

Abstract
Despite their clinical importance, the mechanism of action of the class C β-lactamases is poorly understood. In contrast to the class A and class D β-lactamases, which contain a glutamate residue and a carbamylated lysine in their respective active sites that are thought to serve as general base catalysts for β-lactam hydrolysis, the mechanism of activation of the serine and water nucleophiles in the class C enzymes is unclear. To probe for residues involved in catalysis, the class C β-lactamase from Enterobacter cloacae P99 was studied by combinatorial scanning mutagenesis at 122 positions in and around the active site. Over 1000 P99 variants were screened for activity in a high-throughput in vivo antibiotic resistance assay and sequenced by 96-capillary electrophoresis to identify residues that are important for catalysis. P99 mutants showing reduced capability to convey antibiotic resistance were purified and characterized in vitro. The screen identified an active-site hydrogen-bonding network that is key to catalysis. A second cluster of residues was identified that likely plays a structural role in the enzyme. Otherwise, residues not directly contacting the substrate showed tolerance to substitution. The study lends support to the notion that the class C β-lactamases do not have a single residue that acts as the catalytic general base. Rather, catalysis is affected by a hydrogen-bonding network in the active site, suggesting a possible charge relay system.
Keywords: Enzyme mechanism, β-lactamase, class C, scanning mutagenesis, high-throughput sequencing
β-Lactam antibiotics, including penicillins and cephalosporins, are among the most prevalent antibacterial agents administered worldwide. The most common bacterial resistance mechanism against these antibiotics is the production of β-lactamase enzymes (Ghuysen 1991), which hydrolyze the β-lactam bond, rendering the antibiotics inactive. As β-lactam resistance has emerged as a significant public health risk, so has interest in understanding the β-lactamases on a molecular level to aid in the design of new classes of antibiotics. The β-lactamases have been divided into four classes on the basis of sequence homology (Bush et al. 1995). The class A, C, and D β-lactamases are all quite similar to one another and are serine hydrolases, whereas the class B enzymes are zinc hydrolases, with distinct sequences, structures, and mechanisms from the serine hydrolases. Serine hydrolase β-lactamases follow the classical serine protease mechanism (Kraut 1977). An active-site serine residue attacks the β-lactam bond, forming a covalent acyl-enzyme intermediate. The acyl-enzyme intermediate is then hydrolyzed by a water molecule, thereby inactivating the antibiotic (Fig. 1 ▶). The challenge in designing β-lactam antibiotics, however, is that the targets of these antibiotics, the penicillin-binding proteins (PBPs), are very similar structurally and mechanistically to the β-lactamases (Massova and Mobashery 1998). The PBPs, however, are inefficient catalysts of the hydrolysis of the acyl-enzyme intermediate and, hence, are effectively inactivated by these antibiotics.
Figure 1.

Proposed mechanism of β-lactam hydrolysis. Following noncovalent binding of the substrate to form the E-S complex (A), the active-site serine residue, activated by a general base, attacks at the β-lactam bond of the antibiotic to form a tetrahedral intermediate. The intermediate collapses, ejecting the nitrogen, which is protonated as it leaves, resulting in the formation of acyl-enzyme intermediate (B). Subsequently, a water molecule, activated by a general base, attacks the acyl-enzyme to form a tetrahedral intermediate. The intermediate collapses, regenerating the serine nucleophile and rendering the antibiotic inactive (C). This mechanism is shared by the serine hydrolase β-lactamases and the PBPs.
All serine hydrolase β-lactamases and PBPs of known structure share the same mixed α/β fold (Ghuysen 1991). These enzymes also feature three conserved motifs that are located in similar positions in the active site (Joris et al. 1988). The first is an SXXK motif, including the active-site serine nucleophile. The second motif, (S/Y)XN, shows some variability along class lines. The class A enzymes and PBPs usually are SXN, whereas the class C enzymes are YXN and the class D enzymes SXV. The third motif is a K(T/S)G triad. The class A and class D enzymes each have an additional conserved general base in the active site. In the class A enzymes, the general base is the Glu 166 residue located on the Ω-loop at the base of the active-site cavity. First noted in crystal structures (Herzberg and Moult 1987), the importance of Glu 166 was confirmed by site-directed mutagenesis (Madgwick and Waley 1987). The class D enzymes have a similar functionality, a carbamyl group on the conserved lysine of the SXXK motif (Maveyraud et al. 2000). Evidence for the role of the carbamate acting as a general base includes the CO2 dependence of β-lactamase activity for these enzymes (Golemi et al. 2001). The class C enzymes, meanwhile, as well as the PBPs, do not appear to have an additional residue that could function in this capacity. For the PBPs, this is consistent with their lack of β-lactamase activity. In the case of the class C enzymes, however, which can hydrolyze β-lactams very efficiently (up to the diffusion limit in some cases [Bulychev and Mobashery 1999]), this deficiency is striking.
A good deal of work has gone into elucidating the mechanism of action of the class C β-lactamases. This includes X-ray crystallography of three different enzymes (Oefner et al. 1990; Lobkovsky et al. 1993; Usher et al. 1998) and a number of mutants (Crichlow et al. 1999; Beadle and Shoichet 2002), alone and in complex with good substrates (Beadle et al. 2002; Trehan et al. 2002), poor substrates (Patera et al. 2000; Powers et al. 2001; Trehan et al. 2001), and covalent (Crichlow et al. 2001) and noncovalent inhibitors (Lobkovsky et al. 1994; Caselli et al. 2001; Tondi et al. 2001; Powers and Shoichet 2002; Powers et al. 2002). A number of different class C enzymes have been purified and characterized kinetically with a number of substrates (Galleni and Frere 1988; Galleni et al. 1988; 1993). Site-directed mutants have also been constructed and characterized extensively (Tsukamoto et al. 1990a,b,c; Jacobs et al. 1992; Dubus et al. 1993, 1994a, b, 1995, 1996; Nukaga et al. 1993; Monnaie et al. 1994a,b; Trepanier et al. 1999; Vakulenko et al. 2002). A recent study has explored the sequence determinants of activity with extended-spectrum substrates (Zhang et al. 2001), but the mechanism of action is still a matter of some debate.
From the various studies, the following picture of the class C mechanism has emerged (Fig. 2 ▶). The first step in catalysis is the reversible noncovalent binding of the β-lactam to form the enzyme-substrate (ES) complex. A crystal structure of the inactive S64G mutant of the class C AmpC β-lactamase with bound cephalothin (Beadle et al. 2002) provides the best model of the ES complex. In this structure, the substrate appears to form hydrogen bonds to Gln 120 and Asn 152 at the C7 amide carbonyl and to the backbone nitrogens of Ala 318 and Gly 64 from the lactam carbonyl. These backbone nitrogens later make up the oxyanion hole that helps to stabilize the tetrahedral intermediate. The C7 amide nitrogen is within hydrogen-bonding distance of the Ala 318 (Ser in P99) backbone carbonyl. The C4 carboxylate appears to form long hydrogen bonds with Tyr 150, Lys 315, and Thr 316; Asn 346 is also nearby and may participate in the binding. The variable R1 side chain appears to make hydrophobic contacts with Val 211, Tyr 221, and Thr 319. Tyr 221 often appears to be involved in π-stacking or cation-π interactions with the R1 side chain, whereas Val 211 and Thr 319 do not always contact the substrate. Leu 119 and Tyr 150 make hydrophobic contacts with the dihydrothiazine ring, and these residues, along with Asn 289 (Ser in P99), make hydrophobic contacts with the variable R2 side chain.
Figure 2.

Representation of class C β-lactamase active site. The acyl-enzyme intermediate of AmpC β-lactamase is shown with cephalothin (CEF) covalently bound to Ser 64. Coordinates are from 1KVM (Beadle et al. 2002). Active-site residues Lys 67, Gln 120, Tyr 150, Asn 152, and Lys 315 are also shown, along with proposed hydrogen-bond interactions. Carbon atoms are colored gray; oxygen atoms, red; nitrogen atoms, blue; and sulfur atoms, yellow. The figure was prepared with Swiss-PDB Viewer (Guex and Peitsch 1997).
The next step in the mechanism is acylation, which is composed of two chemical steps with a short-lived tetrahedral intermediate. Ser 64 attacks the β-lactam bond, cleaving it and forming an acyl enzyme intermediate. Although the role of the serine is generally accepted, there is no consensus as to which other residues are involved in the acylation process and to what extent. It is assumed that, as in the classic serine protease mechanism, a general base is required to activate the serine residue and abstract a proton to facilitate formation of the tetrahedral intermediate. A proton donor is then required for the departing lactam nitrogen in the breakdown of the tetrahedral intermediate. This may be the same proton abstracted from the serine, or it may be from another source.
Two main routes have been suggested for the general base-catalyzed acylation. Oefner et al. (1990) first proposed on the basis of the crystal structure that Tyr 150 acts as a general base in the acylation step, activating and abstracting a proton from Ser 64. The proton removed by the tyrosine is later transferred to the ring nitrogen of the cleaved lactam bond, either directly or via a lysine residue. This presupposes that the tyrosine exists as a phenoxide—a rare but not unprecedented occurrence (Bohren et al. 1994). The phenoxide is stabilized by the two positively charged lysines (Lys 67 and Lys 315), which help to lower the pKa of the tyrosine. Evidence to support this possibility has been obtained from studies on tyrosine–lysine analogs in solution (Kato-Toma and Ishiguro 2001) and from molecular modeling of the protein (Lamotte-Brasseur et al. 2000). The substrate carboxylate has also been proposed to assist in the formation of the phenolate (Beadle et al. 2002). The second proposed acylation mechanism identifies Lys 67 as the general base (Dubus et al. 1994a), similar to the proposed role of Lys 70 in the acylation of class A enzymes (Strynadka et al. 1992). This requires that the lysine is neutral in the active enzyme. The proton it abstracts from Ser 64 is then shuttled via the tyrosine to the ring nitrogen.
Each of these proposals draws support from site-directed mutagenesis studies. However, whereas one such study indicates that the Y150F and Y150S mutants are extremely impaired (Dubus et al. 1996), another study shows that they retain significant activity (Dubus et al. 1994a). The Tyr 150 mutants showed greatly decreased activity (500- to 5000-fold) with substrates such as cephalothin and PADAC. However, Y150F showed >10% of wild-type activity with common substrates such as benzylpenicillin, nitrocefin, and CENTA according to one study. The Y150S mutant was also shown to have significant (>10%) β-lactamase activity with many substrates such as FAP, benzylpenicillin, cefotaxime, cephalexin, and cefadroxil. Y150S should also be significantly impaired if it is a phenoxide that acts as the general base. The effect of mutation of Lys 67 has also been studied. Mutation to Gln results in a large decrease in activity; results with the K67R mutant, however, are more difficult to interpret (Monnaie et al. 1994b). These results have been used to support the claim that the role of Lys 67 is electrostatic, rather than that of a general base. Another study of mutants at Asn 152 showed a large drop in activity (500- to 5000-fold) with all mutations and substrates examined (Dubus et al. 1995). This may indicate a more prominent role for Lys 67, as Asn 152 interacts directly with Lys 67, but not with Tyr 150.
The final step, and the crucial one that distinguishes β-lactamases from PBPs, is deacylation of the acyl serine intermediate. This step involves a nucleophilic water molecule, presumably activated by a nearby group. Tyr 150 and Lys 67 have each been proposed to act as a general base in deacylation, parallel to the proposals of their roles in acylation. A more recent proposal has introduced the possibility of substrate-assisted catalysis in the deacylation by the ring nitrogen (and possibly the carboxylate as well). First proposed on the basis of crystal structures and molecular models, some evidence for substrate-assisted catalysis has come from work with substrate analogs (Bulychev et al. 1997). The Thr 316 residue has also been suggested to activate the water molecule for attack (Beadle et al. 2002). Another proposal has the hydrolysis reaction proceeding uncatalyzed (Lobkovsky et al. 1993).
None of these proposals fully explains the drastic decrease in activity of Asn 152 mutants. Mutating the Asn 152 residue to Glu, His, or Leu had a significant effect on catalytic activity with the substrates tested (Dubus et al. 1995), approaching or greater than that of mutations at Tyr 150, Lys 67, and Lys 315. Furthermore, none of these explanations fully account for the difference in activity between the class C β-lactamases and the PBPs. All of the functionalities that have been proposed to participate in the hydrolysis by β-lactamases are also present in the PBPs and occupy similar positions in the respective active sites (Knox et al. 1996). Yet, the β-lactamases efficiently catalyze hydrolysis of β-lactams, whereas the PBPs do not.
To determine whether any other residues play a role in catalysis, and to provide a framework for future studies of the catalytic mechanism of the class C β-lactamases, we engaged in a scanning mutagenesis study of the P99 β-lactamase. Alanine scanning is a commonly used technique for pinpointing important residues in a protein (Cunningham and Wells 1989). Although traditional alanine scanning can be labor intensive, use of a combinatorial method for constructing mutants when coupled to an in vivo assay and sequencing can help to alleviate this problem (Gregoret and Sauer 1993; Morrison and Weiss 2001). Further modification of the system to allow more conservative amino acid replacements can help reduce some of the ambiguity that accompanies any mutagenesis study. Although alanine is used traditionally for scanning studies, any mutation at a position that is crucial for activity should significantly reduce the catalytic efficiency—even a very conservative mutation (for example, see Straus et al. 1985). We constructed combinatorial libraries of P99 mutants at 122 positions in and around the active site, screened them for activity using an in vivo growth assay based on antibiotic resistance, and sequenced the mutant genes. Variants with reduced activity were purified and characterized in vitro. Results of this study are presented herein and discussed in the context of the mechanism of the class C β-lactamases.
Results
Despite their clinical importance, few mutants of the class C β-lactamases have been characterized. Thus, to probe the mechanism of these enzymes, we chose to begin by constructing mutants at all positions in and around the active site of the class C β-lactamase from Enterobacter cloacae P99, and determining their effect on the catalytic activity of the enzyme using a high-throughput screen for antibiotic resistance in vivo (Fig. 3 ▶). The mutants were constructed by overlap-extension PCR with doped oligonucleotides, assayed for growth on ampicillin and cephalothin, and sequenced to allow assessment of the effect of mutation at each of the studied positions. Mutants that were seen to have a significant effect on antibiotic resistance were then purified and their kinetic activities determined (Fig. 4 ▶).
Figure 3.

Ribbon diagram of the P99 β-lactamase, highlighting the residues analyzed in this study. Coordinates are from 2BLT (Oefner et al. 1990; Lobkovsky et al. 1993; Usher et al. 1998). Positions that were included in the scanning mutagenesis are shown in blue. Residues at which mutations had a significant effect on in vivo antibiotic resistance are in orange. Active-site residues Ser 64, Lys 67, Tyr 150, and Asn 152 are shown as stick representations with coloring as in Figure 2 ▶. The image was prepared with WebLab Viewer Lite (Accelrys Inc.).
Figure 4.
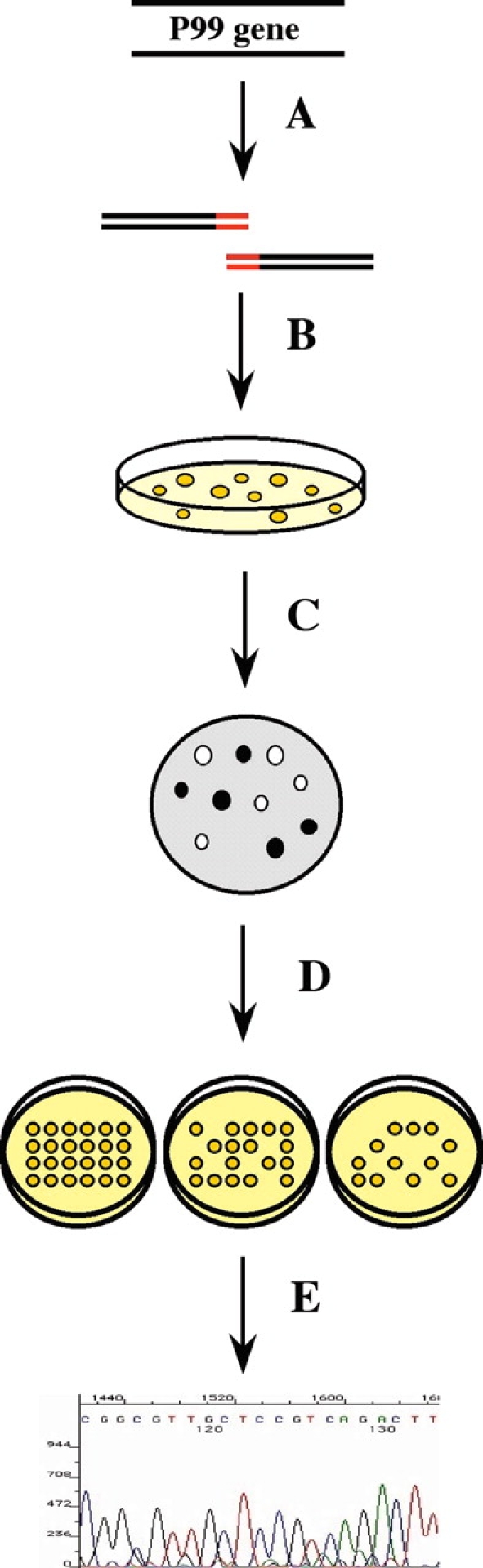
Activity Screen. (A) P99 mutant libraries were constructed by overlap-extension PCR, ligated into an expression vector, and (B) transformed into E. coli and grown on nonselective medium. (C) Plated bacteria was assayed for expression of His-tagged protein by colony blotting and immunodetection. (D) Colonies expressing His-tagged protein were then assayed for antibiotic resistance on agar plates containing varying concentrations of antibiotic, and (E) the P99 variant expressed in each colony was determined by high-throughput sequencing.
Library design
The library was designed to generate mutations at all of the positions in and around the active site, as these are the amino acids that are the most likely to have direct involvement in catalysis. The residues to be included in the library were chosen by analysis of the crystal structure 2BLT (Lobkovsky et al. 1993) and were subjected to scanning mutagenesis to determine their contribution to catalytic activity. The library was designed to include all residues within 5 Å of the active-site cavity. The CastP algorithm (Liang et al. 1998) was used to define the active-site cavity, and Maestro (Schrodinger, Inc.), to determine which residues fall within 5 Å of this cavity. This yielded a list of 122 amino acids—still more than one-third of the total residues in the protein. These residues were divided into 9 libraries of 10 to 16 contiguous positions each.
Rather than traditional alanine scanning, in which residues are probed for their effect on catalysis by mutating them to Ala, specific counterparts were designated for each amino acid. This choice was based on improved conservation of structure. Rather than only using Ala, amino acids with similar chemical and structural properties to the wild-type amino acid were chosen. The choice of amino acid replacements was also constrained to those that could be coded for with only one base change from the wild-type residue. This restriction is necessitated by the combinatorial nature of the experiment, in which mutations are generated simultaneously at multiple positions. The amino acid replacement scheme is detailed in Table 1. The 9 libraries were constructed by overlap-extension PCR, using oligonucleotides doped at the relevant positions with an 85:15 mixture of wild type to mutagenic base. This ratio was chosen because it should give a high percentage of single and double mutants.
Table 1.
Amino acid replacement scheme
| Residue | Codon | Replacement | Codon |
| Ala | GCN | Gly | GGN |
| Asp | GAY | Asn | AAY |
| Glu | GAR | Gln | CAR |
| Phe | TTY | Tyr | TAY |
| Gly | GGN | Ala | GCN |
| His | CAY | Asn | AAY |
| Ile | ATH | Leu | CTH |
| Lys | AAR | Arg | AGR |
| Leu | CTH | Ile | ATH |
| TTR | ATR | ||
| Met | ATG | Leu | CTG |
| Asn | AAY | Asp | GAY |
| Pro | CCN | Ala | GCN |
| Gln | CAR | Glu | GAR |
| Arg | AGR | Lys | AAR |
| Ser | TCN | Thr | ACN |
| AGY | ACY | ||
| Thr | ACN | Ser | TCN |
| Val | GTN | Leu | CTN |
| Trp | TGG | Leu | TTG |
| Tyr | TAY | Phe | TTY |
Amino acids were mutated to the closest homologs that can be reached with only one base change. Codons used are shown above, with relevant degeneracies indicated with IUPAC nomenclature (N = A/C/G/T; Y = C/T; R = A/G; H = A/C/T). Changed base is indicated in bold. Cysteine is not shown, as there are no cysteines in the P99 enzyme.
Activity screen
The screen was designed to allow enzymes with impaired β-lactamase activity to be identified solely on the basis of their in vivo behavior, without having to purify and characterize each protein. To this end, plasmid pSG393 was designed to functionally express P99, giving an antibiotic-resistance phenotype. In this vector, the enzyme is expressed with its native signal peptide, which directs it to the periplasm. It is expressed from the TEM-1 promoter (Sutcliffe 1978)—a weak but constitutive Escherichia coli promoter from the TEM-1 β-lactamase. The plasmid was constructed by inserting a 77-bp piece of the TEM promoter between the NdeI and XhoI sites of the pET26 plasmid. E. coli expressing wild-type P99 from pSG393 grows well at 160 μg/mL ampicillin, slowly at 320 μg/mL, and not at all at 640 μg/mL. With cephalothin, the cells grow well up to 1280 μg/mL and more slowly at 2560 μg/mL. Tests with P99 mutants that are known to have reduced activity showed that they grow only at lower antibiotic concentrations (data not shown). Libraries of mutants were constructed in this vector system and tested for antibiotic resistance. The mutants were screened on both ampicillin and cephalothin, generally considered to be model penicillin and cephalosporin substrates, respectively, to reduce substrate-specific effects.
Library construction was followed by a prescreening step, immunodetection, to remove all constructs that were not expressed well. The immunodetection was performed immediately after subcloning and transformation into bacteria, using antibodies to the carboxy-terminal His-tag. The immunodetection gave a clear map that was used to avoid picking colonies that were not expressing protein. In addition to screening out defective constructs—such as those plasmids that did not get an insert—this screen also presumably eliminated proteins that are poorly expressed and/or quickly degraded in vivo.
The colonies were then assayed for antibiotic resistance by frogging onto agar plates (Fig. 5 ▶; data not shown) containing 30 μg/mL kanamycin and varying concentrations of ampicillin (5, 10, 20, 40, 80, or 160 μg/mL) or cephalothin (20, 40, 80, 160, 320, 640, 1280, or 2560 μg/mL). The highest antibiotic concentration that allowed growth was recorded for all 9 plates (consisting of 96 colonies each), and this data was combined with the sequencing data to assign an MPC to each mutant. Those mutants that appeared to have significantly decreased antibiotic resistance were constructed individually by site-directed mutagenesis, and the MPC was confirmed for each. Additionally, some of the mutations for which the screen did not provide sufficient information to assign an MPC were constructed by site-directed mutagenesis, and assayed.
Figure 5.

Representative antibiotic resistance screen. Antibiotic resistance of colonies expressing P99 mutants was assessed by frogging onto plates containing varying concentrations of ampicillin or cephalothin. The highest antibiotic concentration at which there was growth was recorded. A series of ampicillin plates is shown, with antibiotic concentrations indicated in microgram/milliliter.
Using the data from the growth assay and the sequencing, we classified each of the 122 mutations as either having a limited effect or having a significant effect on resistance (Table 2). The cutoff chosen was growth on plates containing 80 μg/mL antibiotic for ampicillin and growth at 1280 μg/mL cephalothin. Any mutation that rendered the bacteria unable to grow at either of these concentrations was classified as having a significant effect. We determined that 92 of the studied mutations had a limited effect on antibiotic resistance, whereas 11 had a significant effect. For the remaining 19, there was not sufficient data to reach any conclusion. The results obtained with ampicillin and cephalosporin showed very close correlation to each other. The relative growth profiles of the 900 individual colonies showed remarkable similarities for the two antibiotics. Although there were differences in the final list of mutations that had a significant effect on growth, the majority of these (if not all) can be explained by an inexact correspondence of the cutoffs chosen for significance.
Table 2.
Effect of mutations on in vivo antibiotic resistance
| Limited Effect |
| L59I, L62I, I65V, S66T, T68S, F69Y, T70S, T111S, Y112F, T113S, G115A, G116A, L117I, L119I, P122A, D123N, E124Q, R148K, L149I, A151G, S154T, I155V, L157I, F158Y, G159A, A160G, E195Q, E196Q, A197G, H198N, Y199F, A200G, W201L, G202A, R204K, D205N, G206A, K207R, V211I, S212T, P213A, G214A, M215L, L216I, D217N, A218G, Q219E, A220G, Y221F, G222A, V223I, K224R, R258K, Y259F, W260L, R261K, I262V, S264T, M265L, Y266F, Q267E, L269I, M273L, E285Q, G286A, S287T, D288N, K290R, V291I, A292G, L293I, A294G, S311T, V313I, H314N, T316S, (T316A), S318T, T319S, F322Y, G323A, S324T, Y325F, A340G, T342S, S343T, Y344F, P345A, P347A, R349K, V350I, A352G, A353G |
| Significant Effect |
| F60Y, E61Q, S64T, K67R, Q120E, Y150F, N152D, Y203F, K315R, N341D, N346D |
| Insufficient Data |
| G63A, G71A, A114G, P118A, V121I, A153G, G156A, G263A, G268A, G270A, W271L, E272Q, S289T, W312L, G317A, G320A, G321A, A348G, E351Q |
Mutations that rendered the bacteria unable to grow on 80 μg/mL ampicillin or 1280 μg/mL cephalothin were classified as significant. Italics denote a previously characterized mutant. Parentheses denote a spontaneous mutation.
Of the 11 mutations that were shown to have a significant effect, 3 have been characterized previously either in P99 or in the closely related AmpC β-lactamase. This group includes the K67R (Monnaie et al. 1994b), Y150F (Dubus et al. 1994a, 1996), and N152D (Dubus et al. 1995) mutants. The Y150F mutant showed growth on 5 μg/mL ampicillin and no growth on cephalothin, whereas the other two mutants did not grow on any concentration of either antibiotic. The K67R, Y150F, and N152D mutants have all been characterized extensively, and the in vivo results correlate well with the in vitro data seen previously with these mutants. The S64T mutant was also significantly impaired, showing no growth on either antibiotic. Although this threonine mutant has not been studied, it is certainly not unexpected that it has very little activity, given that Ser 64 is the active-site serine nucleophile.
Another of these mutants, K315R, is noteworthy in that its effect on activity is quite small. Although the Lys 315 residue is part of the KT(S)G triad, which is universally conserved across all PBPs and β-lactamases, the MPC with ampicillin is 80 μg/mL and is 640 μg/mL with cephalothin. A similar result was seen with the T316S mutant of the same triad, which showed no significant effect on antibiotic resistance in vivo. Furthermore, the T316A mutant, which arose spuriously in the screen, had a significant effect only on cephalothin resistance, and the MPC was still 640 μg/mL. The T316A (Dubus et al. 1994b) mutant has been studied in AmpC, yielding comparable results.
Thus, six novel mutants were identified that had a significant effect on antibiotic resistance: F60Y, E61Q, Q120E, Y203F, N341D, and N346D. The MPCs for these mutants are 10, 20, 40, 40, 5, and 20 μg/mL with ampicillin, respectively. Although all six had significant effects on ampicillin resistance, only F60Y, N341D, and N346D were classified as having a significant effect on cephalothin resistance. With cephalothin, the MPCs are 160, 1280, 1280, 1280, 40, and 320 μg/mL.
High-throughput sequencing
The P99 gene from each colony that was screened for antibiotic resistance was sequenced in a 96-capillary MegaBACE 1000 DNA sequencing system (Kheterpal and Mathies 1999) by the energy transfer dideoxyterminator method. Energy transfer-sequencing chemistry greatly enhances overall fluorescence detection sensitivity, producing distinct spectral output, more normalized peak heights between different fluorescent tags, and decreased mobility shift artifacts (Ju et al. 1995; Rosenblum et al. 1997). Sequences meeting the chosen quality threshold (read length of at least 100 bp and Phred score of at least 17) were obtained with at least one primer for >90% of the colonies. The data had an average Phred quality score of 27.6, with an average read length of 556 nucleotides per sequence sample. This data was used to determine the sequence variation of the mutants assayed in the library screen.
In vitro characterization of select P99 variants
The six novel mutations that showed a significant effect on antibiotic resistance were constructed in the pSG430 expression vector by site-directed mutagenesis for characterization in vitro. The K315R mutant was also constructed, as were the S64T and Y150F mutants to serve as negative controls, and the D217N mutant to serve as a positive control. The controls were chosen on the basis of their performance in the growth assay; D217N showed near wild-type growth, whereas S64T and Y150F showed virtually no antibiotic resistance at all.
These 11 proteins—wild-type P99 and the 10 mutants—were overexpressed in E. coli and purified on nickel-affinity spin columns via their carboxy-terminal His-tags. The proteins were all >95% pure as judged by Coomassie Blue staining. Whereas all of the proteins expressed at similar levels to the wild-type P99, there were variations in the amount of soluble protein that could be obtained. The E61Q and Y203F mutants yielded a lower percentage of soluble protein than wild-type P99. The N341D and F60Y mutants, meanwhile, gave no detectable soluble protein at all, and could not be refolded and further characterized.
Circular dichroism wavelength scans of all of the purified proteins were acquired to ensure that none of the mutants had any large structural perturbations from the wild-type protein. All proteins measured showed spectra nearly identical to that of the wild-type protein, with minima at 217 nm (data not shown).
The enzymatic activity of the proteins was analyzed. Steady-state kinetic constants for the reaction of the purified proteins with ampicillin or cephalothin were determined by monitoring the change in UV absorbance upon cleavage of the β-lactam bond, at 236 and 265 nm, respectively. Initial velocity was determined at eight different substrate concentrations for cephalothin and four for ampicillin, and this data was fit to the Michaelis-Menten equation by nonlinear regression analysis. The kinetic constants kcat and Km could both be determined for cephalothin from the fitting of the curve. For ampicillin, however, due to the very low Km (Galleni and Frere 1988) coupled with the low Δɛ at the monitored wavelength, only kcat could be determined from the initial rates.
The in vivo antibiotic resistance is expected to correlate with both kcat and (inversely) Km, although not necessarily linearly (Nikaido and Normark 1987). Thus, mutants with a decrease in kcat/Km should be identifiable in vivo by lowered antibiotic resistance. This approach was successful with mutants that are known (or expected) to have greatly reduced catalytic activity, as all of these showed greatly decreased antibiotic resistance. The screen was successful to a lesser degree at identifying mutations having a smaller effect on activity. Mutants with 10- to 100-fold decreases in kcat/Km with cephalothin were identified, but a mutant with almost no effect on catalysis was also picked out in the screen. This anomaly can likely be attributed to differences in the concentration of active enzyme in the cell.
Kinetic constants were measured for the wild-type enzyme and eight mutants (Table 3). The two enzymes that served as negative controls, S64T and Y150F, showed very weak β-lactamase activity in vitro. Both showed a decrease in kcat/Km of more than five orders of magnitude with cephalothin as compared with wild-type P99. The kinetic constants with ampicillin were not fully determined, but it was noted that they are both extremely slow, and it appears that the Kms are significantly higher than that of the wild-type or any of the other mutants (data not shown; see supplemental material). The positive control, D217N, showed near wild-type activity with both cephalothin and ampicillin. The K315R mutant was 20-fold less active than the wild-type enzyme with cephalothin, with a 3-fold reduction in kcat and 7-fold increase in Km. With ampicillin, however, kcat was comparable with that of wild-type P99.
Table 3.
Kinetic characterization
| Enzyme | kcat (s−1) | Km (μM) | kcat/Km (M−1s−1) | % Wild-type | |
| WT | AMP | 0.41 ± 0.01 | — | — | — |
| CEF | 153 ± 6 | 5.7 ± 1.5 | 2.7 × 107 | — | |
| E61Q | AMP | 0.340 ± 0.009 | — | — | 83 |
| CEF | 110 ± 3 | 44 ± 4 | 2.5 × 106 | 9 | |
| S64T | AMP | ND | — | — | — |
| CEF | 0.07 ± 0.01 | 880 ± 230 | 80 | 0.0003 | |
| Q120E | AMP | 0.079 ± 0.004 | — | — | 19 |
| CEF | 30.0 ± 0.6 | 10.9 ± 1.3 | 2.7 × 106 | 10 | |
| Y150F | AMP | ND | — | — | — |
| CEF | 0.107 ± 0.008 | 750 ± 100 | 1.4 × 102 | 0.0005 | |
| Y203F | AMP | 0.32 ± 0.007 | — | — | 78 |
| CEF | 127 ± 3 | 5.8 ± 1.1 | 2.2 × 107 | 81 | |
| D217N | AMP | 0.31 ± 0.004 | — | — | 76 |
| CEF | 164 ± 5 | 8.3 ± 1.6 | 2.0 × 107 | 74 | |
| K315R | AMP | 0.42 ± 0.005 | — | — | 102 |
| CEF | 47 ± 1 | 39 ± 3 | 1.2 × 106 | 4 | |
| N346D | AMP | 0.27 ± 0.007 | — | — | 66 |
| CEF | 34 ± 3 | 130 ± 28 | 2.6 × 105 | 1 |
β-lactamase activity with ampicillin (AMP) and cephalothin (CEF) was measured for the P99 β-lactamase or P99 variant in sodium phosphate buffer pH 7.0 at room temperature. Steady-state kinetic parameters were determined by fitting initial rates at different substrate concentrations to the Michaelis-Menten rate equation. The kinetic constants kcat and Km were determined for cephalothin; only kcat was determined for ampicillin as the Km is too low to be measured by initial velocity kinetics (Galleni and Frere 1988). Kinetic parameters were not determined for S64T and Y150F with ampicillin, as they showed nonideal behavior.
Of the four remaining mutants analyzed, N346D had the weakest catalytic activity with cephalothin. Both kcat and Km were affected, and kcat/Km was 1% that of wild-type P99. With ampicillin, meanwhile, kcat was close to wild-type levels. E61Q and Q120E both had 10% of wild-type activity, but the effect was mainly on Km in E61Q, whereas kcat was affected in the Q120E. The kcat of these mutants with ampicillin was consistent, with E61Q showing near wild-type activity and Q120E decreased nearly 10-fold. The Y203F mutant, meanwhile, showed near wild-type activity with both substrates.
Discussion
Unlike the class A and class D β-lactamases, it appears that the class C enzymes do not have a lone residue that acts as the catalytic general base. No general base has been identified that abrogates enzyme activity when mutated, as is observed for the Glu 166 in the class A β-lactamases (Madgwick and Waley 1987). Rather, the catalytic activity appears to be extended across an active site hydrogen-bonding network, possibly with assistance from the β-lactam substrate. Asn 152, Lys 67, and Tyr 150 form a hydrogen-bonding network, and mutation of any of these residues reduces the activity of the enzyme by several orders of magnitude (Dubus et al. 1994a, 1995, 1996; Monnaie et al. 1994b). This hydrogen-bonding network is flanked by Gln 120 and Lys 315, and these residues also contribute to catalysis, although less so. The residue that technically serves as the general base, accepting the proton in acylation and deacylation, can likely be narrowed down to one of Lys 67, Tyr 150, and the substrate ring nitrogen. It is apparent, however, that other residues making up the hydrogen-bonding network play a crucial role in catalysis, either as part of a proton shuttle or in an electrostatic role.
In contrast to the hydrogen-bonding network, the remaining active-site residues appear to be largely unnecessary for catalysis. Although some of these positions may be involved in recognition, which may not be perturbed much by conservative mutations, a crucial role in catalysis can likely be ruled out. This includes residues that appear to make hydrophobic contacts to the substrate, such as Leu 119, Asn 289, Leu 293, Tyr 221, Val 211, and Thr 319, as well as those such as Thr 316 and Asn 346 that appear to be in position to form hydrogen bonds or electrostatic interactions. Although the N346D mutant shows reduced activity, the Ala and Ile mutants have shown near wild-type behavior (Zhang et al. 2001). This would indicate that it is the addition of a negative charge, rather than subtraction of the wild-type amino acid, that produces the effect. These mutagenesis studies also confirm that the hydroxyl of the conserved K(T/S)G triad is not necessary for catalysis in the class C β-lactamases, but rather appears to be a vestigial residue that is no longer necessary for this class of enzyme.
The remaining residues studied, with the exception of one cluster, do not appear to have a significant effect on enzyme activity. Whereas much has been made of recent examples of mutations distant from the active site that have a surprisingly large effect on enzymatic activity (for example, see Shimotohno et al. 2001), these mutagenesis studies recapitulate our understanding that it is the residues in the active site directly contacting the substrate that are the most likely to affect catalysis. None of the 100+ residues mutated in this study that do not directly contact the substrate had a significant effect on catalysis. Interestingly, however, mutations at Phe 60, Glu 61, Tyr 203, and Asn 341, all positions buried in the α/β sandwich domain of the enzyme in close proximity to one another did affect antibiotic resistance. The behavior of these mutants suggests that this cluster is important for the stability rather than the activity of the protein. The mutations studied at these positions all affected the solubility of the protein, with the F60Y and N341D proving insoluble under all conditions tested in vivo. The E61Q mutant did show a slight decrease in activity, however, mostly affecting Km. On the basis of the three-dimensional structure of the enzyme, Glu 61 may affect the positioning of the active site nucleophile Ser 64.
Thus, it appears that the class C β-lactamases have a charge relay system, rather than a lone residue, acting as a general base catalyst. Gln 120, Asn 152, Lys 67, Tyr 150, and Lys 315 form a hydrogen-bond network that directly interacts with the substrate. Even conservative mutations at any of these positions affect the catalytic activity of the enzyme, with mutations at Asn 152, Lys 67, and Tyr 150 affecting activity by several orders of magnitude. Based simply on inspection of high-resolution structures of class C β-lactamases, both Lys 67 and Tyr 150 are in a suitable position to abstract a proton from Ser 64 or the hydrolytic water molecule and interact with the hydrogen-bond network. It is less clear how the β-lactam nitrogen would interact with this network if there is substrate-assisted catalysis. Further studies are underway to determine the mechanism of the proton relay system more precisely. A puzzle that remains to be explained is how the reactivity of the class C β-lactamases can differ from that of PBPs such as the Streptomyces R61 DD-Transpeptidase (Kuzin et al. 1995), given that this hydrogen-bonding network is conserved between the two.
Materials and methods
General methods
Standard protocols for molecular biology were used. Restriction enzymes, Vent Polymerase, and T4 DNA ligase were purchased from New England Biolabs. Pfu Turbo Polymerase and the E. coli TG1 strain were from Stratagene. Taq polymerase was purchased from Promega. The E. coli Tuner (DE3) strain, the pET26b plasmid, and the BugBuster protein extraction reagent were purchased from Novagen. Oligonucleotides were purchased from the Great American Gene Company. Falcon 96-well plates with U-shaped wells were used for growing bacteria. The Frogger used to transfer cells into 96-well plates or onto petri plates containing agar medium was purchased from Dan-Kar Corp. Penta-His anti-His tag antibody was purchased from QIAGEN, as were the Ni-NTA spin columns used to purify the proteins. Slide-a-Lyzer dialysis cassettes were purchased from Pierce. Isopropyl-β-D-thiogalactoside (IPTG) was from American Bio-Organics. All other chemicals were purchased from Aldrich or Sigma, as was Accutaq polymerase. Circular nitrocellulose membranes were from Osmonics. The MegaBACE 1000 capillary sequencer was purchased from Amersham Biosciences, as were the ECL-Plus chemiluminescence reagent, the Hyperfilm MP autoradiography film, and the ET terminator premix. The anti-mouse IgG secondary antibody conjugated to horseradish peroxidase was from Cell Signaling Technology. N,N,N′,N′-Tetramethylethylenediamine (TEMED) and acrylamide:bis-acrylamide (37.5:1) for making 10% acrylamide/bis-acrylamide gels were purchased from Fisher. For protein molecular weight detection, the Kaleidoscope SDS-PAGE molecular weight marker from Bio-Rad was used. An MJ Research PTC-200 Peltier Thermal Cycler was used for all PCR reactions. UV/Vis measurements were taken using a Molecular Devices 384 SpectraMax Plate Reader. Quartz cuvettes for UV spectrometry were purchased from Fisher. CoStar UV-transparent 96-well plates were from Corning. Electroporation was carried out using a Bio-Rad E. coli Gene Pulser and Bio-Rad 0.1-cm electroporation cuvettes. An M35A-M X-OMAT Processor from Kodak was used to develop the film for the immunodetections. Sequencing of individual plasmid constructs was performed by GeneWiz. Plasmid pNU602 (Dubus et al. 1993) encoding the E. cloacae P99 class C β-lactamase was kindly provided by J. Frere. The P99 gene on this plasmid contained five point mutations, I16V, A88P, and A299V, and two silent mutations Thr 42 ACA→ACG and Ala 292 GCA→GCG.
Plasmid construction
All plasmids used in this work were derived from pET26b (kanR), a T7 overexpression plasmid (Table 4). An 1107-bp fragment encoding the E. cloacae P99 class C β-lactamase without its signal peptide and with a carboxy-terminal 6-His tag, was amplified by PCR from plasmid pNU602 using primers VWC167 (5′-GCATACGTCCATATGACGCCAGTGTCAGAAAAA) and VWC69 (5′-GCATTGCTGAAGCTTAGTGGTGGTGGTGGTGGTGCTGTAGCGCCTCGAGG). This fragment was inserted between the NdeI and Hind III sites in pET26 to generate the plasmid pSG430. The constructs for expression of mutant enzymes were all generated from this plasmid by QuickChange site-directed mutagenesis according to the manufacturer’s protocol (Stratagene). The primers used for mutagenesis were as follows:
Table 4.
Plasmids used in this study
| Name | Description | Source/reference |
| pNU602 | P99 β-lactamase | (Dubus et al. 1993) |
| pET26b | PT7 kanR pBR ori | Novagen |
| pSG397 | PTEM kanR pBR ori | This study |
| pSG422 | PTEM/P99 β-lactamase-His6 kanR pBR ori | This study |
| pSG430 | PT7/P99 β-lactamase-His6 kanR pBR ori | This study |
| pSG736 | No promoter, Sfi sites, kanR pBR ori | This study |
| pSG1132 | pSG430 with F60Y mutation | This study |
| pSG1133 | pSG430 with E61Q mutation | This study |
| pSG1134 | pSG430 with S64T mutation | This study |
| pSG1135 | pSG430 with Q120E mutation | This study |
| pSG1136 | pSG430 with Y150F mutation | This study |
| pSG1137 | pSG430 with Y203F mutation | This study |
| pSG1138 | pSG430 with D217N mutation | This study |
| pSG1139 | pSG430 with K315R mutation | This study |
| pSG1140 | pSG430 with N341D mutation | This study |
| pSG1141 | pSG430 with N346D mutation | This study |
VWC972 (5′-TTACGCCTCAGACCCTGTACGAGCTCGGTTCTATAAGTAAAACC) and VWC973 (5′-GGTTTTACTTATAGAACCGAGCTCGTACAGGGTCTGAGGCGTAA) to generate plasmid pSG1132; VWC953 (5′-TTACGCCTCAGACCCTGT TCCAATTGGGTTCTATAAGTAAAACC) and VWC954 (5′-GGTTTTACTTATAGAACCCAATTGGAACAGGGTCTGAGGCGTAA) to generate plasmid pSG1133; VWC1039 (5′-GACC CTGTTCGAGCTGGGTACCATAAGTAAAACCTTCACCG) and VWC1040 (5′-CGGTGAAGGTTTTACTTATGGTACCCAGCTCGAACAGGGTC) to generate plasmid pSG1134; VWC1043 (5′-CCGCTGGCGGCCTGCCGCTCGAGGTACCGGATGAGGTCAC) and VWC1044 (5′-GTGACCTCATCCGGTACCTCG AGCGGCAGGCCGCCAGCGG) to generate plasmid pSG1135; VWC560 (5′-GGCACAACGCGTCTTTTTGCCAACGCCAGCATC) and VWC561 (5′-GATGCTGGCGTTGGCAAAAAGACGCGTTGTGCC) to generate plasmid pSG1136; VWC955 (5′-GCGCATTACGCCTGGGGCTTTCGCGACGGTAAAGCGGTGCGCG) and VWC956 (5′-CGCGCACCGCTTTACCGTCGCGAAAGCCCCAGGCGTAATGCGC) to generate plasmid pSG1137; VWC1000 (5′-GCGTTTCGCCGGGTATGCTGAATGCGCAAGCCTATGGCGTGAAAAC), and VWC1001 (5′-GTTTTCACGCCATAGGCTTGCGCATTCAGCATACCCGGCGAAACGC) to generate plasmid pSG1138; VWC1047 (5′-CAAAGCGTCCTGGGTCCATAGAACGGGTTCTACTGGCGGGTTTGGCAG), and VWC1048 (5′-CTGCCAAACCCGCCAGTAGAACCCGTTCTATGGACCCAGGACGCTTTG) to generate plasmid pSG1139; VWC957 (5′-CAGATCGGTATTGTGATGCTAGCGGATACAAGCTATCCGAACCCG) and VWC958 (5′-CGGGTTCGGATAGCTTGTATCCGCTAGCATCACAATACCGATCTG) to generate plasmid pSG1140; VWC1049 (5′-CGCGAATACAAGCTATCCGGATCCGGCACGCGTTGAGGCGG) and VWC1050 (5′-CCGCCTCAACGCGTGCCGGATCCGGATAGCTTGTATTCGCG) to generate plasmid pSG1141. Each set of primers incorporated a restriction site in addition to the given mutation to expedite the screening for mutants.
The T7 promoter in pET26 was replaced with a 77-bp fragment from the TEM-1 β-lactamase promoter (Sutcliffe 1978). This fragment was generated by annealing two complementary oligonucleotides, VWC169 (5′-GATCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTCA) and VWC170 (5′-TATGACTCT TCCTTTTTCAATATTATTGAAGCATTTATCAGGGTTATTGTCTCATGAGCGGATACATATTTGAATGTATTTA), to produce a cassette with single-stranded overhangs. The cassette was inserted between the BglII and NdeI sites of pET26 to generate pSG393. An 1189-bp fragment encoding the full-length P99 gene with a carboxy-terminal 6-His tag was amplified from pNU602 with primers VWC68 (5′-GCATACGTCCATATGATGAGAAAATCCCTTTG) and VWC69 (see above). This fragment was inserted between the NdeI and Hind III sites in pET26 to generate the plasmid pSG422.
The T7 promoter and MCS in pET26 were replaced by a 60-bp cassette containing a similar MCS flanked by two SfiI sites. This cassette was generated by annealing the overlapping oligonucleotides VWC509 (5′-GATCTGGCCCCCGGGGCCATGGATCCGCGGAGCTCGAATTCGTCGACAAGCTTGGCCCTGCCGGC C) and VWC510b (5′-TCGAGGCCGGCAGGGCCAAGCTTGTCGACGAATTCGAGCTCCGCGGATCCATGGCCCCGGGGG CCA). The cassette was inserted between the BglII and XhoI sites to generate plasmid pSG736.
Library construction
A total of 9 libraries were constructed, each containing mutations concentrated in a region of 10 to 16 residues. The libraries were made by overlap-extension PCR, using oligonucleotides doped with 15% mutant base at the relevant positions. The outside primers, VWC562 (5′-CGGCAGACTGGATTCGAGATCTAAATA CATTCAAATATG) and VWC497 (5′-CAGACCCAGTTACGTCTAGCAGCCGGATCTCAG), were the same for all nine libraries. The internal primers used were: QscanAs: 5′-TCGQTUCTITAAQTAIAFCCTUCFCCGQCGTTTTAGGTGGGGATGCC; QscanAa: 5′-GGUTETAOTTAEAGFAOCGAJCTOGFAGAJGGTCTGAGGCGTAACG; QscanBs: 5′-CIACGOCAQCITCGQTLTTTUTGQTGOGCTGGCGGTCAAACCTTC; QscanBa: 5′-AOCGAEGOTGQCGTEGQCGUAAAJCZTCGTTGTGCCAGGCTTC; QscanCs: 5′-ATTFCGOCTJGGQCTFTAPAPACGQTAIAGCGGTGCGCGTTTCG; QscanCa: 5′-TAUAGOCCLAGQCGUAATJCOCCTOTTOCGCTTTCGGCACGTTAA; QscanDs: 5′-TGLTCPATGOAOAAGOCTFTGQCPTCAIAACCAACGTGCAGGATATG; QscanDa: 5′-AGQCTTQTQCATZGAJCAIAOCCGQCGFAAZGCGCACCGCTTTACCG; QscanEs: 5′-GQGUCAZTGTFTOAGGQTLTTGQCTJGQAGZTGCTCAACTGGCCCGTGG; QscanEa: 5′-AOCCTZAUACAITGFCOCGAETZTCLAGUACZTCGACTGCGCCAGCGC; QscanFs: 5′-CAQCPACAQTAIGPTAGOGLTAGOGCCGTTGCCCGTGGTAG; QscanFa: 5′-CQCTAZCETAOTGTZGOTGOCCTOGACCACCGTGTTGGCC; QscanGs: 5′-CGGQCUCTFCTGQCGQGTUTGQCAQCTFCGTGGCCTTTATTCCTGAA; QscanGa: 5′-CGOCAGUAGUGOCCGUTETATJGAZCLAGGFCGCTTTGACCGGGGG; QscanHs: 5′-TOCGIACOCGGOAAPGPTTQAGGOGGOATACCA TATCCTCGAGGC; QscanHa: 5′-CZTTQCCGQGTECGQAUAGOTTGUATECQCGAGCATCACAATACCGAT; in which E = 85% T, 15% G; F = 85% A, 15% T; I = 85% A, 15% G; J = 85% G, 15% T; L = 85% C, 15% A; O = 85% C, 15% G; P = 85% G, 15% A; Q = 85% G, 15% C; U = 85% T, 15% A; Z = 85% A, 15% C.
For each library, the gene was amplified in two pieces using primers VWC562 and the appropriate Qscan-a (antisense) primer in one reaction and primer VWC497 with the appropriate Qscan-s (sense) primer in the other. The genes were amplified from the pSG422 vector. The PCR reactions were run with Vent polymerase under standard conditions. The resulting fragments were gel purified on 2% agarose and used as templates for the fusion PCR.
The fusion PCR was run with primers VWC564 (5′-GCATACGTCGGCCCCCGGGGCCGGCAGAC GGATTCG) and VWC565 (5′-GCATACGTCGGCCGGCAG GGCCAGACCCAGTTACG TCT), which contain the SfiI sites and are designed to amplify only fragments that were made with primers VWC497 and VWC562. This PCR was run with Taq Polymerase under standard conditions, using approximately equimolar amounts of each half. The resulting PCR fragments were digested with SfiI, ligated into the pSG736 vector, and transformed into E. coli TG1 cells.
Immunodetection
Libraries were plated to a density of 200 to 500 colonies per plate on LB agar supplemented with kanamycin (30 μg/mL). Colonies were grown 8–10 h, at 37°C, and then lifted with a nitrocellulose membrane. The plate was regrown for 4–6 h, and stored for later use. The cells were lysed on the membrane following a published protocol (QIAGEN), and then assayed for protein expression with the Penta-His anti-His tag antibody, followed by secondary antibody-HRP conjugate and chemiluminescence detection (ECL-Plus) according to standard protocols. Chemiluminescence proved to be more suitable than chromogenic detection, despite the antibody manufacturer’s recommendation to the contrary. The resulting image was then used as a guide to select the colonies to be included in the library.
Growth assay
A total of 92 colonies from each library that showed expression in the immunodetection assay were picked and grown to saturation in 96-well plates. Each plate also contained positive and negative controls. Culture was then frogged onto LB agar plates containing kanamycin (30 μg/mL) and different concentrations of ampicillin (5, 10, 20, 40, 80, or 160 μg/mL) or cephalothin (20, 40, 80, 160, 320, 640, 1280, or 2560 μg/mL). The plates were grown at 37°C for 10–12 h, after which the MPC (Maximal Permissive Concentration), or the highest antibiotic concentration at which cells could grow, was recorded for each well in the plate. The remaining culture was used for sequencing.
High-throughput DNA sequencing
One microliter of bacterial culture from each of the wells tested for antibiotic resistance was diluted with 20 μL of water and heated for 4 min at 94°C to lyse the cells. PCR amplification was performed in 20-μL reaction volumes containing 2 μL 10× Accutaq PCR Buffer, 4 nmole each dATP, dGTP, dTTP, and dCTP, 4 pmole forward primer VWC706 (5′-GATGCGTCCGGCG TAGAG G), 4 pmole reverse primer VWC 422 (5′-CAAGACCC GTTTAGAGGC), 0.5 U of Accutaq DNA polymerase, and 5 μL of lysed cells. The PCR profile used was 96°C for 30 sec, 30 cycles of 94°C for 15 sec, 55°C for 30 sec, and 72°C for 5 min, and finally 30 min at 68°C. Cycle sequencing (20-μL final reaction volume) was performed with 5 μL of PCR product, 8 μL ET terminator premix, and 5 pmole primer (the forward primer used in PCR reactions above for 5′ reactions or the reverse primer for 3′ reactions). Forward reactions were subjected to 30 cycles of 95°C for 20 sec, 54°C for 20 sec, and 60° C for 1 min. Reverse reaction conditions were 30 cycles of 95°C for 20 sec, 55°C for 20 sec, and 60°C for 1 min. Following isopropanol precipitation and 70% ethanol washes, DNA sequencing samples were suspended in MegaBACE formamide loading solution and loaded onto a MegaBACE 1000 capillary sequencer via electrokinetic injection by applying 3000 v for 80 sec. The samples were separated electrophoretically by applying 8000 v for 120 min. The resulting raw sequencing data was transformed into electropherograms, and base calling was performed using the Cimmaron Slim Phredify 1.53 Basecaller (Amersham Biosciences). Quality scores for the resulting electropherograms were calculated using Phred (Ewing and Green 1998; Ewing et al. 1998). All sequences with a read length of <100, or not maintaining an average Phred score of at least 17, were removed from subsequent analysis.
Protein purification
P99 β-lactamase engineered to have a carboxy-terminal His6 tag was purified from the E. coli strain Tuner(DE3) carrying plasmid pSG430. A cell pellet from 100 μL of culture known to express protein based on analysis by SDS-PAGE was resuspended in 100 μL of LB medium. A 1:350 dilution of this suspension was then used to inoculate 35 mL of LB medium containing 30 μg/mL kanamycin. This culture was grown at 37°C with vigorous shaking to mid-log phase (an OD600 of 0.6–0.8), at which time expression of the enzyme was induced by adding IPTG to a final concentration of 0.1 mM. After growth for an additional 3 h, the cells were harvested by centrifugation and stored at −80°C. The pellet was thawed at room temperature, and the cells lysed by adding 5 mLs of BugBuster Protein Extraction Reagent per gram of cell paste, and following the protocol supplied by the manufacturer. The protein was purified under native conditions using a Ni-NTA Spin Kit. Elution fractions containing protein were then dialyzed against PBS (phosphate-buffered saline; 137 mM NaCl, 2.7 mM KCl, 4.3 mM NaH2PO4, 1.4 mM KH2PO4 at pH 7.2). The enzyme concentration was determined on the basis of the A280 assuming an ɛ = 71000 cm−1M−1 as reported (Joris et al. 1985). The purified protein was stored at 4°C at concentrations of 10–65 μM. An identical procedure was followed for purification of the P99 mutants (with cells carrying the appropriate plasmids). Yields of 130 to 1000 μg of protein were obtained from 35 mL of culture. The proteins were judged to be >95% pure based on Coomassie staining of an SDS–polyacrylamide gel.
Kinetics
β-Lactamase activity with ampicillin and cephalothin was detected by a decrease in A236 and A265, respectively. The extinction coefficients used were 900 M−1 cm−1 for ampicillin (Sideraki et al. 2001) and 8790 M−1 cm−1 for cephalothin (Crowder et al. 1998). Activity was assayed in a solution composed of a 1:1 mixture of enzyme in PBS and antibiotic in 10 mM sodium phosphate (pH 7.0). The final volume of the reaction was 200 μL (except for Y150F and S64T reactions, which were done in 100-μL volumes), and the pH was 7.0. Enzyme concentrations were 1 or 2 nM for cephalothin reactions, except for Y150F and S64T, which were done at 500 nM. All enzymes were 500 nM in ampicillin reactions. Antibiotic concentrations were 10–300 μM for cephalothin reactions (except for Y150F and S64T, which were done at 50–1000 μM) and 250–1000 μM for ampicillin reactions. Reactions were started by addition of 100 μL of the enzyme solution (2× concentrated) to 100 μL of 2× antibiotic solution in the wells, and the progress monitored by UV. All assays were conducted at room temperature (25 ± 1°).
Kinetic constants were determined by initial velocity nonlinear regression analysis. For cephalothin, initial velocities were measured at 8–10 substrate concentrations, and then fit to the Michaelis-Menten equation, υo = Vmax • [S]/(Km + [S]), using Kaleidagraph (Synergy Software). With ampicillin, only the kcat was determined, as Km is too low to be measured by initial velocity kinetics. The kcat was calculated by measuring rates at four substrate concentrations well above Km (0.25–1 mM) and using these values to determine Vmax. Detailed kinetic data is shown for the wild-type P99 enzyme, as well as the S64T and Y150F mutants, in the supplemental material.
Circular dichroism
Wavelength scans were measured for the purified proteins to ensure that the mutants’ overall structure is similar to the wild-type enzyme. Proteins were diluted to 1 μM in PBS buffer (pH 7.2). CD wavelength scans were measured on a Jasco J-810 spectropolarimeter. Spectra were recorded from 190 to 250 nm, every 1 nm, with eight accumulations for each sample.
Electronic supplemental material
Detailed kinetic data is shown for the wild-type P99 enzyme as well as the S64T and Y150F mutants in the file SuppMat.doc.
Acknowledgments
We are grateful for financial support for this work from the NIH (RO1-GM62867) and Columbia University. V.W.C. is a recipient of a Sloan Foundation Fellowship, a Beckman Young Investigator Award, a Burroughs Wellcome Fund New Investigator Award in the Toxicological Sciences, a Camille and Henry Dreyfus New Faculty Award, and a NSF CAREER Award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: See www.proteinscience.org.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0302903.
References
- Beadle, B.M. and Shoichet, B.K. 2002. Structural bases of stability-function tradeoffs in enzymes. J. Mol. Biol. 321 285–296. [DOI] [PubMed] [Google Scholar]
- Beadle, B.M., Trehan, I., Focia, P.J., and Shoichet, B.K. 2002. Structural milestones in the reaction pathway of an amide hydrolase: Substrate, acyl, and product complexes of cephalothin with AmpC β-lactamase. Structure 10 413–424. [DOI] [PubMed] [Google Scholar]
- Bohren, K.M., Grimshaw, C.E., Lai, C.J., Harrison, D.H., Ringe, D., Petsko, G.A., and Gabbay, K.H. 1994. Tyrosine-48 is the proton donor and histidine-110 directs substrate stereochemical selectivity in the reduction reaction of human aldose reductase: Enzyme kinetics and crystal structure of the Y48H mutant enzyme. Biochemistry 33 2021–2032. [DOI] [PubMed] [Google Scholar]
- Bulychev, A. and Mobashery, S. 1999. Class C β-lactamases operate at the diffusion limit for turnover of their preferred cephalosporin substrates. Antimicrob. Agents Chemother. 43 1743–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulychev, A., Massova, I., Miyashita, K., and Mobashery, S. 1997. Nuances of mechanisms and their implications for evolution of the versatile β-lactamase activity: From biosynthetic enzymes to drug resistance factors. J. Am. Chem. Soc. 119 7619–7625. [Google Scholar]
- Bush, K., Jacoby, G.A., and Medeiros, A.A. 1995. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39 1211–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caselli, E., Powers, R.A., Blasczcak, L.C., Wu, C.Y., Prati, F., and Shoichet, B.K. 2001. Energetic, structural, and antimicrobial analyses of β-lactam side chain recognition by β-lactamases. Chem. Biol. 8 17–31. [DOI] [PubMed] [Google Scholar]
- Crichlow, G.V., Kuzin, A.P., Nukaga, M., Mayama, K., Sawai, T., and Knox, J.R. 1999. Structure of the extended-spectrum class C β-lactamase of Enterobacter cloacae GC1, a natural mutant with a tandem tripeptide insertion. Biochemistry 38 10256–10261. [DOI] [PubMed] [Google Scholar]
- Crichlow, G.V., Nukaga, M., Doppalapudi, V.R., Buynak, J.D., and Knox, J.R. 2001. Inhibition of class C β-lactamases: Structure of a reaction intermediate with a cephem sulfone. Biochemistry 40 6233–6239. [DOI] [PubMed] [Google Scholar]
- Crowder, M.W., Walsh, T.R., Banovic, L., Pettit, M., and Spencer, J. 1998. Overexpression, purification, and characterization of the cloned metallo-β lactamase L1 from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 42 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, B.C. and Wells, J.A. 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244 1081–1085. [DOI] [PubMed] [Google Scholar]
- Dubus, A., Monnaie, D., Jacobs, C., Normark, S., and Frere, J.M. 1993. A dramatic change in the rate-limiting step of β-lactam hydrolysis results from the substitution of the active-site serine residue by a cysteine in the class-C β-lactamase of Enterobacter cloacae 908R. Biochem. J. 292 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubus, A., Normark, S., Kania, M., and Page, M.G. 1994a. The role of tyrosine 150 in catalysis of β-lactam hydrolysis by AmpC β-lactamase from Escherichia coli investigated by site-directed mutagenesis. Biochemistry 33 8577–8586. [DOI] [PubMed] [Google Scholar]
- Dubus, A., Wilkin, J.M., Raquet, X., Normark, S., and Frere, J.M. 1994b. Catalytic mechanism of active-site serine β-lactamases: Role of the conserved hydroxy group of the Lys-Thr(Ser)-Gly triad. Biochem. J. 301 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubus, A., Normark, S., Kania, M., and Page, M.G. 1995. Role of asparagine 152 in catalysis of β-lactam hydrolysis by Escherichia coli AmpC β-lactamase studied by site-directed mutagenesis. Biochemistry 34 7757–7764. [DOI] [PubMed] [Google Scholar]
- Dubus, A., Ledent, P., Lamotte-Brasseur, J., and Frere, J.M. 1996. The roles of residues Tyr150, Glu272, and His314 in class C β-lactamases. Proteins 25 473–485. [DOI] [PubMed] [Google Scholar]
- Ewing, B. and Green, P. 1998. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8 186–194. [PubMed] [Google Scholar]
- Ewing, B., Hillier, L., Wendl, M., and Green, P. 1998. Base-calling of automated sequencer traces using Phred. I. Accuracy Assessment. Genome Res. 8 175–185. [DOI] [PubMed] [Google Scholar]
- Galleni, M. and Frere, J.M. 1988. A survey of the kinetic parameters of class C β-lactamases. Penicillins. Biochem. J. 255 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galleni, M., Amicosante, G., and Frere, J.M. 1988. A survey of the kinetic parameters of class C β-lactamases. Cephalosporins and other β-lactam compounds. Biochem. J. 255 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghuysen, J.M. 1991. Serine β-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 45 37–67. [DOI] [PubMed] [Google Scholar]
- Golemi, D., Maveyraud, L., Vakulenko, S., Samama, J.P., and Mobashery, S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. 98 14280–14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoret, L.M. and Sauer, R.T. 1993. Additivity of mutant effects assessed by binomial mutagenesis. Proc. Natl. Acad. Sci. 90 4246–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Herzberg, O. and Moult, J. 1987. Bacterial resistance to β-lactam antibiotics: Crystal structure of β-lactamase from Staphylococcus aureus PC1 at 2.5 Å resolution. Science 236 694–701. [DOI] [PubMed] [Google Scholar]
- Jacobs, C., Dubus, A., Monnaie, D., Normark, S., and Frere, J.M. 1992. Mutation of serine residue 318 in the class C β-lactamase of Enterobacter cloacae 908R. FEMS Microbiol. Lett. 71 95–100. [DOI] [PubMed] [Google Scholar]
- Joris, B., De Meester, F., Galleni, M., Reckinger, G., Coyette, J., Frere, J.M., and Van Beeumen, J. 1985. The β-lactamase of Enterobacter cloacae P99. Chemical properties, N-terminal sequence and interaction with 6 β-halogenopenicillanates. Biochem. J. 228 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joris, B., Ghuysen, J.M., Dive, G., Renard, A., Dideberg, O., Charlier, P., Frere, J.M., Kelly, J.A., Boyington, J.C., and Moews, P.C. 1988. The active-site penicillin-recognizing enzymes as members of the Streptomyces R61 DD-peptidase family. Biochem. J. 250 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju, J., Ruan, C., Fuller, C.W., Glazer, A.N., and Mathies, R.A. 1995. Fluorescence energy transfer dye-labeled primers for DNA sequencing and analysis. Proc. Natl. Acad. Sci. 92 4347–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato-Toma, Y. and Ishiguro, M. 2001. Reaction of Lys-Tyr-Lys triad mimics with benzylpenicillin: Insight into the role of Tyr150 in class C β-lactamase. Bioorg. Med. Chem. Lett. 11 1161–1164. [DOI] [PubMed] [Google Scholar]
- Kheterpal, I. and Mathies, R.A. 1999. Capillary array electrophoresis DNA sequencing. Anal. Chem. 71 31A–37A. [DOI] [PubMed] [Google Scholar]
- Knox, J.R., Moews, P.C., and Frere, J.M. 1996. Molecular evolution of bacterial β-lactam resistance. Chem. Biol. 3 937–947. [DOI] [PubMed] [Google Scholar]
- Kraut, J. 1977. Serine proteases: Structure and mechanism of catalysis. Annu. Rev. Biochem. 46 331–358. [DOI] [PubMed] [Google Scholar]
- Kuzin, A.P., Liu, H., Kelly, J.A., and Knox, J.R. 1995. Binding of cephalothin and cefotaxime to D-Ala-D-Ala-peptidase reveals a functional basis of a natural mutation in a low-affinity penicillin-binding protein and in extended-spectrum β-lactamases. Biochemistry 34 9532–9540. [DOI] [PubMed] [Google Scholar]
- Lamotte-Brasseur, J., Dubus, A., and Wade, R.C. 2000. pKa calculations for class C β-lactamases: The role of Tyr-150. Proteins 40 23–28. [DOI] [PubMed] [Google Scholar]
- Liang, J., Edelsbrunner, H., and Woodward, C. 1998. Anatomy of protein pockets and cavities: Measurements of binding site geometry and implications for ligand design. Protein Sci. 7 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobkovsky, E., Moews, P.C., Liu, H., Zhao, H., Frere, J.M., and Knox, J.R. 1993. Evolution of an enzyme activity: Crystallographic structure at 2-Å resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc. Natl. Acad. Sci. 90 11257–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobkovsky, E., Billings, E.M., Moews, P.C., Rahil, J., Pratt, R.F., and Knox, J.R. 1994. Crystallographic structure of a phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: Mechanistic interpretation of a β-lactamase transition-state analog. Biochemistry 33 6762–6772. [DOI] [PubMed] [Google Scholar]
- Madgwick, P.J. and Waley, S.G. 1987. β-lactamase I from Bacillus cereus. Structure and site-directed mutagenesis. Biochem. J. 248 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova, I. and Mobashery, S. 1998. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 42 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maveyraud, L., Golemi, D., Kotra, L.P., Tranier, S., Vakulenko, S., Mobashery, S., and Samama, J.P. 2000. Insights into class D β-lactamases are revealed by the crystal structure of the OXA10 enzyme from Pseudomonas aeruginosa. Structure 8 1289–1298. [DOI] [PubMed] [Google Scholar]
- Monnaie, D., Dubus, A., Cooke, D., Marchand-Brynaert, J., Normark, S., and Frere, J.M. 1994a. Role of residue Lys315 in the mechanism of action of the Enterobacter cloacae 908R β-lactamase. Biochemistry 33 5193–5201. [DOI] [PubMed] [Google Scholar]
- Monnaie, D., Dubus, A., and Frere, J.M. 1994b. The role of lysine-67 in a class C β-lactamase is mainly electrostatic. Biochem. J. 302 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, K.L. and Weiss, G.A. 2001. Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 5 302–307. [DOI] [PubMed] [Google Scholar]
- Nikaido, H. and Normark, S. 1987. Sensitivity of Escherichia coli to various β-lactams is determined by the interplay of outer membrane permeability and degradation by periplasmic β-lactamases: A quantitative predictive treatment. Mol. Microbiol. 1 29–36. [DOI] [PubMed] [Google Scholar]
- Nukaga, M., Tanimoto, K., Tsukamoto, K., Imajo, S., Ishiguro, M., and Sawai, T. 1993. A survey of a functional amino acid of class C β-lactamase corresponding to Glu166 of class A β-lactamases. FEBS Lett. 332 93–98. [DOI] [PubMed] [Google Scholar]
- Oefner, C., D’Arcy, A., Daly, J.J., Gubernator, K., Charnas, R.L., Heinze, I., Hubschwerlen, C., and Winkler, F.K. 1990. Refined crystal structure of β-lactamase from Citrobacter freundii indicates a mechanism for β-lactam hydrolysis. Nature 343 284–288. [DOI] [PubMed] [Google Scholar]
- Page, M.G. 1993. The kinetics of non-stoichiometric bursts of β-lactam hydrolysis catalysed by class C β-lactamases. Biochem. J. 295 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patera, A., Blasczcak, L.C., and Shoichet, B.K. 2000. Crystal structures of substrate and inhibitor complexes with AmpC β-lactamase: Possible implications for substrate-assisted catalysis. J. Am. Chem. Soc. 122 10504–10512. [Google Scholar]
- Powers, R.A. and Shoichet, B.K. 2002. Structure-based approach for binding site identification on AmpC β-lactamase. J. Med. Chem. 45 3222–3234. [DOI] [PubMed] [Google Scholar]
- Powers, R.A., Caselli, E., Focia, P.J., Prati, F., and Shoichet, B.K. 2001. Structures of ceftazidime and its transition-state analogue in complex with AmpC β-lactamase: Implications for resistance mutations and inhibitor design. Biochemistry 40 9207–9214. [DOI] [PubMed] [Google Scholar]
- Powers, R.A., Morandi, F., and Shoichet, B.K. 2002. Structure-based discovery of a novel, noncovalent inhibitor of AmpC β-lactamase. Structure 10 1013–1023. [DOI] [PubMed] [Google Scholar]
- Rosenblum, B.B., Lee, L.G., Spurgeon, S.L., Khan, S.H., Menchen, S.M., Heiner, C.R., and Chen, S.M. 1997. New dye-labeled terminators for improved DNA sequencing patterns. Nucleic Acids Res. 25 4500–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimotohno, A., Oue, S., Yano, T., Kuramitsu, S., and Kagamiyama, H. 2001. Demonstration of the importance and usefulness of manipulating non-active-site residues in protein design. J. Biochem. 129 943–948. [DOI] [PubMed] [Google Scholar]
- Sideraki, V., Huang, W., Palzkill, T., and Gilbert, H.F. 2001. A secondary drug resistance mutation of TEM-1 β-lactamase that suppresses misfolding and aggregation. Proc. Natl. Acad. Sci. 98 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus, D., Raines, R., Kawashima, E., Knowles, J.R., and Gilbert, W. 1985. Actitve site of triosephosphate isomerase: In vitro mutagenesis and characterization of an altered enzyme. Proc. Natl. Acad. Sci. 82 2272–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strynadka, N.C., Adachi, H., Jensen, S.E., Johns, K., Sielecki, A., Betzel, C., Sutoh, K., and James, M.N. 1992. Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 Å resolution. Nature 359 700–705. [DOI] [PubMed] [Google Scholar]
- Sutcliffe, J.G. 1978. Nucleotide sequence of the ampicillin resistance gene of Escherichia Coli plasmid pBR322. Proc. Natl. Acad. Sci. 75 3737–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondi, D., Powers, R.A., Caselli, E., Negri, M.C., Blazquez, J., Costi, M.P., and Shoichet, B.K. 2001. Structure-based design and in-parallel synthesis of inhibitors of AmpC β-lactamase. Chem. Biol. 8 593–611. [DOI] [PubMed] [Google Scholar]
- Trehan, I., Beadle, B.M., and Shoichet, B.K. 2001. Inhibition of AmpC β-lactamase through a destabilizing interaction in the active site. Biochemistry 40 7992–7999. [DOI] [PubMed] [Google Scholar]
- Trehan, I., Morandi, F., Blasczcak, L.C., and Shoichet, B.K. 2002. Using steric hindrance to design new inhibitors of class C β-lactamases. Chem. Biol. 9 971–980. [DOI] [PubMed] [Google Scholar]
- Trepanier, S., Knox, J.R., Clairoux, N., Sanschagrin, F., Levesque, R.C., and Huletsky, A. 1999. Structure-function studies of Ser-289 in the class C β-lactamase from Enterobacter cloacae P99. Antimicrob. Agents Chemother. 43 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto, K., Kikura, R., Ohno, R., and Sawai, T. 1990a. Substitution of aspartic acid-217 of Citrobacter freundii cephalosporinase and properties of the mutant enzymes. FEBS Lett. 264 211–214. [DOI] [PubMed] [Google Scholar]
- Tsukamoto, K., Nishida, N., Tsuruoka, M., and Sawai, T. 1990b. Function of the conserved triad residues in the class C β-lactamase from Citrobacter freundii GN346. FEBS Lett. 271 243–246. [DOI] [PubMed] [Google Scholar]
- Tsukamoto, K., Ohno, R., and Sawai, T. 1990c. Extension of the substrate spectrum by an amino acid substitution at residue 219 in the Citrobacter freundii cephalosporinase. J. Bacteriol. 172 4348–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usher, K.C., Blaszczak, L.C., Weston, G.S., Shoichet, B.K., and Remington, S.J. 1998. Three-dimensional structure of AmpC β-lactamase from Escherichia coli bound to a transition-state analogue: Possible implications for the oxyanion hypothesis and for inhibitor design. Biochemistry 37 16082–16092. [DOI] [PubMed] [Google Scholar]
- Vakulenko, S.B., Golemi, D., Geryk, B., Suvorov, M., Knox, J.R., Mobashery, S., and Lerner, S.A. 2002. Mutational replacement of Leu-293 in the class C Enterobacter cloacae P99 β-lactamase confers increased MIC of cefepime. Antimicrob. Agents Chemother. 46 1966–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z., Yu, Y., Musser, J.M., and Palzkill, T. 2001. Amino acid sequence determinants of extended spectrum cephalosporin hydrolysis by the class C P99 β-lactamase. J. Biol. Chem. 276 46568–46574. [DOI] [PubMed] [Google Scholar]