

Abstract
The stability of Rhodobacter capsulatus bacterioferritin, a 24-meric homopolymer, toward denaturation on variation in pH and temperature, and increasing concentrations of urea and guanidine.HCl was investigated with native PAGE, and CD and fluorescence spectroscopies. With temperature and urea, the wild-type protein denatured without discernible intermediates in the equilibrium experiments, but with guanidine.HCl (Gnd.HCl) one or more intermediate species were apparent at relatively low Gnd.HCl concentrations. Dissociated subunit monomers, or aggregates smaller than 24-mers containing the high α-helical content characteristic of the native protein were not obtained at any pH without a high proportion of the 24-mer being present, and taken together with the other denaturation experiments and the construction of stable subunit dimers by site-directed mutagenesis, this observation indicates that folding of the bacterioferritin monomer could be coupled to its association into a dimer. Glu 128 and Glu 135 were replaced by alanine and arginine in a series of mutants to determine their role in stabilizing the 24-meric oligomer. The Glu128Ala, Glu135Ala and Glu135Arg variants retained a 24-meric structure, but the Glu128Ala/Glu135Ala and Glu128Arg/Glu135Arg variants were stable subunit dimers. CD spectra of the Glu135Arg, Glu128Ala/Glu135Ala, and Glu128Arg/Glu135Arg variants showed that they retained the high α-helical content of the wild-type protein. The 24-meric Glu135Arg variant was less stable than the wild-type protein (Tm, [Urea]50% and [Gnd.HCl]50% of 59°C, 4.9 M and 3.2 M compared with 73°C, ~8 M and 4.3 M, respectively), and the dimeric Glu128Arg/Glu135Arg variant was less stable still (Tm, [Urea]50% and [Gnd.HCl]50% of 43°C, ~3.2 M and 1.8 M, respectively). The differences in stability are roughly additive, indicating that the salt-bridges formed by Glu 128 and Glu 135 in the native oligomer, with Arg 61 and the amino-terminal amine of neighboring subunits, respectively, contribute equally to the stability of the subunit assembly. The additivity and assembly states of the variant proteins suggest that the interactions involving Glu 128 and Glu 135 contribute significantly to stabilizing the 24-mer relative to the subunit dimer.
Keywords: Bacterioferritin, oligomer, subunit dimer, denaturation, unfolding
BFR is a member of the ferritin protein family, catalyzing the oxidation and hydrolysis of Fe2+ (Stiefel and Watt 1979; Harrison and Arosio 1996; Le Brun et al. 1997; Andrews 1998; Chasteen 1998). Most ferritins consist of 24 subunits of 18–21 kD, each of which fold into four-helix bundles. These associate to create an approximately spherical shell of 20–25 Å thickness that surrounds a cavity of ~80 Å diameter, in which the iron mineral is formed. Channels through the protein shell created by the subunit packing may allow entry of Fe2+ to the cavity (Harrison and Arosio 1996), with electrostatic gradients coincident with the channels guiding the Fe2+ (Douglas and Ripoll 1998). Listeria innocua ferritin is unusual in that it consists of only 12 subunits with a central cavity of ~40 Å diameter (Ilari et al. 2000), which enables it to hold ~500 Fe3+ ions (Ilari et al. 2000), compared with the ~4500 24-mer ferritins can accumulate (Ford et al. 1984). This 12-mer ferritin has a similar structure to Escherichia coli Dps (Grant et al. 1998), which can also hold 500 Fe3+ ions (Ilari et al. 2002; Zhao et al. 2002). BFR is unique among the ferritins in containing 12 bis-Met coordinated hemes in intersubunit sites (Stiefel and Watt 1979; Cheesman et al. 1990; Frolow et al. 1994), although these do not play a role in the aerobic oxidative uptake of Fe2+ (Andrews et al. 1995). BFR also has intrasubunit dinuclear iron centres, called ferroxidase centers, which are needed for rapid formation of a ferric oxide mineral core (Le Brun et al. 1995; Yang et al. 2000). Similar ferroxidase centers are present in H-chain ferritins from eukaryotes and the heme-free bacterial ferritin FTN (Harrison and Arosio 1996; Andrews 1998; Yang et al. 1998; Ha et al. 1999), but they are lacking in L-chain ferritins, although these proteins are still able to lay down a ferric oxide core (Trikha et al. 1995; Harrison and Arosio 1996; Gallois et al. 1997).
The multiplicity of possible reaction sites in 24-mer ferritins creates problems for defining mechanism(s) of iron mineral formation. For example, ferritin core minerals have not been detected by single-crystal X-ray diffraction because although the protein shell is ordered in crystals, the ferric oxide cores are not (Ford et al. 1984; Powell 1998). Also, although in the initial stage of reaction each oligomer may contain many nucleation centers, core formation is rapidly dominated by a small number of nucleation centers (Clegg et al. 1980). Thus, complete mechanistic characterization of 24-mer ferritins, in which averaging between sites occurs and large clusters are formed even at relatively low iron loadings, may not be possible. To circumvent this complication, attempts have been made to dissociate 24-mer ferritins with retention of ferroxidase activity.
Although the native form of most ferritins is a 24-mer, X-ray structures show that the repeating structural unit is a subunit dimer (Fig. 1A,B ▶; Ford et al. 1984; Frolow et al 1994; Trikha et al. 1995; Harrison and Arosio 1996; Gallois et al. 1997; Ha et al. 1999; Cobessi et al. 2002). Disaggregation of wild-type ferritin oligomers into mono-disperse smaller assemblies is only possible by lowering pH or by the addition of denaturants, and these approaches cause the subunits themselves to denature (Listowsky et al. 1972; Crichton and Bryce 1973; Stefanini et al. 1987; Santambrogio et al. 1997). Hence, attempts to stabilize subunit dimer forms of ferritins need to be made by mutagenesis and chemical modifications. Arosio and colleagues have described the preparation of an H-chain ferritin dimer that retained some ferroxidase activity (Levi et al. 1993; Santambrogio et al. 1997). Our construction of a BFR subunit dimer active as a ferroxidase is the only other case to be reported (Spiro et al. 1999). The X-ray structures of E. coli and R. capsulatus BFR (Frolow et al. 1994; Cobessi et al. 2002) show that there are numerous intersubunit contacts that could stabilize association of subunit dimers into high-order oligomers, including interactions between Glu 128 and Glu 135 of one subunit and Arg 61 and the amino-terminal amine, respectively, of neighboring subunits (Fig. 1C ▶). We substituted Glu 128 and Glu 135 of R. capsulatus BFR with arginine, and found that the altered protein, at a range of concentrations up to 160 μM, was a mono-disperse dimeric species (Kilic 1999; Spiro et al. 1999). In the present work, we report comparative studies of the stabilities of wild-type and mutant 24-mer and subunit dimer forms of R. capsulatus BFR, with the aim of characterizing intersubunit interactions important for assembly of a 24-mer.
Figure 1.
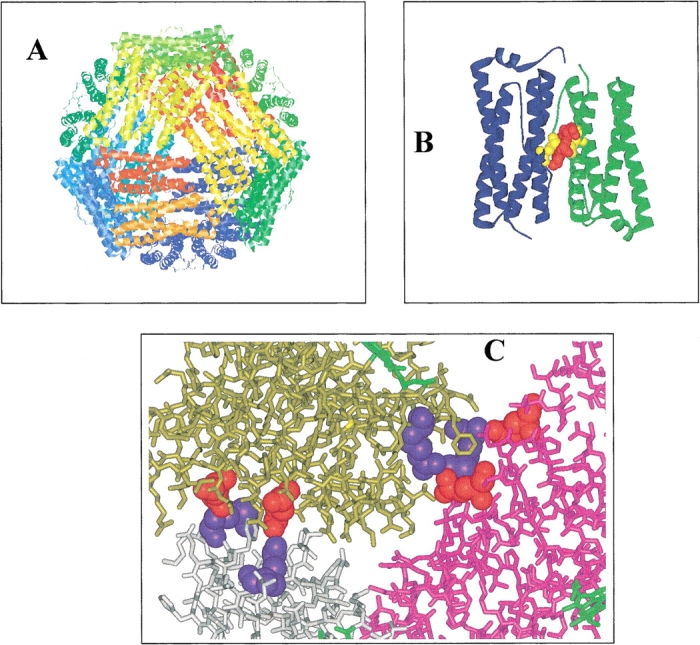
The X-ray structure of R. capsulatus BFR (Cobessi et al. 2002). (A) View of the 24-mer protein shell with a threefold channel opening in the center of the front surface. (B) A subunit dimer with the heme (red) and Met 52 (yellow) iron ligands indicated. (C) Close-up view of a threefold channel opening. Three subunits are shown (purple, gold, and gray) with Glu 128 and Glu 135 (red) of two interacting with the amino-terminal amine and Arg 61 (blue) of neighboring subunits. Parts of three heme groups (green) are visible. Note that the recombinant bacterioferritins studied in this work were essentially heme-free (see Materials and Methods).
Results and Discussion
Biochemical and biophysical characterization of wild-type and mutant bacterioferritins
Wild-type BFR and its variants were investigated by gel-permeation chromatography to determine their states of assembly (Table 1). The wild-type BFR and variant BFRs with one amino acid change per subunit behaved as 100% 24-meric oligomers, and the variant BFRs with two amino acid changes per subunit behaved exclusively as dimers, having molecular weights of 39–40 kD compared with the 36.4 kD expected from the amino acid sequence. Nondenaturing PAGE confirms that the variant BFRs with two amino acid changes per subunit are smaller than the 24-mer BFR (Fig. 2 ▶). The migration rates of the 24-meric wild-type and Glu135Arg BFRs are different (Fig. 2 ▶, lanes 1 and 2) because the Glu135Arg BFR has a net charge difference of +2 per subunit, whereas the faster migration rate of the Glu128Arg/Glu135Arg BFR, which has a net charge difference of +4 per subunit compared with wild-type BFR, results from its smaller size. The Glu128Ala/Glu135Ala variant had low solubility compared with the other proteins, which led to low yields of purified material (typically 2–6 mg per 2 liter culture).
Table 1.
Assembly state and stability of R. capsulatus bacterioferritin
| Bacterioferritin | Assembly state at pH 7.2 | Tm °C | [Urea]50% (M) | [Gnd.HCl]50% (M) |
| Wild-type | 24-mer | 73.2 ± 0.4 | ~8 | 4.3 |
| Glu128Ala | 24-mer | n.d. | n.d. | n.d. |
| Glu135Ala | 24-mer | n.d. | n.d. | n.d. |
| Glu135Arg | 24-mer | 59.2 ± 0.3 | 4.9 | 3.2 |
| Glu128Ala/Glu135Ala | dimer | 47.1 ± 0.3 | n.d. | n.d. |
| Glu128Arg/Glu135Arg | dimer | 43.3 ± 0.3 | 3.2 | 1.8 |
Figure 2.
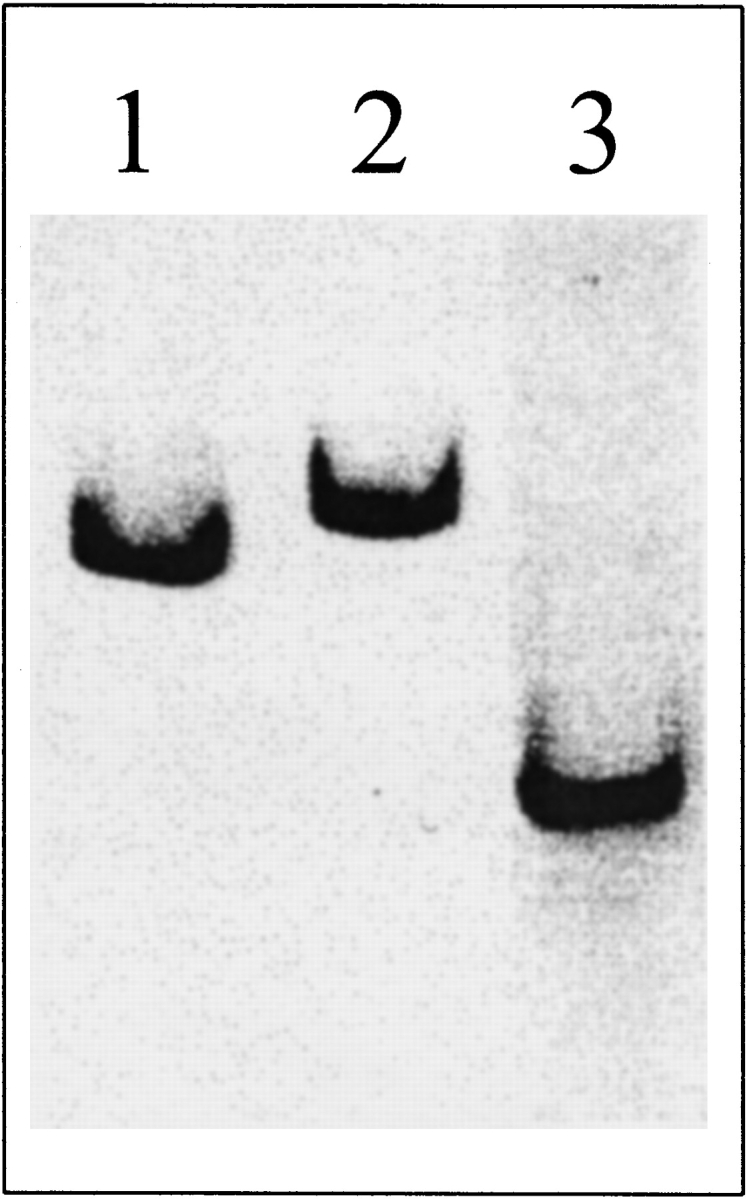
Nondenaturing PAGE of 10 μg of wild-type (lane 1), Glu135Arg (lane 2), and Glu128Arg/Glu135Arg (lane 3) BFR on 6% gels at pH 7.2 (60 mM HEPES and 40 mM imidazole buffer).
The far-UV CD spectrum of wild-type BFR indicates the protein is predominantly α-helical (Fig. 3A ▶), consistent with the X-ray structure (Fig. 1 ▶). Far-UV CD spectra of Glu128Arg/Glu135Arg BFR and Glu128Ala/Glu135Ala BFR were essentially the same as that for wild-type BFR (Fig. 3A ▶), but their near-UV CD spectra (Fig. 3B ▶), which monitor tryptophan environments, are different from those of wild-type and the 24-mer Glu135Arg BFR. Tryptophan fluorescence spectra of the dimeric proteins were also slightly different from the spectrum of wild-type BFR (Fig. 4 ▶). The similarity of far-UV CD spectra shows that the mutations do not significantly perturb the secondary structure content of BFR, and the near-UV CD and fluorescence spectra reflect the assembly states of the proteins determined by gel-permeation chromatography and nondenaturing PAGE. The environments of the two tryptophan residues of BFR, Trp 26, and Trp 35, are affected by both the tertiary and the quaternary structure of 24-mer BFR; Trp 26 is located at the twofold monomer–monomer interfaces, which are present in both the dimer variants and the 24-mer proteins, and Trp 35 is located in a turn of one subunit that makes contact with another subunit in the 24-meric BFR (Frolow et al. 1994). This contact is lost for the dimeric variants and Trp 35 will then be exposed to solvent unless there is a substantial difference between the structure of the dimer on its own and as part of the 24-mer. The retention of ferroxidase activity and the ability to bind heme stoichiometrically at a site providing bis-methionine coordination (Spiro et al. 1999; S. Malone, U. Jayasooriya, and G. Moore, unpubl.), together with conserved high α-helix content (Fig. 3 ▶), all suggest that the tertiary structure will not be much affected by whether the subunit dimer is assembled into a 24-mer or not. This indicates that the difference in the fluorescence spectra between wild-type and dimer variants (Fig. 4 ▶) is a consequence of the change in quaternary structure. The fluorescence emission maximum for 24-meric wild-type BFR is at 339 nm, and for the dimer variants at 343 nm, consistent with Trp 35 being in a less polar environment in the 24-mer than in the dimer. Furthermore, as the aromatic π |Zy π* transitions of tryptophan do not exhibit a strong CD signal unless the ring is located in an asymmetric environment (Schmid 1997), a reduction in the near-UV CD intensity for Trp 35 is expected on disassembly of the 24-mer into subunit dimers, as is observed (Fig. 3 ▶).
Figure 3.
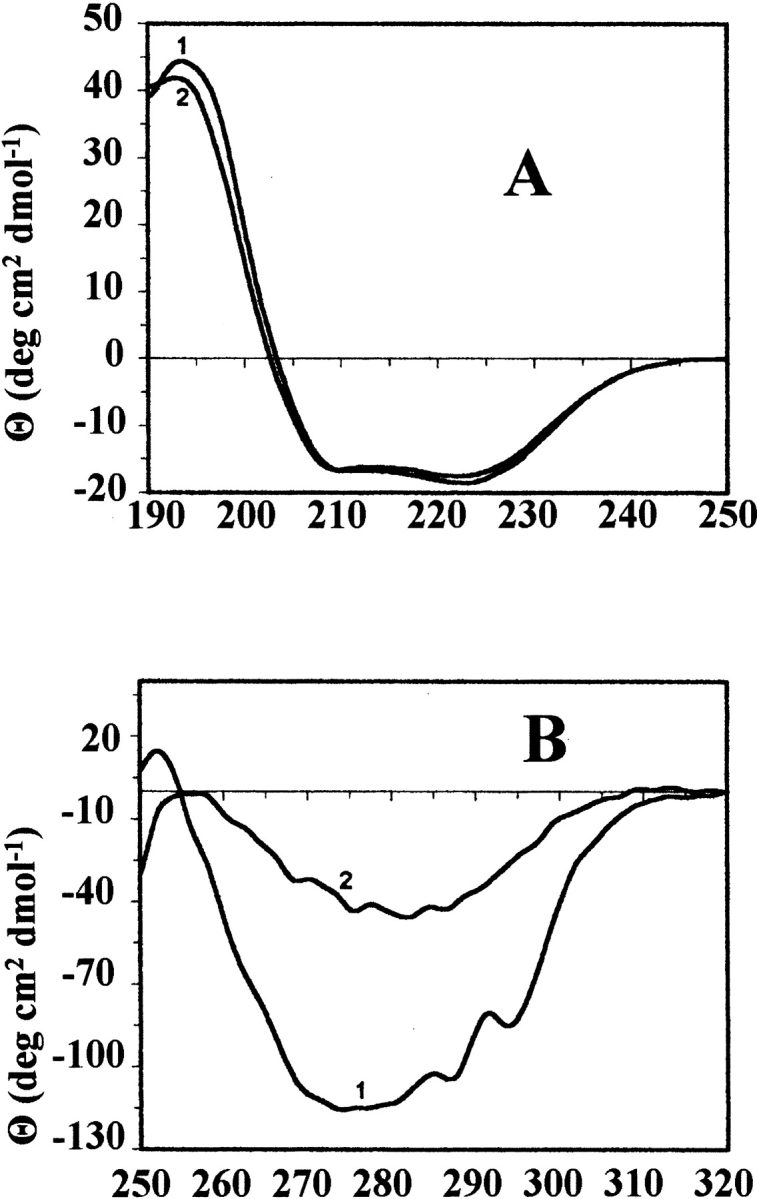
(A) Far-UV and (B) near-UV CD spectra of wild-type BFR (1) and its Glu128Arg/Glu135Arg variant (2). Protein concentrations were 1 mg ml−1 in 25 mM potassium phosphate (pH 7.2), containing 20 mM NaCl and the measurements were made at 25 °C.
Figure 4.

Fluorescence emission spectra of wild-type BFR (1) and its Glu128Arg/Glu135Arg variant (2) on excitation at 295 nm. Protein concentrations were 50 μg ml−1 in 50 mM potassium phosphate buffer (pH 7.2), and the measurements were made at 25°C.
pH-dependent behavior of wild-type bacterioferritin
The effect of variation in pH on the structure and assembly of R. capsulatus BFR was studied with nondenaturing gels, CD and fluorescence spectroscopies, and gel-permeation on Sephacryl S300HR. Nondenaturing gels were used because they allowed small amounts of protein to be investigated. However, when the pH of the running buffer and the sample buffer were not the same, such gels do not provide quantitative data because reassembly or disassembly may occur as the protein passes through the running buffer. However, such gels do provide important qualitative data. Gel-permeation chromatography and nondenaturing gels with sample and running buffers at the same pH showed that BFR was 100% 24-meric over the pH range 5.5–9.5 (data not shown). It was also largely a 24-meric species at pH 10 (Fig. 5 ▶, lane 1), but at higher pH values, disassembly occurred and with a sample pH of 12 and running buffer of pH 10, a 24-meric species was not detected (Fig. 5 ▶, lane 3). The pH 10 lane showed a strong band from 24-meric BFR and fainter bands corresponding to a disassembled form and higher molecular weight species. These latter bands must have come from aggregated BFR, which probably was the origin of the protein that did not migrate out of the wells for the samples at pH 11 and pH 12 (Fig. 5 ▶). Poor solubility over the pH range 3.5–5.4, probably resulting from aggregation of BFR close to its isoelectric point of 4.6, prevented clear data from being obtained, but nondenaturing gels with sample and running buffers at pH 3 (20 mM lactic acid and 20 mM alanine) showed that BFR remained a 24-meric structure (data not shown). At a sample pH of 2 and running buffer of pH 3, BFR ran on nondenaturing gels as a mixture of a 24-mer and a disassembled species (data not shown). CD spectra at pH 3 and over the pH range 5.5–7 showed that the secondary structure and quaternary structure characterized by near-UV CD was constant (Fig. 6 ▶). At pH 2, although the secondary structure appeared unchanged, the decrease in near-UV CD intensity indicates a change in the way subunits pack together that affects the environment of one or more of the tryptophan residues. At pH 1.5, a small decrease in the far-UV CD signal indicates loss of some of the α-helix content. Tryptophan fluorescence emission spectra gave similar results on acidification (data not shown). For pH 5.5–7 and at pH 3, the emission maximum and intensity remained the same, but at pH 2 and below the maximum, shifted from 338 to 342 nm, and its intensity decreased. Just as the CD indicates that the protein is not completely unfolded at pH 1.5, so do the fluorescence data as the emission maximum would be expected to be ~350 nm for an unfolded protein. Similar CD (Fig. 6 ▶) and fluorescence (data not shown) experiments at alkaline pH agree with the nondenaturing gel data (Fig. 5 ▶) showing BFR remains a 24-mer from pH 7 to pH 9.5–10, and suggests that at pH 11, the protein has disassembled with a substantial loss of α-helix content. There is a further loss of secondary structure at pH 12. The tryptophan fluorescence intensity decreased above pH 10 and the emission maximum shifted from 338 nm at pH 7 to 348 nm at pH 12. Thus, the spectroscopic data indicate that BFR at pH 12 is completely unfolded.
Figure 5.

Nondenaturing PAGE of wild-type BFR with a running buffer at pH 10 (20 mM ammonia and 40 mM CAPS) using a 6% (top) and 12% (bottom) gel. (Lanes 1–3) The 10 μg samples are at pH 10, 11, and 12, respectively.
Figure 6.

(A,C)Far-UV and (B,D) near-UV CD spectra of wild-type BFR at different values of pH, as indicated. The protein concentration was 1 mg ml−1 in 50 mM potassium phosphate.
In summary, the data for wild-type R. capsulatus BFR indicate that it is predominantly a 24-mer over the pH range 3–10, and that outside of this range, at least some of the BFR is in a disassembled state and is completly unfolded at pH 12. This indicates that the carboxylates and amino-terminal amine forming the key salt-bridges, Glu 128–Arg 61 and Glu 135-amino-terminal amine, must have perturbed pKa values, as otherwise, the salt-bridges would have been disrupted at a pH close to their intrinsic pKa values (4.3 for Glu and ~7.8 for the amino-terminal amine), leading to disaggregation of the 24-mer. The 24-mer does not lose all its α-helical structure until pH 12, although at both pH 10 and pH 3, where it is predominantly a 24-mer with high α-helical content, some perturbation of the way subunits pack together affects tryptophan fluorescence. The fully disassembled protein appears to lack secondary structure. It is not known whether the extreme pH stability of R. capsulatus BFR is a general property of bacterioferritins, although it has been shown that E. coli BFR consists of a mixture of largely 24-meric, dimer and monomer species at around neutral pH, with the proportion of disassembled species increasing at acidic and alkaline pH values (Andrews et al. 1991,1993; Kilic 1999). However, it is similar to the stabilities of horse spleen ferritin, which is oligomeric over the pH range 2.8–10.6 (Crichton and Bryce 1973; Stefanini et al. 1987; Martsev et al. 1998) with some of its subunits being monomeric and having properties analogous to molten globules below pH 3 (Santambrogio et al 1992).
Temperature-dependent behavior of bacterioferritin
Thermal unfolding of the wild-type and variant BFRs was monitored by far-UV CD spectroscopy (Fig. 7 ▶). In all cases, unfolding was manifested by a temperature-induced reduction in the α-helix content that fitted a two-state unfolding model (Pace and Scholtz 1997; Fig. 7E ▶), indicating that a stable intermediate is not populated during the unfolding process. The temperature at which 50% of the protein was unfolded (Tm) was calculated in each case from the fitted curves (Table 1). Wild-type BFR had the highest Tm, which decreased markedly on replacing Glu 135 with Arg, even though this variant retained a 24-meric structure. The dimeric BFRs had even lower Tm values. The high thermal stability of wild-type R. capsulatus BFR was not unexpected because a heat step is included in its purification (Materials and Methods), as with other ferritins. This high thermal stability does not solely derive from the BFR multimeric assembly, as Glu135Arg BFR is a 24-mer and its Tm is 14° lower than that of the wild-type protein, and the Tm values of the dimer proteins are only 12–16° lower than this. Thus, the reduction in the heat stability of the dimeric variants compared with wild-type BFR suggests that the high thermal stability of the 24-meric proteins arises in part from the charge–charge interactions, Glu 128–Arg 61 and Glu 135–Met 1, along the three- and fourfold symmetry interfaces, rather than the intrinsic stability of the subunits themselves. On the basis of the observation that the near-UV CD spectra of BFR reflects association of subunits into a 24-mer, the temperature dependence of the near-UV CD spectra of wild-type and Glu128Arg/Glu135Arg BFRs indicates that the quaternary structure of wild-type BFR is disrupted at the same time as the α-helix content is lost (Fig. 7E ▶); the Tm value for the 291-nm change is 71.5 ± 0.2, little altered from the Tm of 73.2 ± 0.4 for the loss of α-helix.
Figure 7.


Temperature dependence of the CD spectra of wild-type and mutant bacterioferritins. All proteins were 50 μg ml−1 in 50 mM potassium phosphate buffer (pH 7.2). (A) Far-UV spectra of wild-type BFR. (B) Near-UV spectra of wild-type BFR. (C) Far-UV spectra of Glu128Arg/Glu135Arg BFR. (D) Near-UV spectra of Glu128Arg/Glu135Arg BFR. (E) Thermal unfolding profiles for wild-type (♦), Glu135Arg (▴), Glu128Ala/Glu135Ala (•), and Glu128Arg/Glu135Arg (▪) BFR. The fractional unfolding of each protein was calculated from the change at 222 nm, and the base lines of the pretransition and post-transition regions were corrected by subtraction of linear least squares fitted lines.
According to the unfolding profiles, the 24-meric proteins and the dimeric BFRs were fully unfolded at ~90 and 60 °C, respectively. Wild-type BFR heated to 73 and 90°C showed ~30% and 15% recovery of secondary structure, respectively, on cooling to 22°C, but recovery for the Glu128Arg/Glu135Arg BFR was slightly higher at ~55% and ~30% for protein heated to 43 and 90°C, respectively (data not shown). At 90°C, the wild-type BFR (1 mg/mL) solution was slightly turbid, indicating that denaturation of the protein produced insoluble material, probably resulting from the aggregation of unfolded polypeptides. This is a common phenomenon with the heat denaturation of multimeric proteins (Jaenicke 1987; Wrba et al. 1990). Irreversible heat denaturation has also been reported for horse spleen and human ferritins, with significant reversibility only being observed when the ferritins were heated to 5– 10°C below their Tm values (Stefanini et al. 1996).
Effect of Gnd.HCl on wild-type and mutant bacterioferritins
CD and fluorescence measurements showed that Gnd.HCl caused complete unfolding of wild-type and variant BFRs, which was largely reversible (data not shown). 10-fold dilution of the unfolded BFRs in 100 mM potassium phosphate (pH 7.2) led to a restoration of the far-UV CD spectra of the native proteins. The fluorescence spectra of the BFRs were also largely restored, although the tryptophan emission maximum of refolded wild-type BFR protein was shifted to 340 nm from the 338 nm of native BFR. Refolded and native Glu128Arg/Glu135Arg BFR had the same fluorescence spectra. For the Glu135Arg and Glu128Arg/Glu135Arg variants, Gnd.HCl-induced unfolding profiles constructed from the changes in 222-nm ellipticity fitted well to a two-state unfolding model (Pace and Scholtz 1997), but the wild-type BFR data did not, having an apparent biphasic character with inflexion points at ~3 and ~4 M Gnd.HCl (Fig. 8 ▶). These inflexion points suggest that there is at least one intermediate during Gnd.HCl unfolding of wild-type BFR. Intermediates have been detected with Gnd.HCl-induced dissociation of other oligomeric proteins, and suggested to involve partially unfolded dissociated subunits (Jaenicke and Rudolph 1986). However, this is unlikely to be the case here, as the dimeric variants became fully unfolded at a Gnd.HCl concentration less than that required for the intermediate(s) to be detected in unfolding of the 24-mer. Therefore, we suggest that the intermediate(s) in the unfolding of wild-type BFR are assemblies of subunits larger than the dimer form. Similar data for horse spleen ferritin and recombinant H-chain ferritin indicate that these proteins also have one or more intermediates during Gnd.HCl unfolding (Gerl and Jaenicke 1987; Santambrogio et al. 1992).
Figure 8.

Unfolding profiles determined from the effect of guanidine HCl on the far-UV CD spectra of wild-type and mutant bacterioferritins for wild-type (•), Glu135Arg (▪), and Glu128Arg/Glu135Arg (▴) BFR. The fractional unfolding of each protein (200 μg ml−1 in 100 mM potassium phosphate buffer, pH 7.2) was calculated from the change at 222 nm, and the base lines of the pretransition and post-transition regions were corrected by subtraction of linear least squares fitted lines.
Effect of urea on wild-type and mutant bacterioferritins
Urea-induced unfolding profiles for wild-type and variant bacterioferritins were constructed from changes in fluorescence emission maxima (Fig. 9 ▶), which shifted from 339 to 353 nm for the 24-mer proteins, and from 342 to 353 nm for the dimeric Glu128Arg/Glu135Arg BFR. Tenfold dilution with 100 mM potassium phosphate (pH 7.2) of wild-type BFR in 10 M urea, and of Glu128Arg/Glu135Arg BFR in 6 M urea, led to the restoration of the fluorescence spectra characteristic of the native proteins, indicating that urea unfolding is a reversible process. The unfolding data for the mutant proteins fitted well to a two-state model (Pace and Scholtz 1997), and wild-type BFR also appears to follow a similar model (Fig. 9 ▶), although its greater stability to urea than the mutants results in the absence of a post-transition plateau region. Therefore, the profile for wild-type BFR may be somewhat misleading. The high stability of wild-type BFR to urea is similar to that of horse spleen and human liver ferritins, which are still largely folded in 10 M urea (Listowsky et al. 1972; Otsuka et al. 1981).
Figure 9.

Urea unfolding profiles for wild-type (•), Glu135Arg (▪), and Glu128Arg/Glu135Arg (▴) bacterioferritins. Fractional unfolding was calculated from changes in the Trp emission maxima and fitted to a two-state mechanism with the emission maxima in 100 mM potassium phosphate (pH 7.2) at 25°C corresponding to the fully folded proteins, and the emission maxima of the unfolded proteins at 353 nm.
Conformational stability of bacterioferritin and its dissociation and unfolding pathways
The 24-meric Glu135Arg variant was less stable than the wild-type protein (Tm, [Urea]50% and [Gnd.HCl]50% of 59°C, 4.9 and 3.2 M compared with 73°C, ~8 M and 4.3 M, respectively), and the dimeric Glu128Arg/Glu135Arg variant was less stable than the Glu135Arg variant (Tm, [Urea]50% and [Gnd.HCl]50% of 43°C, ~3.2 M and 1.8 M, respectively). The differences in stability are roughly equal, indicating that the salt-bridges formed by Glu 128 and Glu 135 in the native oligomer contribute similarly to the stability of the subunit assembly. The similarity in stability contributions taken together with the assembly states of the variant proteins (Table 1) further suggests that the interactions involving Glu 128 and Glu 135 play a role in stabilizing the 24-mer relative to the subunit dimer. Such behavior appears to be different from that of animal ferritin, for which disrupting intersubunit electrostatic interactions alone is not sufficient for disassembly of the oligomer (Gerl and Janicke 1988).
The dimeric mutant BFRs unfold with increasing temperature, urea concentration, and Gnd.HCl by a two-state mechanism without intermediates detectable by the equilibrium methods used in this work. In such cases of two-state unfolding, the free energy of unfolding can be estimated by assuming that the linear dependence of ΔGU-F on denaturant concentration observed in the transition region continues to zero denaturant concentration and fits Equation 1 (Pace 1986; Bolen and Santaro 1988; Santaro and Bolen 1988; Pace et al. 1990; Jackson and Fersht 1991; Johnson and Fersht 1995):
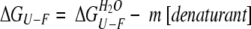 |
(1) |
in which ΔGU-F is −RT ln.(fraction unfolded/fraction folded); ΔGU-FH2O is the free energy of unfolding in the absence of denaturant; and m is the slope of the transition, which is a measure of the increase in the degree of exposure of the protein upon denaturation.
Graphs of ΔGU-F against urea and Gnd.HCl concentrations for Glu128Arg/Glu135Arg BFR and Glu135Arg BFR show that there are good linear relationships between the free energy changes of unfolding and the denaturant concentrations (Fig. 10 ▶), allowing the ΔGU-FH2O and m values to be obtained (Table 2). For each mutant, there is excellent agreement between the ΔGU-FH2O values obtained with the two denaturants, and there is also good agreement in the m values for the different mutants with the same denaturant, consistent with both Glu128Arg/Glu135Arg BFR and Glu135Arg BFR adopting the same unfolded state ensembles. The differences between the m values for the urea and Gnd.HCl unfolding profiles is similar to that of other proteins, for example, phenylmethanesulfonyl chymotrypsin, for which the m value for Gnd.HCl denaturation is about twice that for urea denaturation (Santaro and Bolen 1988), and although m values are specific for each protein (Johnson and Fersht 1995), their variation with denaturation conditions reflects differences in the way unfolded proteins interact with denaturants (Pace et al. 1990). A similar treatment of ΔGU-F to that presented in Figure 10 ▶ for the mutant BFRs is not reliable for wild-type BFR because Gnd.HCl denaturation did not follow a two-state model (Fig. 8 ▶), and it may not have been fully unfolded in 10 M urea, as a post-transition plateau region was not observed (Fig. 9 ▶). However, assuming that in 10 M urea, wild-type BFR is 100% unfolded, a similar analysis to those of Figure 10 ▶ gives its ΔGU-FH2O as ≈5.3 kcal mole−1.
Figure 10.
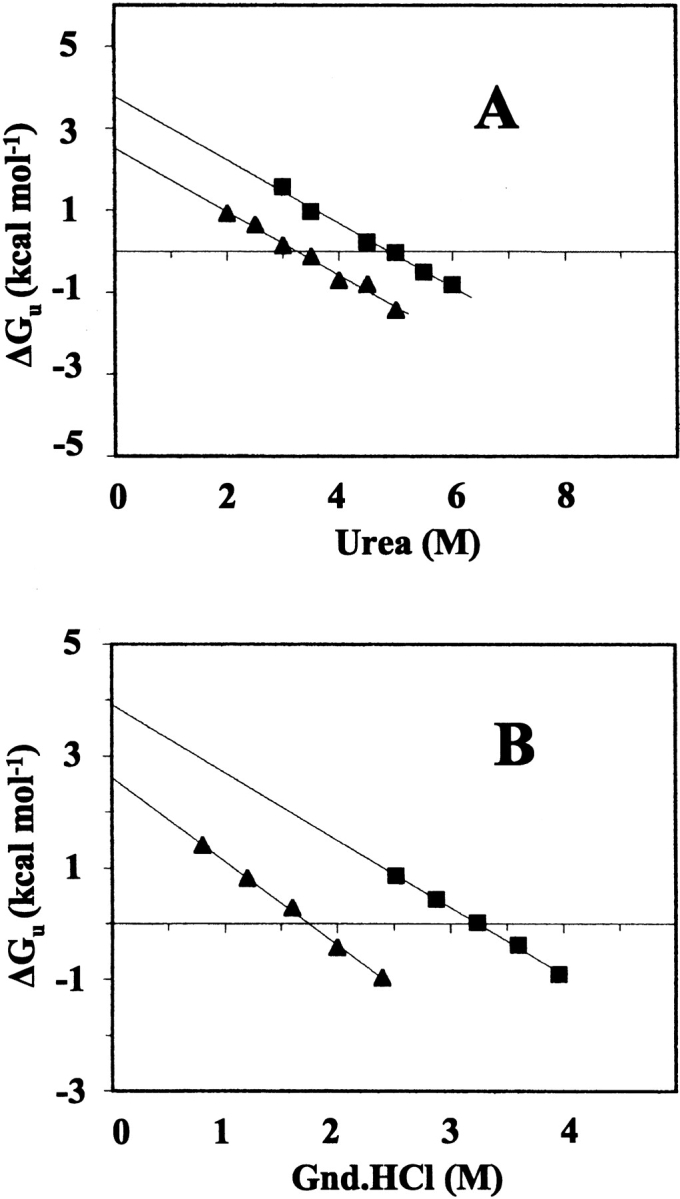
Dependence of the free energy for unfolding of Glu135Arg BFR (▪) and Glu128Arg/Glu135Arg (▴) as a function of the concentration of urea (A) and guanidine HCl (B). Linear extrapolation to 0 M urea or guanidine HCl gives the free energy change of denaturation in the absence of denaturant. The solid lines were obtained from least squares fitting.
Table 2.
Free energy changes for unfolding of R. capsulatus bacterioferritin
| Urea | Gnd.HCl | |||||
| Bacterioferritin | ΔGappH2O (kcal mole−1) | ΔGU-FH2O (kcal mole−1) | m-value (kcal mole−1 M−1) | ΔGappH2O (kcal mole−1) | ΔGU-FH2O (kcal mole−1) | m-value (kcal mole−1 M−1) |
| Wild-type | ~6.2 | n.d. | n.d. | ~5.4 | n.d. | n.d. |
| Glu135Arg | ~3.5 | 3.78 (±0.17) | 0.77 (±0.03) | ~4.3 | 3.91 (±0.12) | 1.20 (±0.03) |
| Glu128Arg/Glu135Arg | n.d. | 2.52 (±0.15) | 0.77 (±0.04) | n.d. | 2.62 (±0.07) | 1.48 (±0.04) |
A rough estimate of the free energy of unfolding of wild-type BFR can be obtained with the assumption that the m values of wild-type BFR are the same as those of the mutant proteins through multiplying the difference between the midpoints of their denaturation curves by the relevant m value (Cupo and Pace 1983). These estimates represent the differences in stability under conditions of relatively high-denaturant concentration because they refer to the 50% unfolding concentration and thus, they may be different from the free-energy values extrapolated to zero denaturant concentration. To distinguish this approach for determining the free energy of unfolding from the graphical linear extrapolation method (Fig. 10 ▶), we have called the extrapolated values ΔGU-FH2O, and the values estimated from the midpoints of the unfolding curves, ΔGappH2O. Values of ΔGappH2O and ΔGU-FH2O for Glu35Arg BFR are in close agreement (Table 2), suggesting that these procedures give comparable results for BFR. For wild-type BFR, ΔGappH2O for denaturation with urea and Gnd.HCl at ~6.2 and ~5.4 kcal mole−1, respectively, are substantially different, probably reflecting the imperfect nature of the calculation. Nevertheless, the calculated values are not so different as to render them unusable. Taken together with the observation that the Tm changes by 14–16°C on replacement of either Glu 128 or Glu 135 with arginine (Table 1), the comparison in unfolding free energies between wild-type and mutant BFR suggests that removal of a salt-bridge between Glu 135 and the terminal amine of Met 1 reduces stability by ~1.6–2.4 kcal mole−1, and removing an additional salt-bridge between Glu 128 and Arg 61 reduces stability by a further ~1.3 kcal mole−1. The energetic contributions of salt-bridges in protein vary according to their location (Fersht 1999), solvent-exposed or partially buried salt-bridges being worth ~1.2–1.5 kcal mole−1, whereas completely buried salt-bridges are worth up to 6 kcal mole−1 (Anderson et al. 1990). The contributions to the stability of wild-type BFR from salt-bridges involving Glu 128 and Glu 135 are consistent with their partial exposure to solvent (Cobessi et al. 2002). The significance of the energetically important roles played by Arg 61, Glu 128, and Glu 135 in maintaining the 24-mer assembly of BFR is reflected by their conservation in all known bacterioferritin sequences except that of Magnetospirillum magnetotacticum BFR2, in which residue 128 is Gln (Bertani et al. 1997). In contrast to this, however, intersubunit salt-bridges corresponding to those involving Glu 128 and Glu 135 are not found in animal ferritin or FTN, although in Dps Arg 61 and Asp/Glu 157, the latter equivalent to Glu 128 of BFR (Kilic 1999) are conserved.
The self-assembly of horse spleen apoferritin subunits into a 24-mer has been shown to proceed via discreet intermediates, including dimer, trimer, dimer of trimers, and octamer species, although some of these only had a transient existence (Gerl and Janicke 1988; Santambrogio et al. 1997). The work reported here on wild-type bacterioferritin is in contrast to this; dissociated subunit monomers, or aggregates smaller than 24-mers, that contained the high α-helical content characteristic of the native protein were not obtained at any pH without a high proportion of the 24-mer being present. Taken together with the other denaturation experiments, this suggests that folding of the wild-type bacterioferritin monomer could be coupled to its association into the oligomer. However, it is clear that folding does not depend upon association into the 24-mer because the Glu128Ala/Glu135Ala and Glu128Arg/Glu135Arg variants are dimeric and structured. Whether folding is coupled to dimerization remains to be established.
Materials and methods
Preparation of bacterioferritin and site-directed mutagenesis
Mutations were introduced by PCR mutagenesis (Hutchings et al. 2000) into the R. capsulatus bfr gene cloned in pET21a (Novagen), and plasmids were transformed into E. coli MAK96 (BL21 bfr::kan). Expression of the bfr gene was induced as described previously (Penfold et al. 1996), and mutant BFR prepared by chromatography of the cell-free extract on a Q-sepharose anion exchange column (50 mM potassium phosphate buffer at pH 7.2), being eluted with a NaCl gradient at 0.22 M NaCl, followed by chromatography on Sephacryl S-100 HR or S-300 HR gel-filtration columns (50 mM potassium phosphate buffer at pH 7.2). The cell-free extracts for wild-type BFR and the variant BFRs with one amino acid change per subunit were heated to 65 and 55°C, respectively, for 15 min, cooled rapidly on ice, and precipitated material removed by centrifugation prior to chromatography.
Analyses of bacterioferritin preparations
ESI-MS of the purified proteins gave masses consistent with the expected amino acid sequences. ESI-MS was carried out with a Micromass platform I mass spectrometer calibrated with horse-heart myoglobin. The solvent used was 1:1 acetonitrile/water containing 0.1% formic acid, and samples were run at a flow rate of 20 μL/min−1. UV/visible electronic absorption spectra were collected with Perkin Elmer Lambda 900 and Hitachi U2000 spectrophotometers, using a 1-cm pathlength. Non-heme iron was determined as the [Fe2+(ferrozine)3] complex on the basis of the method of Stookey (1970). The average level of iron loading was found to be 4–110 and 5–10 Fe3+ per molecule for the 24-mer and subunit dimer, respectively, as prepared, and <<1 per molecule after treatment to remove non-heme iron. Iron-free BFR samples were prepared by anaerobic dialysis of the isolated protein against 100 mM HEPES (pH 7) at 4°C and treatment with 2,2′-bipyridyl and sodium dithionite, followed by dialysis against the required buffer. Heme contents were determined from the 419 absorbance band of the non-heme iron-free protein and the extinction coefficient of the heme Soret band (Ringeling et al. 1994), ɛ419 = 1.4 × 10−5 M−1 cm−1. Samples of 24-mer BFR and the BFR subunit dimer contained 0.1–0.8 and 0.05–0.15 heme groups per molecule, respectively, as prepared.
Molecular weights of wild-type and mutant proteins were determined with calibrated gel-permeation columns. Sephacryl 300 HR was used with 24-mer BFR and sephacryl 100 HR with the subunit dimers. The columns were calibrated with protein standards obtained from Sigma. All samples were run in 50 mM phosphate buffer (pH 7.2), containing 100 mM NaCl, with a flow rate of 0.4 mL/min−1. The molecular weight of wild-type BFR was determined to be 410 kD by this method. Nondenaturing PAGE was done with 6% linear or 6% and 12% gradient gels (Goldenberg 1997; Makowski and Ramsby 1997).
Biophysical measurements
Fluorescence spectra were recorded with Shimadzu RF-5000 and Perkin Elmer LS55 spectrofluorimeters, and CD spectra were measured with a JASCO J-710 spectropolarimeter. Each CD spectrum was collected as an average of three successive scans with buffer background scans subtracted. Mean residue ellipticities were calculated as described by Schmid (1997).
Acknowledgments
We thank the Turkish Government for a studentship to M.A.K.; the Biotechnology and Biology Science Research Council and Wellcome Trust for their support of our work on bacterioferritin; the Joint Infrastructure Fund (062178) for equipment; Prof. D. Cobessi (Lawrence Berkeley National Laboratory, California) for generously supplying coordinates of the R. capsulatus bacterioferritin X-ray structure prior to publication; and Dr. N. Le Brun and Dr. A. Lewin for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BFR, Bacterioferrin
CAPS, 3-(cyclohexylamino)propanesulphonic acid
CD, circular dichroism
Dps, DNA-binding protein from starved cells
ESI-MS, electrospray mass spectrometry
FTN, heme-free bacterial ferritin
Gnd, guanidine
HEPES, N-2-hydroxyethylpiperazine-N-2-ethanesulphonic acid
PCR, polymerase chain reaction
PAGE, polyacrylamide gel electrophoresis
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0301903.
References
- Anderson, D.E., Becktel, W.J., and Dahlquist, F.W. 1990. pH-induced denaturation of proteins—a single salt bridge contributes 3–5 kcal mol−1 to the free energy of unfolding of T4-lysozyme. Biochemistry 29 2403–2408. [DOI] [PubMed] [Google Scholar]
- Andrews, S.C. 1998. Iron storage in bacteria. Adv. Microb. Physiol. 40 281–351. [DOI] [PubMed] [Google Scholar]
- Andrews, S.C., Findlay, J.B.C., Guest, J.R., Harrison, P.M., Keen, J.M., and Smith, J.M.A. 1991. Physical, chemical and immunological properties of the bacterioferritins of Escherichia coli, Pseudomonas aeruginosa and Azotobacter vinelandii. Biochim. Biophys. Acta 1078 111–116. [DOI] [PubMed] [Google Scholar]
- Andrews, S.C., Smith, J.M.A., Hawkins, C., Williams, J.M., Harrison, P.M., and Guest, J.R. 1993. Overproduction, purification and characterization of the bacterioferritin of Escherichia coli and a C-terminally extended variant. Eur. J. Biochem. 213 329–338. [DOI] [PubMed] [Google Scholar]
- Andrews, S.C., Le Brun, N.E., Barynin, V., Thomson, A.J., Moore, G.R., Guest, J.R., and Harrison, P.M. 1995. Replacement of the coaxial-heme ligands of bacterioferritin generates heme-free variants. J. Biol. Chem. 270 23268–23274. [DOI] [PubMed] [Google Scholar]
- Bertani, L.E., Huang, J.S., Weir, B.A., and Kirschvink, J.L. 1997. Evidence for two types of subunits in the bacterioferritin of Magnetospirillum magnetotacticum. Gene 201 31–36. [DOI] [PubMed] [Google Scholar]
- Bolen, D.W. and Santaro, M.M. 1988. Unfolding free energy changes determined by the linear extrapolation method. 2. Incorporation of ΔG°N-U values in a thermodynamic cycle. Biochemistry 27 8069–8074. [DOI] [PubMed] [Google Scholar]
- Chasteen, N.D. 1998. Ferritin. Uptake, storage, and release of iron. Met. Ions Biol. Syst (eds. H. Sigel and A. Sigel). Marcel Dekker Inc., New York. 35 479–514. [PubMed] [Google Scholar]
- Cheesman, M.R., Thomson, A.J., Greenwood, C., Moore, G.R., and Kadir, F.H.A. 1990. Bis-methionine axial ligation of haem in bacterioferritin from Pseudomonas aeruginosa. Nature 346 771–773. [DOI] [PubMed] [Google Scholar]
- Clegg, G.A., Fitton, J.E., Harrison, P.M., and Treffry, A. 1980. Ferritin: Molecular structure and iron-storage mechanisms. Prog. Biophys. Mol. Biol. 36 56–86. [PubMed] [Google Scholar]
- Cobessi, D., Huang, L.-S., Ban, M., Pon, N.G., Daldal, F., and Berry, E.A. 2002. The 2.6 Å resolution structure of Rhodobacter capsulatus bacterioferritin with metal-free dinuclear site and heme iron in a crystallographic ‘special position’. Acta Crystallogr. D Biol. Crystallogr. 58 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton, R.R. and Bryce, C.F.A. 1973. Subunit interactions in horse spleen apoferritin. Biochem. J. 133 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupo, J.F,. and Pace, C.N. 1983. Conformational stability of mixed disulfide derivatives of b-lactoglobulin B. Biochemistry 22 2654–2658. [DOI] [PubMed] [Google Scholar]
- Douglas, T. and Ripoll, D.R. 1998, Calculated electrostatic gradients in recombinant human H-chain ferritin. Protein Sci. 7 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht, A.1999. Structure and mechanism in protein science, pp. 530–532. W.H. Freeman and Company, New York.
- Ford, G.C., Harrison, P.M., Rice, D.W., Smith, J.M.A., Treffry, A., White, J.L., and Yariv, J. 1984. Ferritin—design and formation of an iron storage molecule. Phil. Trans. R. Soc. Lond. B304 551–565. [DOI] [PubMed] [Google Scholar]
- Frolow, F., Kalb, A.J., and Yariv, J. 1994. Structure of a unique twofold symmetric haem-binding site. Nat. Struct. Biol. 1 453–460. [DOI] [PubMed] [Google Scholar]
- Gallois, B., d’Estaintot, B.L., Michaux, M.-A., Dautant, A., Granier, T., Précigoux, G., Soruco, J.-A., Roland, F., Chavas-Alba, O., Herbas, A., et al. 1997. X-ray structure of recombinant horse L-chain apoferritin at 2.0 Å resolution: Implications for stability and function. J. Biol. Inorg. Chem. 2 360–367. [Google Scholar]
- Gerl, M. and Jaenicke, R. 1987. Assembly of apoferritin from horse spleen. Biol. Chem. Hoppe-Seyler 368 387–396. [DOI] [PubMed] [Google Scholar]
- Goldenberg, D.P. 1997. Measuring the conformational stability of a protein. In Protein structure: A practical approach, 2nd ed. (ed. T.E. Creighton), pp. 187–218. IRL Press, Oxford, UK.
- Grant, R.A., Filman, D.J., Finkel, S.E., Kolter, R., and Hogle, J.M. 1998. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 5 294–303. [DOI] [PubMed] [Google Scholar]
- Ha, Y., Shi, D., Small, G.W., Theil, E.C., and Allewell, N.M. 1999. Crystal structure of bullfrog M ferritin at 2.8 Å resolution: Analysis of subunit interactions and the bunuclear metal center. J. Biol. Inorg. Chem. 4 243–256. [DOI] [PubMed] [Google Scholar]
- Harrison, P. and Arosio, P. 1996. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275 161–203. [DOI] [PubMed] [Google Scholar]
- Hutchings, M.I., Shearer, N., Wastell, S., van Spanning, R.J.M., and Spiro, S. 2000. Heterologous NNR-mediated nitric oxide signaling in Escherichia coli. J. Bact. 182 6434–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilari, A., Stefanini, S., Chiancone, E., and Tsernoglou, D. 2000. The dodecameric ferritin from Listeria innocua contains a novel intersubunit iron-binding site. Nat. Struct. Biol. 7 38–43. [DOI] [PubMed] [Google Scholar]
- Ilari, A., Ceci, P., Ferrari, D., Rossi, G., and Chiancone, E. 2002. Iron incorporation into E. coli Dps gives rise to a ferritin-like microcrystalline core. J. Biol. Chem. 277 19–23. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. and Fersht, A.R. 1991. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 30 10428–10435. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. 1987. Folding and association of proteins. Prog. Biophys. Mol. Biol. 49 117–237. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. and Rudolph, R. 1986. Refolding and association of oligomeric proteins. Methods Enzymol. 31 218–250. [DOI] [PubMed] [Google Scholar]
- Johnson, C.M. and Fersht, A.R. 1995. Protein stability as a function of denaturant concentration: The thermal stability of barnase in the presence of urea. Biochemistry 34 6795–6804. [DOI] [PubMed] [Google Scholar]
- Kilic, M.1999. “Structural and functional characterization of Rhodobacter capsulatus bacterioferritin.” Ph.D. thesis, University of East Anglia, Norwich, UK.
- Le Brun, N.E., Andrews, S.C., Guest, J.R., Harrison, P.M., Moore, G.R., and Thomson, A.J. 1995. Identification of the ferroxidase center of Escherichia coli bacterioferritin. Biochem. J. 312 385–392. [PMC free article] [PubMed] [Google Scholar]
- Le Brun, N.E., Thomson, A.J., and Moore, G.R. 1997. Metal centers of bacterioferritins or non-haem-iron-containing cytochromes b557. Struct. Bonding 88 103–138. [Google Scholar]
- Levi, S., Santambrogio, P., Albertini, A., and Arosio, P. 1993. Human ferritin H-chains can be obtained in non-assembled stable forms which have ferroxidase activity. FEBS Lett. 336 309–312. [DOI] [PubMed] [Google Scholar]
- Listowsky, I., Blauer, G., Englard, S., and Betheil, J.J. 1972. Denaturation of horse spleen ferritin in aqueous guanidinium chloride solutions. Biochemistry 11 2176–2181. [DOI] [PubMed] [Google Scholar]
- Makowski, G.S. and Ramsby, M.L. 1997. Measuring the conformational stability of a protein. In Protein structure: A practical approach, 2nd ed. (ed. T.E. Creighton), pp. 1–27. IRL Press, Oxford, UK.
- Martsev, S.P., Vlasov, A.P., and Arosio, P. 1998. Distinct stability of recombinant L and H subunits of human ferritin: Calorimetric and ANS binding studies. Protein Eng. 11 377–381. [DOI] [PubMed] [Google Scholar]
- Otsuka, S., Maruyama, H., and Listowsky, I. 1981. Structure, assembly, conformation and immunological properties of the two subunit classes of ferritin. Biochemistry 20 5226–5232. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure: A practical approach, 2nd ed. (ed. T.E. Creighton), pp. 299–321. IRL Press, Oxford, UK.
- Pace, C.N., Laurents, D.V., and Thomson, J.A. 1990. pH dependence of the urea and guanidine hydrochloride denaturation of ribonuclease A and ribonuclease T1. Biochemistry 29 2564–2572. [DOI] [PubMed] [Google Scholar]
- Penfold, C.N., Ringeling, P.L., Davy, S.L., Moore, G.R., McEwan, A.G., and Spiro, S. 1996. Isolation, characterization and expression of the bacterioferritin gene of Rhodobacter capsulatus. FEMS Microbiol. Lett. 139 143–148. [DOI] [PubMed] [Google Scholar]
- Powell, A.K. 1998. Ferritin. Its mineralization. Met. Ions Biol. Syst. 35 515–561. [PubMed] [Google Scholar]
- Ringeling, P.L., Davy, S.L., Monkara, F.A., Hunt, C., Dickson, D.P.E., McEwan, A.G., and Moore, G.R. 1994. Iron metabolism in Rhodobacter capsulatus. Characterization of bacterioferritin and formation of non-haem iron particles in intact cells. Eur. J. Biochem. 223 847–855. [DOI] [PubMed] [Google Scholar]
- Santambrogio, P., Levi, S., Arosio, P., Palagi, L., Vecchio, G., Lawson, D.M., Yewdall, S.J., Artymiuk, P.J., Harrison, P.M., Jappelli, R., et al. 1992. Evidence that a salt bridge in the light chain contributes to the physical stability difference between heavy and light human ferritins. J. Biol. Chem. 267 14077–14083. [PubMed] [Google Scholar]
- Santambrogio, P., Pinto, P., Levi, S., Cozzu, A., Rovida, E., Albertini, A., Artymiuk, P., Harrison, P.M., and Arosio, P. 1997. Effects of modifications near the 2-, 3- and 4-fold symmetry axes on human ferritin renaturation. Biochem. J. 322 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1997. Optical spectroscopy to characterize protein conformation and conformational changes. In Protein structure: A practical approach, (ed. T.E. Creighton) pp. 261–297. IRL Press, Oxford, UK.
- Spiro, S., Kilic, M., Lewin, A., Dobbin, S., Thomson, A.J., and Moore, G.R. 1999. Interactions of heme and other metal ion complexes with the bacterial Fe-uptake regulatory protein and with bacterioferritin. In Iron metabolism, inorganic biochemistry and regulatory mechanisms, (eds. G.C. Ferreira et al.), pp. 211–226. Wiley-VCH, Germany.
- Stefanini, S., Vecchini, P., and Chiancone, E. 1987. On the mechanism of horse spleen apoferritin assembly. Biochemistry 26 1831–1837. [DOI] [PubMed] [Google Scholar]
- Stefanini, S., Cavallo, S., Wang, C.-Q., Tataseo, P., Vecchini, P., Giartosio, A., and Chiancone, E. 1996. Thermal stability of horse spleen apoferritin and human recombinant H apoferritin. Arch. Biochem. Biophys. 325 58–64. [DOI] [PubMed] [Google Scholar]
- Stiefel, E.I. and Watt, G.D. 1979. Azotobacter cytochrome b557.5 is a bacterioferritin. Nature 279 81–83. [DOI] [PubMed] [Google Scholar]
- Stookey, L.L. 1970. Ferrozine—a new spectrophotometric reagent for iron. Anal. Chem. 42 779–781. [Google Scholar]
- Trikha, J., Theil, E.C., and Allewell, N.M. 1995. High resolution crystal structures of amphibian red-cell L ferritin: Potential roles of structural plasticity and solvation in function. J. Mol. Biol. 248 949–967. [DOI] [PubMed] [Google Scholar]
- Wrba, A., Schweiger, A., Schultes, V., and Jaenicke, R. 1990. Extremely thermostable D-glyceraldehyde-3-phosphate dehydrogenase from the eubacterium Thermotoga maritima. Biochemistry 29 7584–7592. [DOI] [PubMed] [Google Scholar]
- Yang, X., Chen-Barrett, Y., Arosio, P., and Chasteen, N.D. 1998. Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry 37 9743–9750. [DOI] [PubMed] [Google Scholar]
- Yang, X., Le Brun, N.E., Thomson, A.J., Moore, G.R., and Chasteen, N.D. 2000. The iron oxidation and hydrolysis chemistry of Escherichia coli bacterioferritin. Biochemistry 39 4915–4928. [DOI] [PubMed] [Google Scholar]
- Zhao, G., Ceci, P., Ilarim, A., Giangiacomo, L., Laue, T.M., Chiancone, E., and Chasteen, N.D. 2002. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells. J. Biol. Chem. 277 27689–27696. [DOI] [PubMed] [Google Scholar]