | 11 | 6 | IV |
| 9A | Bovine trypsin | APPI | 1TAW | A | B | PH1VV009 | 6 | 3 | II |
| 9B | Bovine trypsin | APPI | 1TAW | A | B | PH1VV010 | 11 | 3 | II |
| 9C | Bovine trypsin | APPI | 1TAW | A | B | PH1VV011 | 12 | 3 | II |
| 10 | Actine | Deoxyribonuclease I | 1ATN | A | D | Jesaitis | 29 | 9 | Ia |
| 11 | Rat trypsin | Ecotin | 1F5R | A | I | PH1BS201 | 14 | 4 | II |
| Dimers | |||||||||
| 12 | ubiquitin | ubiquitin | 1AAR | B | A | PH1NE004 | 6 | 5 | IV |
| 13A | glutathione transferase | glutathione transferase | 1GSD | B | A | PH1NE003 | 20 | 5 | II |
| 13B | glutathione transferase | glutathione transferase | 1GSD | B | A | PH1NE003 | 6 | 15 | II |
| 14 | SH3 | SH2 | 1G83 | A | B | PH1BS005 | 18 | 11 | I |
| Peptide–Protein | |||||||||
| 15 | Peptide | Plasminogen activator | 1C5X | A | B | PH1VV018 | 91 | 6 | I |
| Antigen–Antibody | |||||||||
| 16A | GP120 | Ab 17B | 1G9M | G | LH | PH1BS014 | 13 | 13 | Ib |
| 16B | GP120 | Ab 17B | 1G9M | G | LH | Ferrer | 10 | 7 | I |
| 17A | VEGF | VEGF Ab (CDR H1) | 1BJ1 | V | LH | PH1PO091 | 6 | 6 | I |
| 17B | VEGF | VEGF Ab (CDR H2) | 1BJ1 | V | LH | PH1PO092 | 5 | 5 | I |
| 17C | VEGF | VEGF Ab (CDR H3) | 1BJ1 | V | LH | PH1PO093 | 31 | 6 | I |
| 18A | A6 | Interferon gamma receptor | 1JRH | LH | I | Lang | 23 | 5 | III |
| 18B | A6 | Interferon gamma receptor | 1JRH | LH | I | Lang | 7 | 5 | III |
| 18C | A6 | Interferon gamma receptor | 1JRH | LH | I | Lang | 36 | 5 | III |
| Antigen–Fc | |||||||||
| 19A | h IGg FC | Fragment B of protein A | 1FC2 | D | C | DeLano | 2 | 20 | I |
| 19B | h IGg FC | Protein G C2 | 1FCC | A | C | DeLano | 2 | 20 | I |
| 19C | h IGg FC | h IGM Rheumatoid factor | 1ADQ | A | LH | DeLano | 2 | 20 | I |
| 19D | h IGg FC | Fc receptor (Neunatanl) | 1FRT | A | B | DeLano | 2 | 20 | I |
| 20 | IgM rheumatoid factor | Human IGg FC | 1ADQ | LH | A | DeLano | 12 | 10 | I |
| Hapten–Antobody | |||||||||
| 21 | Fluorescein | 4-4-20 FAb | 4FAB | Hetero | LH | PH1PO122 | 4 | 4 | II |
| 22A | Digoxin | 26-10 FAb | 1IGJ | X | AB | PH1VV021 | 20 | 6 | II |
| 22B | Digoxin | 26-10 FAb | 1IGJ | X | AB | PH1VV022 | 2 | 6 | II |
| 22C | Digoxin | 26-10 FAb | 1IGJ | X | AB | PH1VV023 | 7 | 6 | II |
| 22D | Digoxin | 26-10 FAb | 1IGJ | X | AB | PH1VV024 | 8 | 6 | II |
| Inhibitor/s–Enzyme | |||||||||
| 23A | S-Benzyl-Glutathione | Glutathione transferase A1 | 1GUH | Hetero | A | PH1NE003 | 11 | 5 | II |
| 23B | S-Benzyl-Glutathione | Glutathione transferase A1 | 1GUH | Hetero | A | PH1NE003 | 8 | 15 | II |
| Receptor–Hormone | |||||||||
| 24A | h Growth hormone | Growth hormone receptor | 3HHR | BC | A | PH1BS006 | 24 | 20 | II |
| 24B | h Growth hormone | Growth hormone receptor | 3HHR | BC | A | PH1BS007 | 16 | 20 | II |
| 24C | h Growth hormone | Growth hormone receptor | 3HHR | BC | A | PH1BS008 | 7 | 20 | II |
The definitions of Target, Template, and Library Types are given in the text and in Figure 1▶. Library size indicates the number of phage display library derived peptides. Sequence length (Seq. leng.) indicates the average number of amino acids in a peptide in the library. Library peptides have similar lengths. Redundant cases (i.e, complexes with more than one library and libraries with more than one complex) are denoted by the same serial number. They are distinguished by capital letters. The complex code refers to the Protein Data Bank (Berman et al. 2000). The ASPD entry is from Valuev et al. (2002). Where not present in the ASPD database, these were searched and taken from the literature. (Ferrer = Ferrer and Harrison 1999; Jesaitis = Jesaitis et al. 1999; Lang = Lang et al. 2000; DeLano = DeLano et al. 2000).
a Library formed in diluted antiserm.
b An antibody library prepared from bone marrow lymphocytes.
Phage display library types
The term Combinatorial phage display library refers to a library in which all amino acids are represented equally in the peptides displayed on one of the phage surface proteins. Here we call such libraries Type I (see Fig. 1▶). Because the data set obtained using exclusively Type I libraries is small, we have supplemented it by Semicom‘binatorial libraries. Semicombinatorial libraries refer to four library types (Fig. 1▶).
Figure 1.
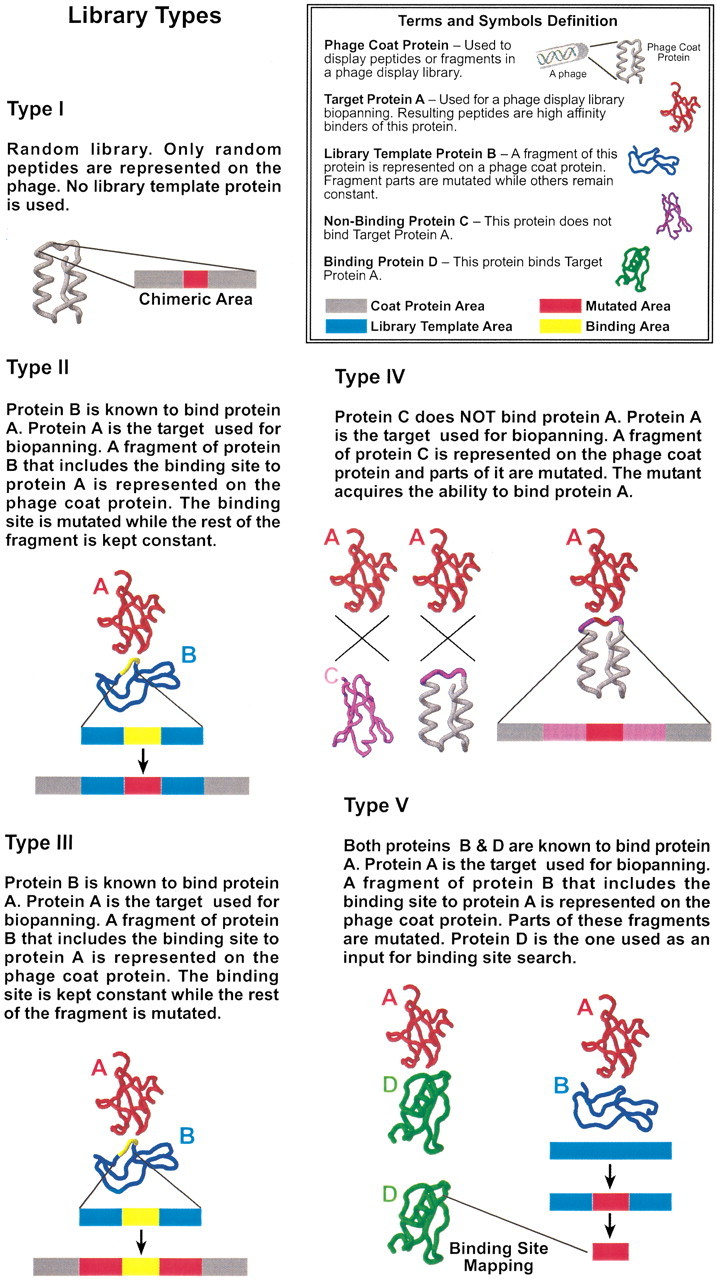
Library types. The different types of phage display libraries used in this study. They are described in detail in Results. Roughly, these types can be divided into two groups: Combinatorial and Semicombinatorial libraries. The term Combinatorial phage display library refers to a library in which peptides are displayed on one of the phage surface proteins. All amino acids are represented equally in these peptides. Here we call such libraries Type I. Semicombinatorial libraries refer to the remaining four library types. The semicombinatorial libraries are not as generic as combinatorial libraries. They were used because the data set obtained using exclusively Type I libraries is small. Semicombinatorial-derived peptides contain mutated and nonmutated parts. If the entire peptide is used, recognition of the binding site could be obvious, because the nonmutated parts would obviously match the interface. Thus, the peptides were parsed to imitate Combinatorial libraries-derived peptides as much as possible. For all library types, only the mutated regions were used (see Materials and Methods).
Type II
The purpose of these libraries is to display mutated variants of the binding site region. A fragment of the library template protein that includes the binding site is displayed on a phage coat protein. Positions within this fragment that are known as hot spots from previous biochemical work are mutated while the rest of the fragment, typically flanking the mutated site, is unchanged. Here we used only the mutated regions for the binding site search.
Type III
The purpose of these libraries is also to display the mutated variants of the binding site region. However, they differ from Type II with respect to the mutated parts. In Type II libraries, the hot spot regions are mutated, whereas in Type III libraries these regions are kept constant, while the flanks are mutated. Both of these approaches try to change the binding affinity to the target protein. Type II libraries fulfill this task by mutating positions that are hypothesized to contribute significantly to the binding, whereas Type III libraries test positions that are presumed not to contribute significantly to the binding.
Type IV
The purpose of this library type is to create novel binding proteins. A protein that binds the Target weakly is used as a Library Template. By mutating this protein it may acquire binding capability. In this molecular evolution process, a nonbinding protein turns into a binding one through mutation and selection. Thus, in this library mutated fragments of the nonbinding protein are displayed on the phage surface.
Type V
This type is used to bypass the limitations of the Types II and III libraries. These libraries are not generic, because the nonmutated regions may play a role in the binding process. First, they may match the Target sterically and be energetically favored. Second, they help the mutated region to acquire a "correct" conformation. Correct conformation may not be established in the absence of the nonmutated regions. Therefore, a local structural environment formed by the nonmutated regions is essential for binding. Although this conclusion was neither confirmed nor refuted by experimental methods, its implications were not overlooked. Further, libraries of Types II and III are not generic because they might produce an epitope composed of both mutated and nonmutated regions. The nonmutated regions are composed of native residues located in the binding site, and therefore might contribute to the interactions. Thus, libraries of Types II and III are less informative than epitopes derived only from the mutated regions. In the Type V libraries our goal is to separate the contribution of the mutated regions to the binding process. To achieve this purpose, we use two different local structural environments. These environments are taken from two proteins that are known to bind the same {it Target}. One of these is used as a Library Template, while the binding site is searched on the second. This is in contrast to the other library types, where the same protein serves both as a Library Template and for binding site searching (see Fig. 1▶). Overall, 14 Type I and 24 Semicombinatorial libraries were used. Among these are 17 Type II, three Type III, three Type IV, and one Type V libraries.
The algorithm
SiteLight seeks to match a phage display derived peptide to a 3D epitope on a protein surface. The peptides are expected to imitate the binding site on the Template protein with respect to amino acid chemical properties and spatial organization. The surface of the protein is divided into overlapping patches. The division is based on geodesic distances between two residues rather than on Euclidean distances. SiteLight examines the potential match of each peptide in the library with each patch. For each potential match a bipartite graph, Graph 2, is created. Its vertices represent patch and peptide residues. Its edges represent similarity between two residues. Residue similarity is determined by a similarity matrix (Table 3▶ in Supplemental Material). The best alignment of a peptide and a patch represented in a bipartite graph is found by the maximal bipartite matching algorithm. The score of each match is determined by the best alignment. Potential matches are sorted by their scores. High scoring matches are iteratively selected until 25% of the Template protein is covered. The minimized surface is expected to include the binding site between the Template and the Target. To assess SiteLight’s success, the "correct" binding site is determined. This site is defined by the Template–Target complex interface (i.e., Template residues that are spatially proximal to the Target). The correctness of each match is represented by the patch overlap with the interface. Figure 2▶ gives a flow chart of the algorithm, and Materials and Methods describes it in further detail.
Table 3.
The results of SiteLight’s prediction of bound and unbound Bag-1/Hsc70 binding site
| No. | Bound/unbound | Peptides type | Peptides length | Predicted residues | Matches | Highest solution | Best solution | N100 |
|---|---|---|---|---|---|---|---|---|
| 1 | Bound | Peptide A | 15 | 17 | 104 (2) | 1 (68) | 1 (68) | 56 (92) |
| 2 | Bound | Peptide A | 7 | 16 | 102 (8) | 1 (100) | 1 (100) | 1 (100) |
| 3 | Bound | Peptide A | 6 | 12 | 102 (8) | 1 (57) | 3 (100) | 3 (100 |
| 4 | Bound | Entire Library | 15 | 13 | 832 (3) | 1 (68) | 1 (68) | 1 (68) |
| 5 | Bound | Entire Library | 7 | 14 | 918 (6) | 1 (100) | 1 (100) | 1 (100) |
| 6 | Bound | Entire Library | 6 | 12 | 306 (12) | 1 (28) | 9 (83) | 21 (100) |
| 7 | Bound | Control | 7 | 0 | 204 (6) | none (0) | none (0) | 82 (88) |
| 8 | Bound | Control | 6 | 3 | 408 (7) | 7 (37) | 7 (37) | 89 (100) |
| 9 | Unbound | Peptide A | 15 | 14 | 107 (3) | 1 (80) | 1 (80) | 21 (85) |
| 10 | Unbound | Peptide A | 7 | 13 | 106 (9) | 1 (87) | 8 (100) | 8 (100) |
| 11 | Unbound | Peptide A | 6 | 9 | 104 (10) | 1 (80) | 3 (87) | 19 (100) |
| 12 | Unbound | Entire Library | 15 | 4 | 856 (2) | 1 (28) | 1 (28) | 87 (85) |
| 13 | Unbound | Entire Library | 7 | 9 | 1060 (9) | 1 (66) | 2 (87) | 90 (100) |
| 14 | Unbound | Entire Library | 6 | 9 | 312 (10) | 1 (80) | 6 (87) | 82 (100) |
The bound data refers to 104 surface residues and 21 interface residues. The unbound data refers to 109 surface residues and 23 interface residues. The table categories are explained in Table 2▶.
Figure 2.

SiteLight algorithm flow chart. This chart gives a schematic description of the SiteLight algorithm. The algorithm is described in further detail in Materials and Methods. SiteLight seeks to match a phage display-derived peptide to a 3D epitope on a protein surface. The surface of the protein is divided into overlapping patches. The division is based on geodesic distances between two residues rather than on Euclidean distances. SiteLight examines the potential match of each peptide in the library with each patch. For each potential match a bipartite graph, Graph 2, is created. Its vertices represent patch and peptide residues. Its edges represent similarity between two residues. Residue similarity is determined by a similarity matrix (Table 2▶ in Supplemental Material). The best alignment of a peptide and a patch represented in a bipartite graph is found by the maximal bipartite matching algorithm. The score of each match is determined according to the best alignment. Potential matches are sorted by their scores. High scoring matches are iteratively selected until 25% of the Template protein is covered. The minimized surface is expected to include the binding site between the Template and the Target. To assess SiteLight’s success, the "correct" binding site is determined. This site is defined by the Template-Target complex interface (i.e., Template residues that are spatially proximal to the Target). The correctness of each match is represented by the patch overlap with the interface.
Algorithm validation
To validate the correctness of the algorithm, we first assess its ability to predict the correct binding site using peptides derived from this site. For each complex, peptides that represent its interface were created, by extracting residues from the binding site, and arbitrarily linking them. The peptides’ length was equal to the average length of the phage display derived peptides. These peptides are referred to as Artificial Interface Peptides. They were then used for binding site prediction on the entire protein. Hence, in this way we test the ability of the Artificial Interface Peptides to remap the interface. Although this may seem as a straightforward experiment at first sight, there are many potential problems that could have prevented its success. The peptides’ length can be as short as 3 to 4 amino acids, while the protein surface can contain more than 400 amino acids. The larger the ratio between the surface size and the peptides’ length, the higher the probability of finding false positive solutions. An additional potential problem is the outcome of the partial exploration of the solution space. To define the complete solution space, let us reduce the problem to a graph theory problem. Each vertex represents a residue. Edges connect structurally defined neighbors (i.e., residues proximate in a 3D space). The complete solution space can be defined as all walks of k steps in this graph, where k is the number of residues in a phage library peptide. If n is the number of residues in graph G1, then the number of walks is proportional to 2n. Therefore, only partial exploration of this space is possible in a reasonable computation time. All of the complexes presented in Table 1▶ were tested. Despite these difficulties, in 82% the correct solution was ranked as the first. In the remaining 18% a correct solution was ranked within the top five solutions. A "correct" solution is defined as one where the peptides optimally matched the interface with an overlap of at least 25%. These results (data not shown) demonstrate the algorithm validity.
Phage display libraries verification
We have tested the validity of the input for SiteLight, that is, the phage display library biopanning results. The purpose is to show that in every library there is at least one peptide, referred to as Peptide A, which can be mapped to the correct binding site. If it does, it should score higher than if it is aligned by SiteLight to other parts of the protein. According to its definition, Peptide A is sufficient for binding site prediction. The method for selecting Peptide A is detailed in Materials and Methods. In 76% of the complexes presented in Table 2▶, a correct solution (over 25% overlap with the interface) was ranked as the first using Peptide A. The best way to estimate this result is by comparing it to the one obtained with the Artificial Interface Peptides. There the result for the Artificial Interface Peptides can be regarded as the best possible solution that can be obtained. Because the data set is the same, these results are comparable. The percent of complexes in which a correct solution was ranked first based on Peptide A is only 6% less than the one obtained with the Artificial Interface Peptides. Only in 2 out of the 43 cases no peptide could be aligned with the binding site for a given complex and a library. The rank of the best match, that is, the solution that overlaps with the interface to the highest extent, is also high in almost all cases (see column 9 in Table 2▶). The exception is in the hapten–antibody category. Overall, the categories with the worst results are the hapten–antibody and the antigen–Fc.
Table 2.
Assessment of SiteLight’s performance on the dataset presented in Table 1
| No. | Fragments | Surface residues | Interface residues (%) | Predicted residues | Matches (selected) | Highest solution | Best solution | N100 |
|---|---|---|---|---|---|---|---|---|
| 1A | 2 | 104 | 21 (20) | 13 | 832 (3) | 1 (68) | 1 (68) | 1 (68) |
| 1B | 2 | 104 | 21 (20) | 12 | 306 (12) | 1 (28) | 9 (83) | 21 (100) |
| 2 | 1 | 58 | 12 (20) | 6 | 990 (3) | 1 (100) | 1 (100) | 1 (100) |
| 4 | 4 | 98 | 14 (14) | 3 | 1692 (3) | 1 (27) | 1 (27) | 65 (90) |
| 5 | 4 | 55 | 12 (21) | 1 | 988 (4) | none | 1 (14) | 50 (75) |
| 6A | 2 | 48 | 11 (22) | 3 | 912 (10) | 1 (50) | 1 (50) | 11 (100) |
| 6B | 2 | 48 | 11 (22) | 10 | 576 (7) | 1 (100) | 1 (100) | 1 (100) |
| 7 | 13 | 305 | 44 (14) | 17 | 19500 (29) | 1 (25) | 27 (66) | 27 (66) |
| 8 | 6 | 55 | 18 (32) | 5 | 624 (3) | 1 (40) | 1 (40) | 65 (60) |
| 9A | 2 | 52 | 12 (23) | 3 | 312 (6) | 1 (100) | 1 (100) | 1 (100) |
| 9B | 2 | 52 | 12 (23) | 8 | 572 (6) | 1 (100) | 1 (100) | 1 (100) |
| 9C | 2 | 52 | 12 (23) | 6 | 624 (6) | 1 (100) | 1 (100) | 1 (100) |
| 10 | 4 | 214 | 21 (9) | 3 | 6400 (11) | 7 (27) | 7 (27) | 29 (70) |
| 11 | 2 | 56 | 11 (19) | 7 | 658 (16) | 1 (40) | 8 (80) | 20 (100) |
| Dimers | ||||||||
| 12 | 5 | 70 | 14 (20) | 6 | 396 (5) | 2 (66) | 2 (66) | 28 (100) |
| 13A | 7 | 205 | 14 (6) | 8 | 3800 (31) | 3 (40) | 6 (75) | 6 (75) |
| 13B | 7 | 205 | 14 (6) | 5 | 1200 (5) | none | 5 (15) | 88 (53) |
| 14 | 3 | 156 | 8 (5) | 0 | 2556 (25) | none | none | 63 (36) |
| Peptide–Protein | ||||||||
| 15 | 6 | 104 | 22 (21) | 8 | 7650 (57) | 7 (60) | 7 (60) | 100 (83) |
| Antigen–Antibody | ||||||||
| 16A | 4 | 406 | 13 (3) | 5 | 2067 (27) | 7 (28) | 7 (28) | 43 (33) |
| 16B | 4 | 406 | 13 (3) | 4 | 1510 (24) | 4 (33) | 4 (33) | 70 (40) |
| 17A | 4 | 205 | 18 (8) | 12 | 1188 (63) | 1 (100) | 1 (100) | 1 (100) |
| 17B | 4 | 205 | 18 (8) | 4 | 985 (22) | none | 3 (16) | 35 (100) |
| 17C | 4 | 205 | 18 (8) | 6 | 6138 (24) | 2 (83) | 2 (83) | 37 (100) |
| 18A | 5 | 94 | 15 (15) | 8 | 2093 (6) | 1 (85) | 1 (85) | 11 (100) |
| 18B | 5 | 94 | 15 (15) | 11 | 637 (10) | 1 (85) | 4 (100) | 4 (100) |
| 18C | 5 | 94 | 15 (15) | 9 | 91 (6) | 1 (33) | 4 (100) | 4 (100) |
| Antigen–Fc | ||||||||
| 19A | 2 | 43 | 11 (25) | 8 | 86 (2) | 2 (38) | 2 (38) | 75 (81) |
| 19B | 1 | 54 | 13 (24) | 12 | 108 (2) | 1 (60) | 1 (60) | 21 (68) |
| 19C | 4 | 223 | 9 (4) | 0 | 406 (3) | none | none | 98 (26) |
| 19D | 5 | 90 | 26 (28) | 12 | 1600 (3) | 2 (57) | 2 (57) | 91 (70) |
| 20 | 3 | 197 | 15 (7) | 2 | 1940 (17) | none | 14 (18) | 63 (41) |
| Hapten–Antibody | ||||||||
| 21 | 4 | 408 | 9 (2) | 4 | 1548 (32) | none | 22 (14) | 85 (25) |
| 22A | 3 | 378 | 9 (2) | 0 | 7260 (22) | none | none | 73 (71) |
| 22B | 3 | 378 | 9 (2) | 4 | 6897 (143) | 1 (60) | 1 (60) | 1 (60) |
| 22C | 3 | 378 | 9 (2) | 1 | 2541 (26) | none | none | 81 (60) |
| 22D | 3 | 378 | 9 (2) | 1 | 2904 (24) | none | none | 59 (100) |
Unlike in Table 2▶ in the Supplemental Material, here the entire corresponding phage display library is used. All of the categories presented in this table refer to 25% coverage of the surface. The number of potential matches used to predict interface residues is determined such that the number of predicted residues will equal 25% of the surface residues.
Fragments, the number of contiguous sequences in the spatially defined interface of the complex; surface residues, the number of residues exposed to the solvent; interface residues, Template residues that are proximate to the Target in space. The interface percent is calculated with regard to surface residues as the 100%; predicted residues, the number of interface residues that were correctly predicted by SiteLight; matches, the number of potential matches. This number equals the number of surface patches multiplied by the number of peptides in the library. The number of the obtained matches for binding site prediction is indicated in parentheses; highest solution, the rank of the first solution that overlaps by 25% or more with the interface out of the obtained matches. The percent of residues that overlap with the interface is indicated in parentheses; best solution, the rank of the match that overlaps with the interface to the largest extent; N100, the rank of a match that overlaps with the interface to the largest extent out of the first 100 solutions. See Halperin et al. (2002). The results refer to 25% coverage of the surface.
Example: in case No. 1A there are 104 surface residues. The interface is composed of 21 residues that are 20% of the surface residues. The interface is made up of two sequential regions that are proximate in space to one another and to the Target. There are 104 potential matches that are tested by SiteLight. Using the threshold that we allow, no more than 25% of the surface of the molecule is to be covered by the matches (otherwise, the prediction of a binding site is too diffuse). Two matches (out of the possible 104) were obtained. Twenty-five percent of the surface is 26 residues. The two matches contain 17 interface residues (out of a possible 21 identified in the interface). Sixty-eight percent of the residues in the highest ranking match (solution no. 1) are interface residues. The second solution overlaps with the interface to a lesser extent. Therefore, in this example, the highest ranking solution is also the best solution with the largest interface coverage. Ninety-two percent of the residues of the 56th ranked match are interface residues. It is the highest overlap with the interface in the first 100 ranked matches.
Assessing SiteLight performance
Table 3▶ describes the results obtained when using all the peptides from a phage library. This is the most critical test of the algorithm. Out of the total number of 43 cases, in 30 the rank of the first solution that overlaps at least 25% of the interface is at the top 5. Out of a total number of 43 cases, in the 27 cases that we have tested, there is at least one binding site prediction that overlaps the interface by at least 50%. Inspection of the different categories suggests that the best results appear to be obtained for the protein–protein complexes. Good results are also obtained for the antigen–antibody and the receptor–hormone, the enzyme–inhibitor, and dimer categories; however, the last categories have a small number of cases. There appears to be a correlation with the interface size. The protein–protein, antibody–antigen, and receptor–hormone have large interfaces. On the other hand, the hapten–antibody category has the smallest number of residues in the interface, and represents a small percentage of the antibody surface. The smaller library size (Table 1▶) also appears to have an effect, possibly explaining the worse cases for the antigen–Fc category. In terms of the best coverage of the interface, in 13 cases within the first 100 solutions (N100) the solutions that cover the interface the most (above 50%) rank at the top 10. Again, the distribution between the categories is similar. Analysis with respect to the type of library (Fig. 1▶; Table 1▶) does not illustrate any significant difference. Figure 3▶ presents the top solution for a few examples.
Figure 3.
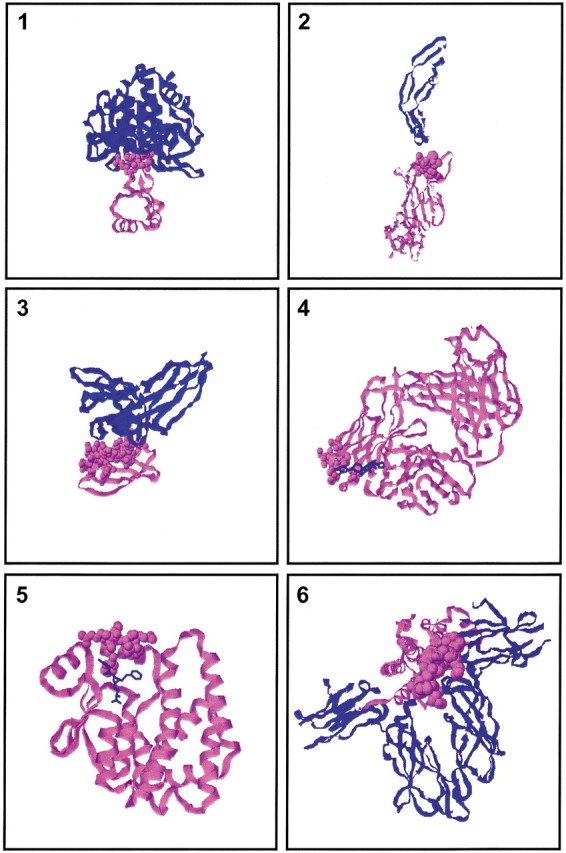
Examples of SiteLight’s binding site prediction for variable complexes. The input in all of the examples presented here includes the Template structure and all library peptides (in contrast to using only one peptide that yields the best results, Peptide A). Although the complex is known, no prior knowledge of the binding site location is used. The first solution predicted by SiteLight to be a binding site is presented for each example in space filling representation. The Target and the Template are colored blue and pink, respectively. Each example represents a different category of the data set (see Table 1▶) on which SiteLight was tested: (1) Subtilisin/EglinC complex (1CSE), example 2. (2) VEGF/VEGF Ab (CDR H1) complex (1BJ1), example 17A. (3) Human IGg Fc/Protein G C2 complex (1FCC), example 19B. (4) Digoxin/26–10 FAb complex (1IGJ), example 22B. (5) S-Benzyl-Glutathione/Glutathione Transferase A1–1 complex (1GUH), example 23A. (6) Human Growth Hormone/Growth Hormone Receptor complex (3HHR), example 24B. The example number refers to Tables 1–2▶▶. The complex code refers to the Protein Data Bank (Berman et al. 2000).
Combined, the results suggest that if the 3D structure of the Template protein is available, for larger interfaces phage display libraries can be used not only to detect which are the binding peptides, but to also use them toward the prediction of the binding site. Furthermore, by using 1D (peptide sequences) to 3D (protein surface) mapping, we are able to detect epitopes that are not necessarily contiguous on the sequence. To illustrate this point, we have characterized the continuous (conformational) nature of each interface. Interface residues were divided into groups according to their sequence continuity. Each group represents a contiguous sequence fragment. The number of these groups is given as the number of fragments in Table 1▶. Our matching procedure completely disregards this type of information. The order of the residues on the chains is not taken into account, making it a general procedure for binding site detection.
Hsc70—detailed result analysis
Hsc70 is a constitutively expressed protein. It prevents misfolding and aggregation of newly synthesized or misfolded proteins. Hsc70 consists of three domains: ATPase, SBD (substrate binding domain), and a C-terminal domain. ATP binding by the ATPase domain regulates substrate binding by an unknown mechanism. Substrate binding promotes ATP hydrolysis. The Hsc70/ADP complex with the substrate is more stable. ATP hydrolysis is also stimulated by Hsp40 proteins. Substrate release is dependent on the exchange of bound ADP for ATP. This reaction is promoted by a nucleotide exchange factor: GrpE in prokaryotes and Bag-1 in euokaryotes. Bag-1 was shown to stimulate the ATPase rate of Hsc70 in an Hsp40-dependent manner and to promote substrate release from the chaperone (Sondermann et al. 2001). Two structures were used for the analysis: the complex of Hsc-70 ATPase doamin and Bag-1, and unbound Bag-1 (PDB codes 1HX1 and 1I6Z, respectively).
Hsc70 phage display libraries
Bovine Hsc70 was used as a Target for screening a 15-mer and a 6-mer phage display random peptide libraries. Each library contained about 108 clones. Three clones were sequenced from the 6-mer library. Ninety-seven clones were sequenced from the 15-mer library after three rounds of selection. These sequences were enriched with lysines, histidines, and aspartic acid. Binding specificity to Hsc70 was confirmed by negative and positive tests. The negative test examined peptide binding to other proteins (BSA, actin, and streptavidin). The positive test examined the peptides’ ability to stimulate ATPase activity in two ways: (1) inorganic phosphate release measurement, and (2) competition with the pigeon cytochrome c peptide, which stimulates ATPase activity (Takenaka et al. 1995).
Previous results have suggested that the heptamers may have an improved affinity compared to hexamers. Therefore, 7-mer peptides were designed based on sequences obtained by biopanning the 15-mer and the 6-mer libraries. Two groups of control sequences of 6 and 7-mer lengths were also constructed. These peptides failed to pass the binding specificity tests (Sondermann et al. 2001).
SiteLight results for the Hsc70–Bag-1 complex
In Table 3▶ it can be seen that there is a correlation between the specificity of peptide binding and its ability to predict the binding site. There is a significant difference between control peptides and specific-binding peptides. Control peptides of both 7 and 6 amino acids (lines 7 and 8 in Table 3▶) were not mapped to the binding site. Either none or 3 residues out of 21 interface residues were found using heptamers and hexamers control peptides, respectively. In comparison, 14 and 12 residues out of 21 interface residues were found using heptamers and hexamers specific-binding peptides, respectively. For the 15-mer peptides, no control was provided. In each of the three libraries (15-mer, heptamer, and hexamer) there is at least one peptide (i.e., Peptide A) that can be mapped to a binding site better than to other parts of the molecule (lines 1–3 in Table 3▶). Bag-1 surface was minimized by 75%. The number of surface residues was reduced from 104 to 26. Using each of the 15-mer, heptamer and hexamer Peptide A’s, a different set of residues was located in the reduced surface. This surface contains 17, 16, and 12 interface residues for the 15-mer, heptamer, and hexamer respective Peptide A’s. The highest ranking solutions (solution no. 1) obtained by the 15-mer, by the heptamer and by the hexamer Peptide A’s overlap with the interface by 68%, 100%, and 57%, respectively.
When the entire libraries (lines 4–6 in Table 3▶) were used, a modest decline in prediction quality is observed compared to the Peptide A results. The reduced interface contains 13, 14, and 12 interface residues for the 15-mer, heptamer, and hexamer entire libraries, respectively. This yields an average decline of 1.6 residues. The rank of the highest solution and the percentage of interface overlap did not change when the 15-mer and heptamer Peptide A’s were replaced by the entire libraries of the 15-mer and heptamers. As in other examined cases, no positive correlation between peptide length and binding site prediction quality was observed.
Bound and unbound Bag-1
Here our goal is to predict the Hsc70/Bag-1 binding site if no such complex is available. In the absence of the Hsc70/Bag-1 complex, the Template is the unbound rather than the bound Bag-1. Therefore, we compared the performance of SiteLight for the bound and unbound Bag-1 structures. The bound structure of Bag-1 (1HX1) is taken from Homo sapiens. The unbound structure of Bag-1 (1I6Z) is from Mus musculus. The sequences of the bound and unbound Bag-1 were aligned using CLUSTAL X (1.81) multiple sequence alignment. They share 85% residue identity and 93.5% similarity. Bag-1 unbound structure (1I6Z) was structurally aligned to bound Bag-1 (1HX1 chain B) using FlexProt (Ma et al. 2002; Shatsky et al. 2002). FlexProt detects the optimal flexible structural alignment of a pair of protein structures. The first structure is assumed to be rigid, while in the second structure potential flexible regions are automatically detected. The root-mean-squared deviation (RMSD) is 2.14 Å for an alignment of 112 residues (the entire length of bound Bag-1) without hinges. The RMSD could not be lowered by insertion of one or two hinges. The structure and sequence alignment are presented in Figure 4. The results obtained with Peptide A for the bound and unbound Bag-1 (Table 3▶) show that the number of predicted residues was smaller for the unbound than for the bound Bag-1. The ranks of the first (highest) solution that overlaps the interface by 25% or more and of the solution that overlaps the interface to the largest extent draw a different picture. The solution that overlaps the interface by at least 25% ranks number one for all six cases: bound 15-mer, heptamer, and hexamer and unbound 15-mer, heptamer, and hexamer. The interface overlap percentage of the highest solution indicates a nonuniform trend. It is higher for the unbound 15-mer and hexamer (80% for both) compared with bound 15-mer and hexamer (68% and 57%, respectively). It is lower for the unbound heptamer compared with bound heptamers (87% and 100%, respectively).
The example of Hsc70/Bag-1 demonstrates SiteLight’s ability to predict a conformational epitope based on phage display peptide sequences. The Hsc70/Bag-1 interface consists of two helices. Library peptides that were mapped to the binding site, including Peptide A, could be mapped to both. This demonstrates a correlation between the specificity of peptide binding and its ability to predict the binding site. Control peptides were poorly mapped to the binding site compared to specific-binding peptides. The length of the specific-binding peptides does not seem to affect the prediction quality. Reasonable results could also be obtained using the unbound structure of Bag-1. This demonstrates the applicability of SiteLight to both bound and unbound structures.
Discussion
SiteLight enables testing the applicability of random phage display libraries to binding site mapping on a 3D structure. SiteLight’s application is primarily directed toward conformational epitope search. Most macromolecular interfaces are conformational (i.e., consist of a few, rather than one, contiguous regions on the polypeptide chain). However, most current studies aiming at phage-based epitope discovery deal with contiguous epitopes. Computational tools, like SiteLight, that enable searches for conformational "real" 3D epitopes, substantially broaden the applicability of peptide libraries. As a primary research tool, we put an emphasis on short running times. Running time is less than 30 sec for all tested cases (on a Red-Hat Linux 7.1, 1 processor, Pentium 4 1.80-GHz, 256-KB cache machine).
One of the initial research goals was to establish a data set on which SiteLight can be tested. Our data set includes 25 complexes and 39 phage display libraries. Although this data set is large and diverse, it is far from being ideal. It needs to be considerably enlarged. Further, ideally, it should primarily consist of the generic {it Type I} libraries. Because the interest in phage display application for computational prediction of binding sites is only beginning, we expect that this limitation will be resolved in the near future. Current available data were not fully exploited yet. In 2002, Valuev et al. (2002), the creators of ASPD, have predicted that its size would double within a year. The enlargement of the available data set will enable application of statistical tools in a meaningful way. In addition to the expected growth in combinatorial phage display data, the data set available to SiteLight may be enlarged by using nonphage-based methods of peptide display and selection. These include both artificial evolution methods that are not phage-based (like ribosome display; Hanes and Pluckthun 1997) and bacterial display (Wikstrom 2000), as well as large scale peptide display methods (like peptide microarrays). From a computational point of view, there is no difference between peptides derived from any of these methods. However, it remains to be determined if SiteLight will perform equally well on such inputs as it does on phage display inputs.
Two types of validation tests, algorithm validation and phage display libraries verification, were carried out. The first confirmed the ability to remap a binding site using peptides derived from this site. The second revealed at least one peptide in 95% of the tested libraries that can be aligned to the binding site better than to other parts of the protein. The existence of such peptides in each of these libraries reinforces the idea that random phage display libraries can be mapped to a 3D binding site. The expected result in the first type of validation is clear—to be able to remap the site from which peptides were derived. Such an experiment can be carried out with various sites regardless of their binding properties. On the other hand, in the second validation test the expected result is not entirely clear because a few types of epitopes can be defined as the "correct" answer.
A binding site can be divided into two types of epitopes: structural and energetic. An energetic epitope consists of amino acids that can be shown to individually contribute significantly to the binding energy. This epitope is also known as hot spots. On the other hand, a structural epitope is expected to be larger than the energetic epitope because not all interface residues are biologically relevant (Valdar and Thornton 2001). Some of the structural nonenergetic residues may be critical for dictating the 3D configuration of the epitope. Thus, a consensus derived from combinatorial phage display peptides may include only hot spots, only critical residues dictating the 3D structure, other structural epitope residues, or their combination. The relative prevalence of these groups in the peptides sequences is unknown. Experimental methods of hot spots determination vary from one study to another and are often incomplete in the sense that only a subset of the positions are examined. Therefore, we have chosen crystallographically defined interfaces. Such a definition is uniform, and appears most applicable to computational studies. Nevertheless, it is unavoidable that sometimes the epitope represented by phage display peptides deviates from this definition.
SiteLight’s performance can be assessed within the framework of the available data set and a "correct" epitope definition. SiteLight can reduce the protein surface by 75% without excluding the binding site. The reduced surface includes at least one solution that overlaps the interface by at least 50% in 63% of the cases. The fact that we do not achieve "correct" solution in the remaining cases can be the outcome of a number of reasons: (1) partial exploration of the solution space. It is possible that the patches that are not explored include the ones that would yield the best results. (2) Site mimicry: sites that have similar amino acid composition as the binding site cannot be distinguished from it. This mimicry can shift the results from the interaction site. The existence of such mimicry is indicated by false positives found using interface-derived peptides. (3) Irrelevant peptides: The library is expected to include both relevant peptides that bind the Target and irrelevant peptides that do not bind. Despite the negative selection steps, there are derived peptide sequences that are intrinsic to components of the biopanning process itself, including the plastic, the immobilization system or the blocking agents (Adey et al. 1995). These irrelevant peptides might mask the "correct" peptides that can be mapped to the binding site. Furthermore, (4) the relevant peptides might not all bind the Target at the same site as the Template. Let us consider a protein with multiple binding partners. The binding sites on this protein can overlap, but can also be distinct. Distal interaction sites can each bind a different set of peptides. (5) Related to the last possible reason for failure is the fact that during affinity selection a single high-affinity binding site might dominate the library. Other binding sites may not be represented at all. Thus, the presence of peptides that mimic the Template binding site depends on the biopanning components, the number of binding sites, and the affinity of the peptides for each site. In this regard, it would be interesting to experimentally examine proteins with a few binding partners, particularly those with different site affinity to their natural ligands. There may be a correlation between the affinity of a site to its natural ligand and its ability to select affinity peptides. Nevertheless, such a correlation does not necessarily exist, because the peptides’ affinity can exceed the affinity of the natural ligand. Fewer rounds of selection might allow selection of peptides for multiple sites. In such a case, the peptides will be mapped to more than one Template, and may be divided into groups according to their similarity. Each group should be aligned to one of the Templates.
Sequence alignment of the peptides may, in principle, help in discriminating between relevant and irrelevant peptides for a single binding protein partner. Relevant phagotope discrimination based on multiple alignment of peptides derived from random phage display libraries was carried out for the primary biliary cirrhosis and type I diabetes (Davies et al. 1999). Although not yet tested on a large data set, the encouraging preliminary results suggest that this procedure may be adopted in the future to improve SiteLight’s performance. Peptides can be divided into groups according to their alignment. This alignment can then be utilized in two ways: First, each group will yield a consensus that will be searched on the Template. Second, weights can be assigned to the peptides and (or) position according to its deviation from the consensus. All of the peptides will be searched on the Template. This may yield improved prediction.
One of the measurements used for phage display peptides characterization is affinity to the Target molecule. Affinity data was not used in this study due to two reasons: First, affinity data are either incomplete or completely missing for most of the libraries in the data set. This problem can be partially resolved by an indirect affinity data. A potential substitution for direct affinity data is the number of appearances of the peptide. This number is assumed to reflect the prevalence of the peptide in the postpanning library. Because Biopanning is based on the principle that high-affinity binders are enriched with selection, if the representative sample of sequenced peptides is big enough, the frequency of the peptide in the library is likely to reflect the affinity. Second, no correlation was found between the affinity and the "goodness" of the binding site mapping. Assuming that such a correlation should exist is not straightforward. In some libraries peptides with improved affinity (i.e., higher affinity than the native substrate) were found (Lang et al. 2000). Such peptides are expected to differ from the native binding site with respect to binding site location and/or residue composition. A good peptide for binding site mapping using SiteLight is one that (1) binds in the same or overlapping location, (2) in a similar conformation, and (3) consists of similar residues as the native Template. Therefore, there is no simple correlation between peptide affinity and similarity to the native binding site. It might be interesting to examine this correlation when the binding location is confirmed, for example, by competitive elution, catalytic panning or structure determination. In such cases, peptides that bind the Target with similar affinity as the Template might mimic better the Template binding site.
The SiteLight algorithm may also be applicable to other biologic problems in addition to phage-based binding site mapping. SiteLight attempts to answer a broader question than mapping a 1D peptide sequence, to a 3D protein structure. Because the sequence order of the peptide is disregarded, SiteLight can also be applied for 3D—3D amino acid similarity detection. The main reason for not using the peptide sequential order is the inclusion of Semicombinatorial libraries in the data set. Some of the peptides were parsed (see Materials and Methods) and their order was lost. This unique feature of SiteLight, performing sequence alignment without sequence order, might be applicable to searching promiscuous activities. These are typically found by a search of a random library of ligands (James and Tawfik 2001). Mimicry of known interaction sites can be searched by creating a series of peptides that represent the amino acid composition of a known binding site. These Virtual Peptide libraries can be used to search a database of structures to discover similar sites in unrelated structures.
Conclusions
Here we illustrate that random phage-display peptide libraries can be applied to binding site mapping on a 3D structure. SiteLight provides a vehicle for such an application. SiteLight maps binding sites consisting both of contiguous residues, and those that constitute "truly" 3D conformational epitopes. The algorithm is highly efficient and effective. It successfully remaps short peptides (3–20 amino acids long) to the sites they were derived from even on large 3D protein structures.
SiteLight was able to reduce the surface by 75% without excluding the binding site. The reduced surface included at least one solution that overlaps with the interface by at least 50% in 63% of the cases. Although some trends appear to occur, nevertheless, unfortunately, no firm conclusions can be drawn regarding the applicability of this method to different molecular groups (antigen–antibody, protein–protein, etc.) due to the current limited data set size. This limitation also holds with respect to the comparison between different phage display peptide library types.
In particular, this study appears to validate the applicability of phage-display libraries for automated binding site prediction on 3D structures, and as such, suggests the feasibility of their further broadened utility.
Materials and methods
Data set creation
The ASPD (Valuev et al. 2002; available at wwwmgs.bionet.nsc.ru/mgs/gnw/aspd/), is a new database that incorporates proteins and peptides that were obtained through in vitro-directed evolution processes. Most of them were obtained through phage display libraries Biopanning. The current version of the ASPD includes 195 selection experiments. It was searched for complexes that fulfilled the following criteria: (1) One of the macromolecules was used as a target for biopanning a phage display library. This molecule is named Target. The types of phage display libraries used is described in detail in the Results and illustrated in Figure 1▶. (2) The other macromolecule is a protein. This molecule is termed Template. Nineteen complexes and 30 libraries were obtained from this procedure. All of them are phage display libraries. Six additional complexes and eight phage display libraries were obtained by manual literature search. A variety of complex types are represented in the data set: receptor–hormone, enzyme–inhibitor/substrate, and other protein–protein complexes. Ten protein–protein complexes with 14 libraries, three dimer complexes with four libraries, one peptide–protein complex with one library, three antigen–antibody complexes with eight libraries, four antigen–Fc complexes with two libraries, two hapten–antibody complexes with five libraries, one enzyme–inhibitor/substrate complex with two libraries, and one receptor–hormone complex with three libraries. There is some redundancy in this data set. Redundant examples (complexes with more than one library and libraries with more than one complex) are denoted in Table 1▶ by the same serial number. They are distinguished by capital letters. Overall there are 24 nonredundant cases in the data set.
Peptides parsing
In combinatorial libraries the entire mutated fragment of each peptide is used without further parsing. However, semicombinatorial libraries contain mutated and nonmutated parts. If the entire peptide is used, recognition of the binding site could be obvious, because the nonmutated parts would obviously match the interface. Thus, the peptides were parsed to imitate combinatorial libraries-derived peptides as much as possible. For all libraries, only the mutated regions were used for the binding site search. Positions that were mutated by less than eight amino acids were removed. Two mutated positions were linked if the sequential distance between them did not exceed four positions. Thus, for example, if position number 1 in a peptide was mutated by 8 residues, position 2 mutated by 2, position 3 by 4 residues, position 4 by 12, and position 5 by 10 residues, the new peptide would consist only of positions 1, 4, and 4. Because our matching is 1D to 3D, the order on the chain can be disregarded. By omitting positions 2 and 3, we do not use information that would otherwise straightforwardly lead to matching to the binding site. Further, if the distance between positions mutated by at least eight residues is larger than four residues, the peptide is cut into short fragments, where each fragment contains strictly the highly mutated residue positions. The minimum fragment length is three residues. If a crystal structure of the Library Template (defined in Fig. 1▶) is available, the peptides are joined if their residues are next to each other in a three dimensional space.
Algorithm description
The SiteLight algorithm can be divided into three main stages: creation of surface patches, matching surface patches with phage libraries-derived peptides, and scoring the solutions to assess their correctness.
Stage 1: Creation of surface patches
There are three steps in the creation of surface patches:
1. Molecular shape representation.
This step computes the molecular surface of the molecule. First, a high density Connolly surface is generated by the Molecular Surface program (Connolly 1983a,b). The Connolly surface is generated by rolling a probe ball over the van der Waal’s surfaces of the atoms of the molecule. Three types of shapes are created: convex, saddle, and concave. A sparse surface representation is computed (Lin et al. 1994) consisting of a limited number of critical points disposed at key locations over the surface. The sparse surface representation is composed of three types of points nicknamed caps, belts, and pits. These correspond to the face centers of the convex, saddle, and concave areas. A cap point belongs to one atom, a belt to two atoms. and a pit to three atoms.
2. Surface-distance calculation.
Based on the set of sparse critical points, we construct a graph (Graph 1 below). The graph represents surface distances between two residues. Each vertex V is an atom center that belongs to a surface residue. A residue is defined as a surface residue if at least one of its atoms is assigned to a critical point. This definition is very loose, and reflects our goal of exploiting crystallographic data in an imprecise manner. It enables application of this algorithm to low-resolution unbound and modeled structures. An edge connects two atoms u and v if they share a "critical" point. Therefore, a cap does not create edges, whereas a belt and a pit create two and three edges, respectively. To create a walk between every two Cα-atoms in the graph, Cα-atoms with a zero degree, that is, unconnected vertices are connected to the closest atom of the same residue that is either a belt or a pit. The geodesic distance between two connected atoms is calculated and assigned to the connecting edge. The surface distance between two residues is calculated as the shortest path between the corresponding Cα-atoms. Graph 1 is then
 |
 |
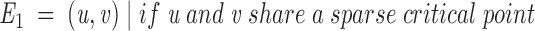 |
An example for Graph 1 is presented in Figure II in the supplemental material.
3. Selecting patch members.
The goal of this stage is to divide the protein surface to overlapping patches. The number of possible patches equals the number of walks of K steps, where K is the number of residues in a phage library peptide in graph G1. If n is the number of residues in graph G1, then the number of walks is proportional to 2n. Therefore, only some of the possible patches are created. Cα-atoms are iteratively used as patch centers. The patch radius is determined with respect to the average peptide length, X, according to Equation 1. All residues with a surface distance from the patch center lower than the patch radius are regarded as members of that patch. Because nonidentical centers can produce identical patches, the patches are processed to remove multiple appearances. Therefore, the number of patches equals to or is smaller than the number of surface residues. This method explores the patch solution space only partially, and creates nearly ball-shaped patches. A correction to this nonuniform space sampling is achieved by Equation 1. The average number of residues in patches cut by this radius is higher than X, the average peptide length. If X = 5.0, the patch radius is 8.775 Å, and can include seven residues. The number of combinations of five residues out of a group of seven residues is 7!/(7–5)! = 2520. In other words, the alignment of a five-residue peptide with a seven-residue patch can be compared with an alignment of a five-residue peptide with 2520 five-residue patches. The patch radius is
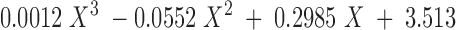 |
With Va, Vb being vertices belonging to the target and imapped peptide.
Stage 2: Matching surface patches with phage libraries derived peptides
There are two steps in this stage:
1. 3D sequence matching.
The goal of this step is to match peptide residues to patch residues. Because the patch lacks sequential order, sequence alignment methods cannot be used. Because the peptides structures are usually unknown, structural alignment methods cannot be used. Phage display-derived peptides are often structured. Although this is not common for peptides, the peptides structures are rarely solved.
To align 1D data from a peptide to 3D data from a surface patch, we use a maximal bipartite graph algorithm. Each surface patch is matched with each peptide. For each match a bipartite graph, Graph 2, is created. The graph is composed of two parts: vertices representing patch residues, and vertices representing peptide residues. All possible patch and peptide residues pairs are connected by edges. Here an edge represents similarity between two residues. The edge score is determined by a similarity matrix detailed below (Table 3▶ in Supplemental Material).The maximal bipartite algorithm is used to determine the best alignment between the patch and the peptide. A set of edges, M, of a graph G(V,E) with no self-loops is called a match if every vertex is incident to at most one edge of M (Horwitz 1989). V is a vertex and E is an edge. The bipartite matching complexity is given by Equation 2, where n is the number of patch residues and m is the number of peptide residues.
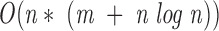 |
Graph 2:
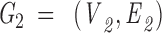 |
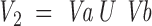 |
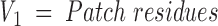 |
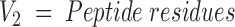 |
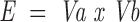 |
with Va, Vb being vertices belonging to the target and mapped peptide.
2. Similarity matrix.
Amino acid similarity can be quantified according to geometric criteria (size, shape), chemical properties, and frequency of replacement in sequences, surfaces, or binding sites. Because we are looking for binding mimicry, we have used chemical similarity to score the amino acids pairs. The matrix we used is based on the one proposed by McLachlan (1972) and presented in Table 3▶ in the Supplemental Material.
Stage 3: Scoring and correctness assessment
The score of each match was determined according to the best alignment found by the maximal bipartite matching algorithm. The scores of the edges that participate in the alignment are summed. This score is expected to reflect the degree of similarity between the peptide and patch matches. The matches were sorted according to this score. A high score is equivalent to a high rank of a solution.
High scoring matches are iteratively selected until 25% of the Template protein is covered. The number of selected matches can therefore vary even between proteins of the same size (i.e., same number of surface residues). If patches corresponding to high scoring matches overlap to a large extent, then the number of selected matches needed to cover 25% of the Template would be larger compared to the modest number of overlapping patches. This method was chosen over a fixed number of selected matches because it guarantees reduction of the effective surface. On the other hand, even a small fixed number of selected matches can include large portions of the Template’s surface. Such a prediction does not contribute to the identification of the binding site location. The now reduced surface is expected to include the binding site between the Template and the Target.
Because the entire Template molecules we use as a data set (presented in Table 1▶) derive from complexes, the "correct" answer can be calculated. Interface residues (i.e., Template residues that are spatially proximal to the Target) are defined using geometric hashing. The atoms of the Template are inserted to a geometric hash of a 0.5 Å3 bin. The hash is queried by Target atoms with a 4 Å threshold. The distance between the Target atoms used for the query and each of the query results is calculated. If it is lower than 4 Å the Template atom is defined as an interface atom. A residue is defined as an interface residue if any of its atoms is an interface atom. The correctness of each match is represented by the patch overlap with the interface that is calculated according to Equation 3. If A is the number of patch residues that are interface residues, and B the number of patch residues, the match correctness is:
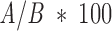 |
Peptide A
Peptide A is defined as a peptide that can be mapped to the correct binding site. If it does, it should score higher than if it is aligned by SiteLight to other parts of the protein. According to its definition, Peptide A is sufficient for binding site prediction. To choose such a peptide for a specific library, a few runs of SiteLight were performed. In each of these the input library consisted of a single peptide. Thus, the number of runs needed for the selection of Peptide A equals the size of the library (i.e., the number of peptides). The selection of Peptide A was based on the following criteria: (1) the number of predicted residues, (2) the highest rank of the solution that overlaps at least 25% of the interface; (3) the highest overlap (percentage) of the solution with the interface; (4) the rank of the solution with the largest overlap with the interface; and (5) the rank of the solution with the largest interface overlap percentage. The definitions of these criteria are given in the legend to Table 2▶.
Acknowledgments
We thank the members of the Structural Bioinformatics group. In particular, we thank Adi Barzilai, Dina Duhovny, Yuval Inbar, and Maxim Shatsky. The research of R.N. and H.J.W. in Israel has been supported in part by the Center of Excellence in Geometric Computing and its Applications, funded by the Israel Science Foundation (administered by the Israel Academy of Sciences). The research of H.J.W. is partially supported by the Hermann Minkowski-Minerva Center for Geometry at Tel Aviv University. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number NO1-CO-12400. The content of this publication does not necessarily reflect the view or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. The publisher or recipient acknowledges the right of the U.S. Government to retain a nonexclusive, royalty-free license in and to any copyright covering the article. This work was performed in partial fulfillment of the requirements for a Ph.D. degree of Inbal Halperin, Sackler Faculty of Medicine, Tel-Aviv University, Israel. The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0237103.
References
- Adey, N.B., Martaragnon, A.H., Rider, J.E., and Kay, B.K. 1995. Characterization of phage that bind plastic from phage-displayed random peptide libraries. Gene 156 27–31. [DOI] [PubMed] [Google Scholar]
- Aloy, P., Querol, E., Aviles, F.X., and Sternberg, M.J.E. 2001. Automated structure-based prediction of functional sites in proteins: Applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J. Mol. Biol. 311 395–408. [DOI] [PubMed] [Google Scholar]
- Armon, A., Grauer, D., and Ben-Tal, N. 2001. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogentic information. J. Mol. Biol. 307 447–463. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The protein data bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliznyuk, A.A. and Gready, J.E. 1999. Simple method for locating possible ligand binding sites on protein surfaces. J. Comp. Chem. 20 983–988. [Google Scholar]
- Bock, J.R. and Gough, D.A. 2001. Predicting protein–protein interactions from primary structure. Bioinformatics 17 455–460. [DOI] [PubMed] [Google Scholar]
- Bogan, A.A. and Thorn, K.S. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280 1–9. [DOI] [PubMed] [Google Scholar]
- Connolly, M. 1983a. Solvent-accessible surfaces of proteins and nucleic acids. Science 221 709–713. [DOI] [PubMed] [Google Scholar]
- ———. 1983b. Analytical molecular surface calculation. J. Appl. Crystallogr. 16 548–558. [Google Scholar]
- Dandekar, T., Snel, B., Huynen, M., and Bork, P. 1998. Conservation of gene order: A fingerprint of proteins that physically interact. Biochem. Sci. 23 324–328. [DOI] [PubMed] [Google Scholar]
- Davies, J.M., Scealy, M., Cai, Y., Whisstock, J., Mackay, I.R., and Rowley, M.J. 1999. Multiple alignment and sorting of peptides derived from phage-displayed random peptide libraries with polyclonal sera allows discrimination of relevant phagotopes. Mol. Immunol. 36 659–667. [DOI] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 12 14–20. [DOI] [PubMed] [Google Scholar]
- DeLano, W.L., Ultsch, M.H., de Vos, A.M., and Wells, J.A. 2000. Convergent solutions to binding at a protein–protein interface. Science 287 1279–1283. [DOI] [PubMed] [Google Scholar]
- DesJarlais, R.L., Sheridan, R.P., Dixon, J.S., Kuntz, I.D., and Venkataraghavan, R. 1986. Docking flexible ligands to macromolecular receptors by molecular shape. J. Med. Chem. 29 2149–2153. [DOI] [PubMed] [Google Scholar]
- Enshell-Seijffers, D., Smelyanski, L., and Gershoni, J.M. 2001. The rational design of a ‘type 88’ genetically stable peptide display rector in the filamentous bacteriophage fd. Nucleic Acids Res. 29 E50, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariselli, P., Pazos, F., Valencia, A., and Casadio, R. 2002. Prediction of protein–protein sites in heterocomplexes with neural networks. Eur. J. Biochem. 269 1356–1361. [DOI] [PubMed] [Google Scholar]
- Ferrer, M. and Harrison, S.C. 1999. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J. Virol. 73 5795–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire, E. 1999. The propagation of binding interactions to remote sites in proteins: Analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc. Natl. Acad. Sci. 96 10118–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommel, C., Peters, K.P., and Fauck, J. 1996. The automatic search for ligand binding sites in proteins of known three dimentional structure using onlygeometric criteria. J. Mol. Biol. 256 201–213. [DOI] [PubMed] [Google Scholar]
- Gabb, H.A., Jackson, R.M., and Sternberg, M.J.E. 1997. Modeling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol. 272 106–120. [DOI] [PubMed] [Google Scholar]
- Geysen, H.M., Rodda, S.J., and Mason, T.J. 1986. A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol. Immunol. 23 709–715. [DOI] [PubMed] [Google Scholar]
- Gilson, M.K. and Honig, B. 1987. Calculation of electrostatic potentials in an enzyme active site. Nature 330 84–86. [DOI] [PubMed] [Google Scholar]
- Goh, C.S., Bogan, A.A., Joachimiak, M.J., Walther, D., and Cohen, F.E. 2000. Co-evolution of proteins with their interaction partners. J. Mol. Biol. 299 283–293. [DOI] [PubMed] [Google Scholar]
- Halperin, I., Ma, B., Wolfson, H., and Nussinov, R. 2002. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 47 409–443. [DOI] [PubMed] [Google Scholar]
- Hanes, J. and Pluckthun, A. 1997. In vitro selection and evolution of functional proteins using ribosome display. Proc. Natl. Acad Sci. 94 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, S., Kitao, A., and Berendsen, H.J.C. 1997. Model-free methods of analyzing domain motions in proteins from simulations of lysozyme. Proteins 27 425–437. [DOI] [PubMed] [Google Scholar]
- Horwitz, E. 1989. Graph algorithms. Computer Science Press, Technion—Israel Institue of Technology, Haifa, Israel.
- Hu, Z., Ma, B., Wolfson, H., and Nussinov, R. 2000. Conservation of polar residues as hot spots at protein–protein interfaces. Proteins 39 331–342. [PubMed] [Google Scholar]
- Huan-Xiang, Z. and Yibing, S. 2001. Prediction of protein interaction sites from sequence profile and residue neighbor list. Proteins 44 336–343. [DOI] [PubMed] [Google Scholar]
- James, L.C. and Tawfik, D.S. 2001. Catalytic and binding poly-reactivities shared by two unrelated proteins: The potential role of promiscuity in enzyme evolution. Protein Sci. 10 2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesaitis, A.J., Gizachew, D., Dartz, E.A., Siemsen, D.W., Stone, K.C., and Burritt, J.B. 1999. Actin surface structure revealed by antibody imprints: Evaluation of phage-display analysis of anti-actin antibodies. Protein Sci. 8 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, B.K., Winter, J., and McCaffery, J. 1996. Phage display of peptides and proteins. Academic Press, New York.
- Kini, R.M. and Evans, H.J. 1995. A hypothetical structural role for proline residues in the flanking segments of protein–protein interaction sites. Biochem. Biophys. Res. Commun. 212 1115–1124. [DOI] [PubMed] [Google Scholar]
- Lang, S., Xu, J., Stuart, F., Thomas, R.M., Vrijboled, J.W., and Robinson, J.A. 2000. Analysis of antibody A6 binding to the extracellular interferon γ receptor α chain by alanine-scanning mutagenesis and random mutagenesis with phage display. Biochemistry 39 15674–15685. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A. 1995. SURFNET: A program for visualizing molecular surfaces, cavities and intermolecular interactions. J. Mol. Graph. 13 323–330. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Luscombe, N.M., Swindells, M.B., and Thornton, J.M. 1996. Protein clefts in molecular recognition and function. Protein Sci. 5 2438–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C., Dowd, C.S., Zhang, W., and Chaiken, I. 2001. Phage randomization in a charybdotoxin scaffold leads to CD4-mimetic recognition motifs that bind HIV-1 envelope through non-aromatic sequences. J. Peptide Res. 57 507–518. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996a. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257 342–358. [DOI] [PubMed] [Google Scholar]
- ———. 1996b. Evolutionarily conserved Gαβγ binding surfaces support a model of the G protein-receptor complex. Proc. Natl. Acad. Sci. 93 7505–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge, O., Yamamoto, K.C., and Cohen, F.E. 1997. Identification of functional surfaces of the binding domains of intracellular receptors. J. Mol. Biol. 274 325–337. [DOI] [PubMed] [Google Scholar]
- Lin, S.L., Nussinov, R., Fischer, D., and Wolfson, H. 1994. Molecular surface representation by sparse critical points. Proteins 18 94–101. [DOI] [PubMed] [Google Scholar]
- Luque, I. and Friere, E. 2000. Structural stability of binding sites: Consequences for binding affinity and allosteric effects. Proteins Suppl. 4 63–71. [DOI] [PubMed] [Google Scholar]
- Ma, B., Shatsky, M., Wolfson, H., and Nussinov, R. 2002. Multiple ligands binding at a single site: A matter of pre-existing conformations. Protein Sci. 11 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ng, H.L., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999. Detecting protein function and protein–protein interactions from genome sequences. Science 285 751–753. [DOI] [PubMed] [Google Scholar]
- McLachlan, A.D. 1972. Repeating sequences and gene duplication in proteins. J. Mol. Biol. 64 417–437. [DOI] [PubMed] [Google Scholar]
- Pazos, F., Helmer-Citterich, M., Ausiello, G., and Valencia, A. 1997. Correlated mutations contain information about protein–protein interaction. J. Mol. Biol. 271 511–523. [DOI] [PubMed] [Google Scholar]
- Pettit, F.K. and Bowie, J.U. 1999. Protein surface roughness and small molecular binding sites. J. Mol. Biol. 285 1377–1382. [DOI] [PubMed] [Google Scholar]
- Shatsky, M., Fligelman, Z., Nussinov, R., and Wolfson, H. 2000. Alignment of flexible protein structures. In Proceedings of the 8th conference on intelligent systems in molecular biology (ISMB) (eds. R. Altman et al.), pp. 329–343. AAAI Press, Menlo Park, CA. [PubMed]
- Shatsky, M., Nussinov, R., and Wolfson, H. 2002. Flexible protein alignment and hinge-bending detection. Proteins 48 242–256. [DOI] [PubMed] [Google Scholar]
- Sidhu, S.S., Fairbrother, W.J., and Deshayes, K. 2003. Exploring protein–protein interactions with phage display. Chem. BioChem. 14 14–25. [DOI] [PubMed] [Google Scholar]
- Sondermann, H., Scheufler, C., Schneider, C., Hohfeld, J., Hartl, F.U., and Moarefil, I. 2001. Structure of a Bag/Hsc70 complex: Convergent functional evolution of Hsp70 nucleotide exchange factors. Science 291 1553–1557. [DOI] [PubMed] [Google Scholar]
- Sowa, M.E., He, W., Slep, K.C., Lichtarge, O., and Kercher, T.G. 2001. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat. Struct. Biol. 8 234–237. [DOI] [PubMed] [Google Scholar]
- Takenaka, I.M., Leung, S.M., McAndrew, S.J., Brown, J.P., and Hightower, L.E. 1995. Hsc70-binding peptides selected from a phage display peptide library that resemble organellar targeting sequences. J. Biol. Chem. 270 19839–19844. [DOI] [PubMed] [Google Scholar]
- Todd, M.J. and Freire, E. 1999. The effect of inhibitor binding on the structural stability and cooperativity of the HIV protease. Proteins 36 147–156. [DOI] [PubMed] [Google Scholar]
- Todd, M.J., Semo, N., and Freire, E. 1998. The structural stability of the HIV-1 protease. J. Mol. Biol. 283 475–488. [DOI] [PubMed] [Google Scholar]
- Tong, A.H.Y., Dress, B., Nardelli, G., Bader, G.D., Brannetti, B., Castagnoli, L., Evangelista, M., Ferraucti, S., Nelson, B., Paoluzi, S., et al. 2002. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295 321–324. [DOI] [PubMed] [Google Scholar]
- Tsai, C.J., Lin, S.L., Wolfson, H.J., and Nussinov, R. 1997. Studies of protein–protein interfaces: A statistical analysis of the hydrophobic effect. Protein Sci. 6 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar, W.S.J. and Thornton, J.M. 2001. Conservation helps to identify biological relevant crystal contacts. J. Mol. Biol. 313 399–416. [DOI] [PubMed] [Google Scholar]
- Valuev, V.P., Afonnikov, D.A., Ponomarenko, M.P., Milanesi, L., and Kolchanov, N.A. 2002. ASPD (Artificially Selected Proteins/Peptides Database): A database of proteins and peptides evolved in vitro. Nucleic Acids Res. 30 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom, W.B. 2000. Peptide display on bacterial flagella: Principles and applications. Int. J. Med. Microbiol. 290 223–230. [DOI] [PubMed] [Google Scholar]
- Wu, S.-J., Li, J., Tsui, P., Cook, R., Zhang, W., Hu, Y., Canzianin, G., and Chaiken, I. 1999. Randomization of the receptor α chain recruitment epitope reveals a functional interleukin-5 with charge depletion in the CD loop. J. Biol. Chem. 274 20479–20488. [DOI] [PubMed] [Google Scholar]
- Wu, S.-J., Tambyraja, R., Zhang, W., Zahn, S., Godillot, A.P., and Chaiken, I. 2000. Epitope randomization redefines the functional role of glutamic acid 110 in interleukin-5 receptor activation. J. Biol. Chem. 275 7351–7358. [DOI] [PubMed] [Google Scholar]
- Xavier, G., Benoit, C., Annick, T., and Robert, B. 2000. A fast method to predict protein interaction sites from sequences. J. Mol. Biol. 302 917–926. [DOI] [PubMed] [Google Scholar]