

Abstract
Mutations in HIV-1 drug targets lead to resistance and consequent therapeutic failure of antiretroviral drugs. Phenotypic resistance assays are time-consuming and costly, and genotypic rules-based interpretations may fail to predict the effects of multiple mutations. We have developed a computational procedure that rapidly evaluates changes in the binding energy of inhibitors to mutant HIV-1 PR variants. Models of WT complexes were produced from crystal structures. Mutant complexes were built by amino acid substitutions in the WT complexes with subsequent energy minimization of the ligand and PR binding site residues. Accuracy of the models was confirmed by comparison with available crystal structures and by prediction of known resistance-related mutations. PR variants from clinical isolates were modeled in complex with six FDA-approved PIs, and changes in the binding energy (ΔEbind) of mutant versus WT complexes were correlated with the ratios of phenotypic 50% inhibitory concentration (IC50) values. The calculated ΔEbind of five PIs showed significant correlations (R2 = 0.7–0.8) with IC50 ratios from the Virco Antivirogram assay, and the ΔEbind of six PIs showed good correlation (R2 = 0.76–0.85) with IC50 ratios from the Virologic PhenoSense assay. ΔEbind cutoffs corresponding to a four-fold increase in IC50 were used to define the structure-based phenotype as susceptible, resistant, or equivocal. Blind predictions for 78 PR variants gave overall agreement of 92% (kappa = 0.756) and 86% (kappa = 0.666) with PhenoSense and Antivirogram phenotypes, respectively. The structural phenotyping predicted drug resistance of clinical HIV-1 PR variants with an accuracy approaching that of frequently used cell-based phenotypic assays.
Keywords: HIV protease inhibitors, drug resistance, molecular modeling, binding energy, structure-based phenotyping
Antiretroviral drugs targeting the RT and PR enzymes of HIV-1 may result in dramatic suppression of viral replication in infected individuals (Palella Jr. et al. 1998; Carpenter et al. 2000). However, when viral replication is incompletely suppressed, drug-resistant variants emerge through the accumulation of mutations in the HIV-1 RT or PR genes, leading to therapeutic failure (Hirsch et al. 2000). Genotypic testing for resistance is a relatively rapid and inexpensive method to identify PR and RT amino acid substitutions leading to drug resistance (Baxter et al. 2000; Schinazi et al. 2000; Shafer 2002). Genotyping is recommended for use in clinical practice (Hirsch et al. 2000). However, rules-based interpretation systems are retrospective in nature and must be frequently updated to accommodate new mutational patterns and new antiretrovirals. As a result, genotypic predictions for complex mutational patterns and for new antiretrovirals may be inaccurate (Baxter et al. 2000). Cell-based viral phenotyping assays (Hertogs et al. 1998; Petropoulos et al. 2000; Qari et al. 2002) have been correlated with favorable clinical outcomes (Cohen et al. 2002) and may offer a more reliable measure of resistance in cases in which mutational patterns are complex or the genotypic resistance pattern has not been determined (Haubrich et al. 2001). However, phenotypic assays are more time-consuming and expensive than genotypic assays, and the clinically significant cutoffs for the change in IC50 for some drugs are still uncertain. A hybrid approach, matching viral sequence against a database of paired viral genotypes and phenotypes, is used to derive a VirtualPhenotype, a computational prediction of viral resistance (Tibotec-Virco; http://www.tibotec-virco.com). This method provides a quantitative estimate of drug resistance, although it may fail to give a prediction when few phenotypic matches for a particular variant are identified in the database, as is the case for rare mutational patterns or newly introduced drugs.
HIV-1 PR, a homodimer containing two identical 99 amino acid polypeptide chains, is a key enzyme involved in the catalysis of HIV protein cleavage in the viral replication cycle (Kohl et al. 1988). Processing by PR is essential for viral maturation and infectivity. Therefore, an intense effort has been made to rationally design and develop HIV-1 PIs (for review, see Wlodawer and Vondrasek 1998; Gulnik et al. 2000; Tomasselli and Heinrikson 2000). Six FDA-approved PIs (Fig. 1 ▶) are currently in clinical use. However, the rapid emergence of resistant HIV-1 variants can result in therapeutic failure (Schinazi et al. 2000; Shafer 2002). Individual mutations and combinations of mutations at more than 20 PR codons are known to contribute to PI resistance (Hirsch et al. 2000), including amino acids directly involved in the inhibitor binding, as well as some residues located up to 20 Å apart from the inhibitor binding site. The structural determinants of resistance for some of the PR variants have been characterized by X-ray crystallography (Baldwin et al. 1995; Hong et al. 2000; Mahalingam et al. 2001; King et al. 2002).
Figure 1.
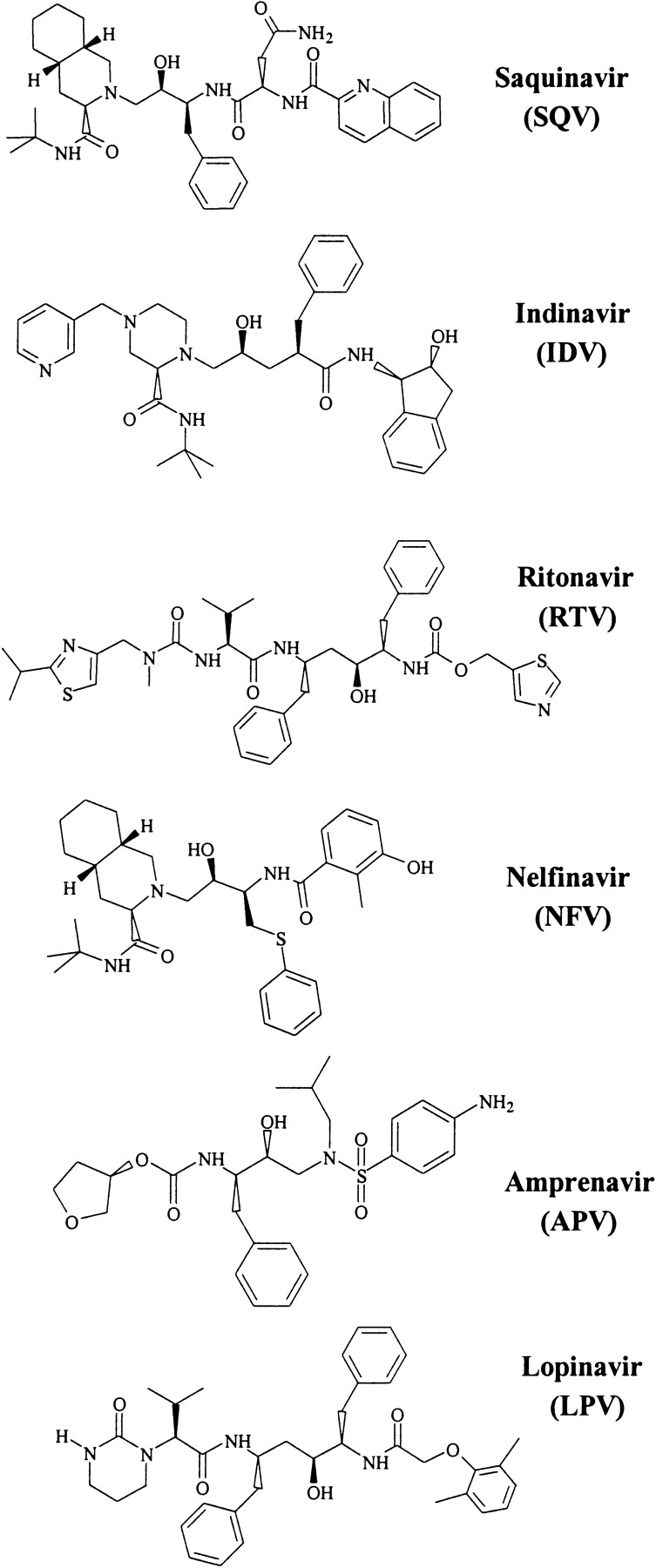
Structures of the FDA-approved HIV-1 PIs used in this study.
Several attempts have been made to develop computational methodologies for molecular modeling of PR–inhibitor complexes, and to build QSAR models for prediction of PI activity (Holloway et al. 1995; Perez et al. 1998; Nair et al. 2002). Molecular mechanics and molecular dynamics approaches have also been used to predict resistance of mutant PR variants to various PIs (Dominy and Brooks III 1999; McCarric and Kollman 1999). The approaches based on calculations of protein–ligand interaction energies resulted in significant correlations with experimental binding energies (Weber and Harrison 1999) or in the correct prediction of 75% of the single amino acid mutations causing resistance to commercial PIs (Wang and Kollman 2001). These computational methods, however, have not been developed to predict resistance of PR variants with multiple amino acid substitutions obtained from clinical HIV-1 isolates.
The present study tests the hypothesis that resistance to PIs results mainly from decreased inhibitor binding affinity to mutant PR variants, as was previously shown by inhibitor binding kinetics measurements for engineered PR variants containing known resistant mutations (Gulnik et al. 1995). We have further assumed that (1) mutant PR–inhibitor complexes can be accurately modeled based on X-ray crystal structures of WT PR complexes, (2) changes in the binding affinity on mutation may be approximated as changes in calculated binding energy of PR–inhibitor complexes, and (3) PI resistance may be predicted from a significant increase in the binding energy of an inhibitor with respect to the WT complex. We describe here a structural approach to predicting the resistance of HIV-1 PR mutants to the six FDA-approved PIs using molecular modeling of the PR complexes in order to calculate changes in the inhibitor binding energy to the mutant PR relative to the reference WT complex. In preliminary studies (Shenderovich et al. 2001a,b), we obtained statistically significant correlations between calculated and experimental values of binding energy of four FDA-approved inhibitors with engineered PR variants containing single and double amino acid mutations. In this study, we show that the changes in binding energy can be directly correlated to the fold changes in PI IC50 values measured in two commercially available phenotypic assays. We propose a model for a structural phenotyping resistance assay based on these correlations.
Results
Accuracy of the models of mutant PR–inhibitor complexes
Accuracy of the models may be estimated by comparison with the X-ray crystal structure of the same or similar complexes. A few structures of PR–inhibitor complexes containing resistance-related mutations are available in the Protein Data Bank (PDB; Baldwin et al. 1995; Mahalingam et al. 2001; King et al. 2002), and only one crystal structure (Hong et al. 2000; PDB entry 1fb7) that involves an FDA-approved inhibitor, SQV, in complex with a highly resistant G48V/L90M PR mutant shows significant structural changes with respect to the WT PR–inhibitor complex (PDB entry 1hxb). In the preliminary study (Shenderovich et al. 2001a,b), we modeled SQV complexes with PR variants containing the G48V mutation. Figure 2 ▶ displays superposition of the crystal structure (yellow) and our model (white) of the mutant G48V PR–SQV complex. The crystal structure of the WT PR–SQV complex is shown in blue. In the WT complex, the quinoline moiety of SQV is tightly bound to the flap residues 47–50, and flexibility of the flap residues is restricted by interactions with the Phe 53 side chain. In the mutant complex, the bulky Val 48 side chain, which appears exactly between the quinoline ring of the ligand and the phenyl ring of Phe 53, causes a steric repulsion of the aromatic moieties that increases the van der Waals component of the binding energy (see Table 1). Both the crystal structure and the computational model show very similar displacements of the aromatic moieties of the ligand and Phe 53. The RMSD between the model and the crystal structure of the mutant complex are 0.5 and 1.0 Å for Cα atoms and all heavy atoms, respectively, of the PR residues located within a 7.0 Å shell from the ligand. It is noteworthy that our model was built from an optimized crystal structure of the WT complex before the crystal structure of the mutant complex was published.
Figure 2.

Superimposed stereo views of the crystal structure (yellow; Hong et al. 2000) and our model (white) of the SQV complex with the G48V/L90M mutant HIV PR. The crystal structure of WT PR–SQV complex (blue) is also shown for comparison. The SQV molecules are shown as tube models. Only PR residues closest to the ligand are displayed.
Table 1.
Calculated and experimental binding energy changes for known resistance mutations of HIV-1 protease
| Mutations | PI | ΔEbind (calc), kcal/mole | ΔEvwa | ΔEhba | ΔEela | Fold resistanceb | ΔEbind (exptl), kcal/molec | Reference |
| G48V | SQV | 3.5 | 2.1 | 0.5 | 0.5 | 160 | 3.1 | Maschera et al. 1996 |
| I50V | SQV | 2.2 | 1.9 | 0.3 | 0.1 | 21 | 1.9 | Markland et al. 2000 |
| APV | 2.9 | 2.9 | 0.1 | 0.1 | 83 | 2.7 | Markland et al. 2000 | |
| M46I/I47V/I50V | APV | 3.6 | 1.5 | 1.8 | 0.4 | 270 | 3.4 | Partaledis et al. 1995 |
| M46I/G48V/I50V/I84L | SQV | 3.8 | 1.6 | 1.3 | 0.6 | 300 | 3.5 | Markland et al. 2000 |
| APV | 0.9 | 1.0 | −0.2 | −0.1 | 2 | 0.4 | Markland et al. 2000 | |
| V82A | IDV | 1.8 | 1.2 | 0.0 | 0.5 | 22 | 1.9 | Gulnik et al. 1995 |
| RTV | 2.4 | 2.8 | 0.2 | −0.5 | 10 | 1.4 | Gulnik et al. 1995 | |
| I84V | SQV | 2.2 | 2.1 | −0.1 | 0.1 | 12 | 1.5 | Partaledis et al. 1995 |
| APV | 2.0 | 2.0 | 0.0 | 0.1 | 23 | 1.9 | Partaledis et al. 1995 | |
| IDV | 1.5 | 1.0 | 0.0 | 0.4 | 20 | 1.8 | Partaledis et al. 1995 | |
| V32I/I84V | IDV | 2.9 | 2.2 | 0.2 | 0.2 | 80 | 2.7 | Gulnik et al. 1995 |
| RTV | 2.0 | 1.7 | 0.2 | 0.1 | 64 | 2.6 | Gulnik et al. 1995 | |
| V82T/I84V | IDV | 2.4 | 1.9 | 0.0 | 0.3 | 59 | 2.5 | Schock et al. 1996 |
| RTV | 2.9 | 2.6 | 0.1 | 0.0 | 158 | 3.1 | Schock et al. 1996 | |
| M46I/I84V | NFV | 1.7 | 1.5 | −0.1 | 0.2 | 5–30d | Patick et al. 1996 | |
| D30N | NFV | 1.5 | −0.9 | 1.6 | 0.8 | 7d | Patick et al. 1996 | |
| L10F/V32I/M46I/ I47V/I84V | LPV | 2.9 | 3.3 | −0.3 | −0.3 | 25–100 | 2.0–2.8 | Carrillo et al. 1998 |
a ΔEvw, ΔEhb, and ΔEel are changes in the van der Waals, hydrogen bonding, and electrostatic components of binding energy (see equation 2 in Materials and Methods).
b Fold changes in Ki or IC50 values.
c ΔEbind(exptl) was calculated using equations 4 or 5 in Materials and Methods.
d Ratios of EC90 values; experimental binding energies were not estimated.
Crystal structures of the mutant L63P/V82T/I84V PR in complex with IDV and its analogs were recently solved (King et al. 2002). The mutant complex with IDV (PDB entry 1k6c) shows relatively small changes with respect to the crystal structure of the WT complex (PDB entry 1hsg) with all Cα atom RMSD = 0.46 Å. Our model of the mutant V82T/I84V PR–IDV complex is also close to the WT complex (all Cα atom RMSD = 0.42 Å). Furthermore, the crystal structure and our model of the mutant complex are reasonably close to each other: Cα atom and all heavy atom RMSD is 0.5 and 0.75 Å, respectively, for residues located in a 7.0 Å shell around the ligand. Both crystal structure and the model predict similar positions of the ligand and conformations of the mutated side chains, and the calculated change in the binding energy (Table 1) is very close to the measured value (King et al. 2002).
Evaluation of resistance-related mutations for HIV-1 PIs
Statistically significant correlations between calculated and experimental binding energy were obtained in our preliminary studies (Shenderovich et al. 2001a,b) for four clinically available PIs—APV, IDV, RTV, and SQV—in complex with genetically engineered PR variants. In the present study, we modeled complexes of six FDI-approved PIs (Fig. 1 ▶) with PR variants containing known resistance-related mutations. The calculated changes in the main components of the binding energy are given in Table 1 in comparison with energy values estimated from experimental Ki or IC50 ratios. It may be noted that the computational mutagenesis procedure predicts significant change in the binding energy (ΔEbind ≥ 1.5 kcal/mole) for the majority of known resistant single and double mutations: G48V for SQV; V82A for IDV and RTV; I84V in various combinations for SQV, NFV, IDV, RTV, and APV; and D30N for NFV. Furthermore, the most resistant mutations (>100-fold change in Ki values) can be reliably predicted by ΔEbind values of ≥3.0 kcal/mole. For the majority of resistance-related single and double mutations, the increase in PI binding energy is due mainly to the van der Waals component of the binding energy function. Most of the conservative mutations in positions 50, 82, and 84 do not change significantly the ligand binding mode and structure of the complex, but only decrease the number of favorable van der Waals contacts between the ligand and the protein side chains. In particular, the crystal structure of IDV in complex with the mutant L63P/V82T/I84V PR (King et al. 2002) was similar to that of the WT PR–IDV complex, whereas thermodynamic measurements showed a 2.5 kcal/mole loss in the binding enthalpy of the mutant complex. This is in good agreement with the increase in the binding energy due to the reduced van der Waals contacts of the mutant side chains predicted for the IDV complex with the V82T/I84V mutant PR (Table 1). The noticeable exception is the complex of NFV with the mutant D30N PR, where substitution of neutral Asn for the charged Asp 30 that directly interacts with the m-phenol group of NFV weakens the hydrogen bonding and increases electrostatic components of the binding energy. More complex mutation patterns may cause significant changes in the 3D structure of the complex that leads to disruption of ligand–PR hydrogen bonding (APV complex with a triple mutant and SQV complex with a quadruple mutant; see Table 1). Only a combination of at least five mutations in residues close to the PR binding site causes a significant increase in the binding energy of the recently approved PI LPV (Table 1). This is consistent with experimental findings that clinical isolates resistant to LPV display complex mutational patterns involving positions 46, 47, and 84 (Carrillo et al. 1998).
Using the computational mutagenesis procedure, we calculated the binding energy profiles of each PI for the set of single, double, and triple mutations that were described in the literature (Gulnik et al. 1995; Markowitz et al. 1995; Maschera et al. 1996; Pazhanisamy et al. 1996; Klabe et al. 1998; Markland et al. 2000). The profiles for SQV and APV shown in Figure 3 ▶ represent the histograms of calculated ΔEbind ranked by increasing binding energy. The right shoulders of these profiles contain potentially resistant mutations. We assumed that ΔEbind = 2.0 kcal/mole was a reliable threshold for resistance prediction from the binding energy profiles. The bars exceeding this threshold are colored gray in the histograms in Figure 3 ▶; the most resistant mutations (ΔEbind ≥ 3.0 kcal/mole) are shown in black. Black bars in the binding energy profile of SQV correspond to PR variants containing the G48V mutation, known to cause a high resistance to SQV (Maschera et al. 1996). Other potentially resistant variants (gray bars in Fig. 3A ▶) contain mutations I84V and I50V. The latter mutation is known to cause resistance to APV (Pazhanisamy et al. 1996) but not to SQV. Indeed, mutation I50V, either alone or in combination with substitutions in positions 10, 46, or 47, comprised the most resistant variants in the binding energy profile of APV with ΔEbind ≥ 3.0 kcal/mole (Fig. 3B ▶). Nonetheless, mutation I50V causes a 20-fold increase in the Ki value for SQV (Pazhanisamy et al. 1996; Markland et al. 2000), which is in good agreement with the calculated ΔEbind of about 2.0 kcal/mole (Table 1, Fig. 3A ▶). The quadruple mutant (M46I/G48V/I50V/I84L) that combines single-residue mutations determining resistance to both inhibitors appears to be highly resistant to SQV but sensitive or only slightly resistant to APV (ΔEbind = 3.8 and 0.9 kcal/mole for SQV and APV, respectively), which is consistent with the experimental observations (Markland et al. 2000). The model of the quadruple mutant complex with APV did not deviate significantly from the model of the WT complex (RMSD of 0.4 Å for heavy atoms of the ligand and binding site residues). Accommodation of the bulkier Leu 84 side chain in complex with APV resulted in tighter ligand packing in the vicinity of Val 50 and Pro 81. These small conformational rearrangements, together with favorable van der Waals interactions of APV with the Val 48 side chains, almost compensate for the ligand–protein van der Waals interactions lost because of the I50V mutation. Superposition of 3D models of the WT and the quadruple mutant PR in complex with SQV is shown in Figure 4 ▶. Conformational changes in the quadruple mutant complex with SQV were similar to those caused by the single G48V mutation (see Fig. 2 ▶) with similar displacements of the SQV quinoline ring. The RMSD of the ligand heavy atoms in both mutants is 0.4 Å compared with the RMSD of 0.7 Å between the ligand positions in WT and quadruple mutant models. However, additional mutations in positions 46 and 50 result in a more significant displacement of the flap residues: RMSD = 0.66 Å between Cα atoms of flap residues 46 to 54 in the single G48V mutant and the quadruple mutant complexes. Furthermore, the simultaneous substitution of bulkier residues in positions 46 and 48 may cause a rotamer transition of the Phe 53 side chain in the PR chain A from χ1 ≈ −60° to χ1 ≈ 60° (Fig. 4 ▶). In this case, unfavorable van der Waals interactions of the Val 48 side chains, combined with the losses in hydrogen bonding and electrostatic energy due to conformational changes in the complex, resulted in a high increase in binding energy consistent with the 300-fold increase in Ki (Markland et al. 2000).
Figure 3.
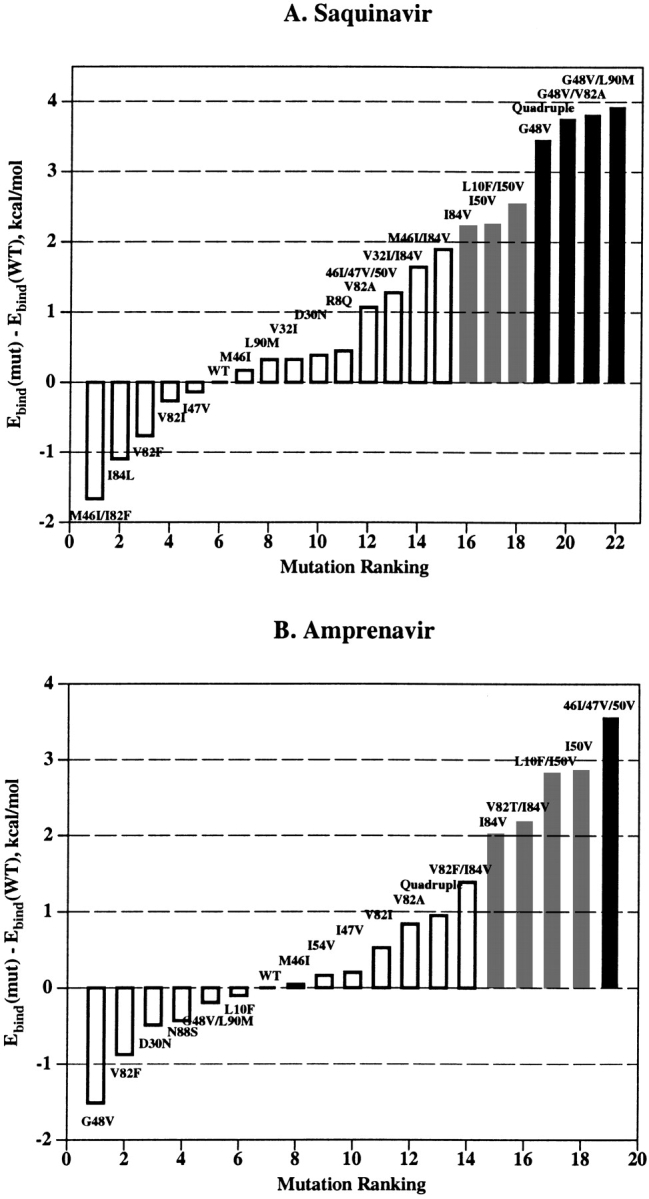
Binding energy profiles for SQV (A) and APV (B). PR mutations described in the literature are ranked by the increasing ΔEbind. Predicted resistant (ΔEbind ≥ 2.0 kcal/mole) and highly resistant (ΔEbind ≥ 3.0 kcal/mole) mutants are shown as the gray and the black bars, respectively.
Figure 4.

Superimposed stereo views of the model SQV complexes with the WT (yellow) and the quadruple mutant (M46I/G48V/I50V/I84L, white) HIV-1 PRs. The SQV molecules are shown as tube models. Hydrogen bonds of the ligand and flap water molecule in the mutant complex are displayed. Mutated residues and other important residues are labeled.
Correlation between calculated binding energies and cell-based phenotypes of PR variants from clinical HIV-1 isolates
The changes in binding energies of mutant versus WT complexes calculated for 65 clinical HIV-1 PR variants with five PIs and 48 PR variants with LPV were correlated with PhenoSense cell-based phenotypic resistance assays from ViroLogic Inc. (Petropoulos et al. 2000). Characteristics of the regression analyses are given in Table 2A, and a cumulative correlation plot for all drugs is shown in Figure 5 ▶. The correlation coefficients R2, ranging from 0.76 for NFV to 0.83 for APV and SQV, demonstrate statistically significant correlation with P < 0.001. Furthermore, the standard errors in prediction for these correlations ranged from 0.4 to 0.6 kcal/mole, corresponding to only a 2.0- to 2.5-fold standard error in predicted IC50 ratios (see the last column in Table 2A). The entire data set of 65 PR variants with six PIs gave a correlation with R2 = 0.76 and about a 2.5-fold standard error of prediction. Changes in binding energy calculated for a group of 63 PR variants were correlated with another commercially available phenotypic assay, the Antivirogram assay from Tibotec-Virco (Hertogs et al. 1998). Correlation coefficients R2 ranging from 0.53 (APV) to 0.81 (LPV) with 2.0- to 2.5-fold standard errors in resistance prediction were obtained for this group (see Table 2B). Generally, the correlations of calculated binding energies with the Antivirogram phenotypes were less significant than the correlations with the PhenoSense phenotypes, especially for APV. To obtain significant correlations for five PIs with the Antivirogram phenotypes, we needed to exclude several outliers, that is, the data points with large discrepancies between calculated and observed resistance measures. Most of the outlying data originated from a small number of PR variants, indicating possible discrepancies between the genotyped sequence and the viral amplicon that was phenotyped in the cell-based system. The correlation coefficients obtained for five of six PIs after outlier exclusion were of the same range as the correlations between two different phenotypic assays, Virco Antivirogram and Viralliance Phenoscript (Dam et al. 2001). Interestingly, the correlation between APV phenotypes obtained in the two cell-based assays also was not significant (R2 = 0.12; Dam et al. 2001).
Table 2.
Regression analysis of calculated ΔEbind and experimental changes in binding energya
| A: ViroLogic PhenoSense | ||||
| Inhibitor | N | R2 | S.E. (kcal/mole) | S.E. (fold)b |
| Amprenavir | 65 | 0.83 | 0.36 | 1.8 |
| Indinavir | 65 | 0.80 | 0.43 | 2.0 |
| Lopinavir | 48 | 0.81 | 0.46 | 2.1 |
| Nelfinavir | 65 | 0.76 | 0.50 | 2.3 |
| Ritonavir | 65 | 0.78 | 0.57 | 2.5 |
| Saquinavir | 65 | 0.83 | 0.45 | 2.1 |
| B: Virco Antivirogram | ||||
| Inhibitor | N (outliers)c | R2 (+outliers) | S.E. (+outliers) | S.E. (fold)b (+outliers) |
| a Experimental changes in the binding energy were estimated as RT ln(IC50mut/IC50wt). The number of data points (N), correlation coefficients (R2), and standard errors (S.E.) in predicted binding energies are given. | ||||
| b The standard error of prediction is expressed as a change in the fold resistance. | ||||
| c Outliers are data points that were excluded to obtain statistically significant correlations. Characteristics of correlations obtained with outliers are given in parentheses. | ||||
| Amprenavir | 63 (6) | 0.53 (0.34) | 0.42 (0.55) | 2.0 (2.4) |
| Indinavir | 63 (8) | 0.72 (0.51) | 0.43 (0.57) | 2.0 (2.5) |
| Lopinavir | 63 (3) | 0.81 (0.70) | 0.35 (0.44) | 1.8 (2.0) |
| Nelfinavir | 63 (2) | 0.70 (0.57) | 0.47 (0.58) | 2.1 (2.6) |
| Ritonavir | 63 (3) | 0.78 (0.68) | 0.54 (0.68) | 2.4 (3.0) |
| Saquinavir | 61 (5) | 0.66 (0.37) | 0.56 (0.77) | 2.5 (3.5) |
Figure 5.

Total correlation between the calculated changes in binding energies (ΔEbind ) of the WT vs. mutant HIV-1 PR–inhibitor complexes and the estimates of the changes in binding free energies obtained from experimental IC50 ratios (see equation 5 in Materials and Methods). The correlation plot includes 35 PR variants in complex with LPV and 65 PR variants in complex with five other PIs (total 360 data points). Experimental IC50 ratios were obtained from PhenoSence resistance assay (ViroLogic Inc., http://www.ViroLogic.com).
Semiquantitative resistance predictions
The ViroLogic PhenoSense data were used to group the variants into susceptible (less than fourfold increase in IC50) and resistant (fourfold or higher increase in IC50) classes for each PI. We determined the ΔEbind distributions and calculated the cutoffs c1 and c2 for semiquantitative prediction of susceptibility and resistance, as described in Materials and Methods (Fig. 6 ▶). PR variants were then classified as resistant, sensitive, or equivocal. Excellent concordances between the structural ratings and the PhenoSense ratings (95% sensitivity, 88% specificity; 0.567 ≤ kappa ≤ 0.907, P < 0.0001) were obtained for this training set of 65 PR variants with six PIs. The same structural cutoff values were then applied to Virco Antivirogram data and were able to predict phenotypic resistance to five of six PIs with a high degree of concordance (79% sensitivity, 88% specificity; 0.561 ≤ kappa ≤ 0.743, P < 0.0001). Good concordance between structural prediction and Antivirogram phenotype was not obtained for APV (50% sensitivity, 87% specificity; kappa = 0.367). For the Antivirogram assay, phenotypic changes in APV resistance also showed the poorest correlation with the calculated binding energies (see earlier).
Figure 6.

A semiquantitative model for a structure-based PI resistance assay. Distributions of calculated ΔEbind for PR variants phenotypically sensitive and resistant to six PIs were used to define the binding energy cutoffs c1 and c2 for prediction of sensitivity (S) or resistance (R) to each PI, respectively. Cases c1 ≤ ΔEbind ≤ c2 are considered equivocal (E).
In order to evaluate the performance of the semiquantitative energy cutoffs, binding energy calculations and blind resistance predictions were performed for an additional set of 78 PR samples. The predictions were then compared with phenotypic data available from PhenoSense (46 samples) and Antivirogram (32 samples) assays. Drug-specific semiquantitative cutoffs and the observed concordances are shown in Table 3. For both data sets, the number of equivocal predictions was small, and rules-based genotypic prediction agreed with the phenotypic predictions in most cases (Table 3). The specificity of the structural predictions was high for all drugs, but sensitivities were better for the PhenoSense phenotypes (Table 4). Overall agreement between the structural predictions was 92% (kappa = 0.756) for the PhenoSense phenotypes and 86% (kappa = 0.666) for the Antivirogram phenotypes. For the 46 discordances, 20 (43%) of the structural predictions were concordant with genotypic rules, whereas for 26 (57%) of the discordances genotypic rules agreed with the phenotypic predictions; the proportions were nearly the same for the PhenoSense and the Antivirogram phenotypes. A comparable concordance of 89% was reported in a study comparing these two experimental phenotypic assays (Qari et al. 2002). Another study comparing the Virco Antivirogram assay with the Viralliance Phenoscript assay demonstrated 87% overall concordance for 30 PR samples and five PIs (Dam et al. 2001). Therefore, we conclude that the qualitative structure-based resistance predictions agree well with the resistance class assignments of cell-based phenotypic assays.
Table 3.
Concordance of structural phenotype predictions with the results of two phenotypic assays
| A: ViroLogic PhenoSense | ||||||||
| ΔEbind cutoffsa | Phenotypic concordanceb | Equivocal predictionsd | ||||||
| PI | c1 | c2 | Sensitivity | Specificity | Kappac | P | Number | Genotypic concordance |
| APV | 0.7 | 1.4 | 62.5% | 100% | 0.730 | <0.0001 | 4/46 | 4/4 |
| IDV | 0.6 | 1.5 | 100% | 94.1% | 0.870 | <0.0001 | 3/46 | 3/3 |
| LPV | 0.3 | 0.7 | 100% | 87.1% | 0.652 | <0.0001 | 0/46 | NA |
| NFV | 0.7 | 1.0 | 64.3% | 96.9% | 0.665 | <0.0001 | 0/46 | NA |
| RTV | 0.7 | 1.4 | 72.7% | 100% | 0.800 | <0.0001 | 2/46 | 1/2 |
| SQV | 0.6 | 1.1 | 71.4% | 100% | 0.809 | <0.0001 | 0/46 | NA |
| B: Virco Antivirogram | ||||||||
| a ΔEbind cutoffs in kcal/mole established as described in Materials and Methods. | ||||||||
| b Sensitivity is the proportion of phenotypically resistant variants predicted to be resistant. Specificity is the proportion of phenotypically sensitive variants predicted to be sensitive. | ||||||||
| cKappa is a measure of inter-assay agreement (Fleiss 1981). Kappa > 0.75, excellent agreement; 0.4 < kappa < 0.75, good agreement; kappa < 0.4, poor agreement. | ||||||||
| d Genotypic predictions were performed in cases of equivocal structure-based predictions. The number of such predictions and their concordance with the phenotypic predictions are shown. | ||||||||
| APV | 0.7 | 1.4 | 85.7% | 95.5% | 0.812 | <0.0001 | 3/32 | 2/3 |
| IDV | 0.6 | 1.5 | 69.2% | 94.7% | 0.664 | <0.0001 | 0/32 | NA |
| LPV | 0.3 | 0.7 | 70.0% | 90.5% | 0.621 | <0.0003 | 1/32 | 1/1 |
| NFV | 0.7 | 1.0 | 72.7% | 88.9% | 0.627 | <0.0004 | 3/32 | 1/3 |
| RTV | 0.7 | 1.4 | 72.7% | 100% | 0.764 | <0.0001 | 4/32 | 2/4 |
| SQV | 0.6 | 1.1 | 50.0% | 95.5% | 0.510 | <0.002 | 0/32 | NA |
Table 4.
Overall concordance of structural phenotype with experimental phenotype for blind predictions
| PhenoSensea | Antivirogramb | |||
| Structural Prediction | S | R | S | R |
| S | 196 | 13 | 112 | 19 |
| R | 7 | 41 | 7 | 43 |
| Ec | 6 | 3 | 4 | 6 |
a 266 comparisons for 46 samples and 6 PIs. The overall agreement between structural phenotype and PhenoSense prediction of PI resistance was 92.2% (kappa = 0.756, P < 0.00001). (S) Sensitive; (R) resistant; (E) equivocal.
b 191 comparisons for 32 samples and 6 PIs. The overall agreement between structural phenotype and Antivirogram prediction of PI resistance was 85.6% (kappa = 0.666, P < 0.00001).
c Equivocal assignments where the change in binding energy was between the lower and the upper cutoffs. For equivocal structural predictions, genotypic resistance assignments agreed with the phenotypic predictions in 8 of 9 cases (PhenoSense) and 6 of 10 cases (Antivirogram).
Discussion
We have developed a computational approach for the prediction of resistance of clinical HIV-1 PR variants to PIs. This approach uses molecular modeling and binding energy evaluation for the WT and mutant PR–inhibitor complexes. The computational mutagenesis procedure can accurately reproduce the changes in 3D structure of the mutant complexes. Our model of the G48V/L90M mutant PR complex with SQV correctly reproduced significant changes in 3D structure with respect to the WT complex, including the ligand movement and conformational changes in the PR flap moiety (see Fig. 2 ▶), observed in the crystal structure of the same complex (Hong et al. 2000). The model of the IDV complex with the mutant V82T/I84V PR was also consistent with the crystal structure of a similar complex (King et al. 2002) and correctly predicted the experimentally observed change in the binding energy.
This study is based on the assumption that PI resistance is primarily determined by a reduction in binding affinity of PIs to mutant PR. This implies that mutations in the PR substrate cleft and in the flap regions are the major factors determining resistance to PIs. The influence of these mutations on affinity of PIs has been intensively investigated by site-directed mutagenesis and enzyme kinetic studies (Gulnik et al. 1995; Markowitz et al. 1995; Maschera et al. 1996; Pazhanisamy et al. 1996; Klabe et al. 1998), which showed significant increases in Ki for PR variants with known resistant mutations.
A second assumption of this study was that the changes in binding affinity might be accurately reproduced by changes in calculated binding energy of PR–inhibitor complexes. Previously we obtained statistically significant correlations with R2 ranging from 0.69 to 0.81 between changes in experimental binding affinities derived from Ki ratios and calculated binding energies for complexes of four PIs with PR variants containing primary resistance mutations (Shenderovich et al. 2001a). It should be noted that in these studies we did not attempt to develop a QSAR procedure to evaluate and compare binding energies of different PIs. We developed a computational procedure that calculated relatively small changes in the binding energy on PR side chain substitutions in reference WT PR–inhibitor complexes derived from respective crystal structures. The binding energy function that included nonweighted van der Waals, hydrogen bonding, side-chain entropy, and electrostatic Coulomb and solvation terms (Schapira et al. 1999; Shenderovich et al. 2001b) was able to identify resistance mutations by a significant increase in binding energy. The primary resistance mutations (G48V for SQV, V82A for IDV and RTV, I50V for APV, D30N for NFV) caused more than a 1.5-kcal/mole increase in the binding energy of the respective PIs (Shenderovich et al. 2001b), whereas a strong (>100-fold) resistance could usually be predicted by more than a 3.0 kcal/mole increase in binding energy.
The majority of resistance-related mutations are conservative substitutions among hydrophobic side chains (Val, Ile, Leu, Met, Ala) that do not change the structure of the complex significantly, but modify van der Waals interactions between the ligand and the side chains of the PR active site. Respectively, changes in the binding affinity of PIs on resistance-related single and double mutations are mostly explained by changes in the van der Waals component of the binding energy (see Table 1). These calculations were able to predict both additive and differential effects of combined primary mutations on the resistance to different PIs. For example, the binding energy profile for IDV (not shown) predicts about 1.0 and 1.5 kcal/mole increases in the binding energy for single mutations V82T and I84V, respectively. A combination of two point mutations results in an additive 2.4 kcal/mole increase in binding energy of IDV (Table 1), which is consistent with the 2.5 kcal/mole increase in the binding enthalpy observed for a PR variant containing V82T/I84V mutations (King et al. 2002). Such additivity often appears when the WT side chains are substituted with less bulky ones and the structure of the mutant complex does not change significantly. On the other hand, a combination of the G48V mutation, which was highly resistant to SQV (Maschera et al. 1996), with the I50V mutation, which was highly resistant to APV and moderately resistant to SQV (Pazhanisamy et al. 1996), resulted in the quadruple mutant, M46I/G48V/I50V/I84L, which was highly resistant to SQV but, surprisingly, sensitive to APV (Markland et al. 2000). This effect was correctly reproduced by computational mutagenesis (see Results).
Generally, accumulation of several resistance-related active site and flap region mutations may cause significant structural changes of mutant PR–inhibitor complexes, which result in a partial disruption of the hydrogen bond network between the ligand and protein, as reflected in the increased hydrogen bonding component of the binding energy function. This is the case for the quadruple mutant complex with SQV and for the triple M46I/I47V/I50V mutant complex with APV (see Table 1). On the other hand, except for rare mutations that involve charged and polar residues located close to the active site (Arg 8, Asp 30, and Asn 88), Coulomb and electrostatic solvation components do not make significant contributions to changes in binding energy of mutant complexes. This is probably due to the specific structure of PR–inhibitor complexes, with a ligand completely buried between the mostly hydrophobic residues of the PR binding cleft and the flap loops (Wlodawer and Vondrasek 1998).
The presence of a deep, well-defined binding pocket, the prevalence of van der Waals and hydrogen bonding ligand–protein interactions, and the availability of numerous crystal structures that can be used both as templates and as benchmarks have made HIV-1 PR complexes a common target for molecular modeling and docking simulations. Quantitative structure–activity relationship studies based on various binding energy functions have been generally successful in obtaining significant correlation equations for a series of structurally related PIs (Holloway et al. 1995; Perez et al. 1998; Nair et al. 2002), but have had a limited predictive power for a more diverse set of inhibitors or for mutant PR variants. Successful attempts have been made to calculate binding free energies of PR substrates and inhibitors using molecular dynamics simulations and thermodynamic cycle calculations with empirical binding energy functions (Dominy and Brooks III 1999; McCarric and Kollman 1999). Although these methods supply relatively accurate binding energies, they are impractical for the routine screening of multiple PR variants against commercially available drugs. Molecular modeling of mutant PR–inhibitor complexes based on the crystal structures of WT complexes and evaluation of changes in the binding energy of mutant versus WT complexes using molecular mechanics binding energy functions were performed by Weber and Harrison (1999). They obtained significant correlations (R = 0.79 and 0.68 for SQV and IDV, respectively) between changes in binding energy and binding affinity calculated from Ki ratios for SQV and IDV complexes with nine single and double mutant PR variants. In our preliminary study (Shenderovich et al. 2001a,b), we modeled IDV and SQV complexes with 17 and 18 PR variants, respectively (including mutations studied by Weber and Harrison), and obtained stronger correlations between changes in binding energy and in binding affinity (R = 0.79 and 0.84, respectively). We also obtained significant correlations (R = 0.90) for two other commercial PIs, RTV and APV.
An attempt to predict HIV-1 PR resistance to five FDA-approved PIs using a complex binding energy function and a simplified molecular modeling approach was recently published by Wang and Kollman (2001). They calculated residue contributions (ΔGres) to the binding free energy of WT complexes, and used a product of ΔGres and residue variability to predict resistance of single residue mutations to five PIs. Mutant PR–inhibitor complexes were not modeled, and therefore any conformational changes that could influence binding energy were neglected. The residue contributions calculated for WT sequences neglected the nature of mutant residues and underestimated the consequences of substitution of a bulky side chain for a smaller one. In particular, this study failed to predict resistance of the G48V mutant PR to SQV. As was shown earlier, introduction of Val 48 in the PR–SQV complex causes significant steric strain that is accompanied by conformational changes in the complex. These effects could not be predicted by considering interactions of the parent Gly 48 residues only.
The primary aim of this study was to develop a relatively simple computational procedure for rapid discrimination between resistant and sensitive PR variants. A typical set of clinical PR variants included 20% to 30% of phenotypically resistant PR–inhibitor complexes that might undergo significant structural changes with respect to the WT complexes. Accumulation of multiple resistance-related mutations that may differentially affect binding modes of various ligands is often the case for clinical PR variants that emerge under therapeutic regimens involving multiple PIs. The total number of amino acids mutated with respect to a consensus WT sequence may amount to up to 20 for each of two 99 amino acid chains of the homodimeric HIV PR, and some clinical PR variants may contain more than 10 side-chain substitutions in the vicinity of the inhibitor binding site. Evaluation of structural and energetic effects of multiple mutations required the development of a stepwise computational procedure, which first introduces and locally optimizes all side-chain substitutions, gradually moving from the mutations closest to the ligand toward the periphery of the protein, and then minimizes the molecular mechanics energy of the complex over a flexible ligand and protein amino acids in the vicinity of the ligand. The minimization shell around the ligand (see Materials and Methods) should be large enough to include all mutations that may directly or indirectly influence ligand binding, but should be relatively narrow to avoid significant deviations from the WT complexes derived from the crystallographic coordinates. The computational protocol was separately optimized for each PI to get the best correlations between calculated and experimental binding energy for the training set totaling 127 PR variants obtained from clinical HIV-1. Furthermore, in the preliminary study (Shenderovich et al. 2001a,b), we found that optimization of the crystal structures of WT complexes that involved a Monte Carlo search and energy minimization (see Materials and Methods) was necessary to achieve significant correlation between experimental and calculated changes in binding energies. Modifications introduced in nonrefined crystallographic structures usually did not reproduce experimental changes in binding energies estimated from Ki ratios for engineered single and double mutant PR variants.
The computational mutagenesis procedure developed in this study was able to reproduce changes in binding affinity of clinical PR variants, as shown by statistically significant correlations between calculated changes in binding energy and experimental measurements of drug resistance. We obtained good overall correlation (R2 = 0.76) between calculated changes in binding energy and experimental measures of phenotypic resistance (ViroLogic PhenoSense assay), with a standard error corresponding to only a 2.5-fold change in resistance, comparable with the standard error of cell-based phenotypic assays. This level of accuracy resulted in well-separated distributions of ΔEbind that were used to determine binding energy cutoffs for phenotypically sensitive and resistant PR variants, allowing the qualitative classification of clinical isolates as susceptible or resistant to the six FDA-approved PIs, with a small number of equivocal assignments.
The remarkable concordance between the structural predictions and the measured resistance in two different cell-based assays indicates that changes in inhibitor binding affinity indeed explain most of the observed PI resistance, as was hypothesized. The failure to predict the phenotypic resistance in some cases may have been caused, in part, by mutations such as L90M (61% and 79% of the false negative calls for SQV and NFV, respectively) that do not greatly alter inhibitor binding affinity but may cause resistance by other mechanisms, such as decreasing the PR dimer stability (Xie et al. 1999; Hong et al. 2000). More than 40% of the predictions that were discordant with phenotype were nevertheless concordant with genotypic rules-based predictions of resistance or susceptibility. The inherent variability of cell-based phenotyping may be responsible for some of these variations. The differences in phenotypic technologies may also produce different results in a small number of cases (Qari et al. 2002). Structural predictions were complicated in some cases by the presence of multiple viral quasi-species encoding more than one amino acid variant at several PR codons that occasionally resulted in significantly different resistance predictions between these variants. In such cases, we arbitrarily selected the variant with the highest calculated ΔEbind for our blind predictions, which was not always the variant in closest agreement with the experimentally measured phenotypic resistance.
Conclusions
We have developed a computational protocol for molecular modeling and binding energy evaluation for the WT and mutant PR–inhibitor complexes, and a structure-based approach for the prediction of resistance of clinical HIV-1 PR variants to PIs. The computational mutagenesis procedure accurately reproduced the changes in 3D structure of mutant PR–inhibitor complexes and correctly predicted mutations in the PR binding site and flap region that produce HIV-1 variants resistant to particular PIs. Applied to clinical HIV variants with multiple mutations, the procedure resulted in statistically significant correlations between calculated changes in the inhibitor binding energies and fold changes in inhibitor IC50 values determined in experimental phenotypic assays. These correlations were used to define the binding energy cutoff values for semiquantitative prediction of HIV variants sensitive or resistant to each PI.
Our structure-based resistance predictions performed as well as the commercially available cell-based assays in categorizing viral variants as resistant or susceptible to PI inhibitors. The structure-based predictions may offer an alternative to rules-based genotyping for current PIs, and may be used as a rapid resistance prediction method for new PIs, for which genotypic rules are not yet known. The availability of a large database of clinical sequences and PR structures (the VARIOME database; Maggio and Ramnarayan 2001) can also enable investigators to perform rapid in silico screening of drugs under development to estimate the frequencies of resistant variants in the patient population. The structural phenotyping technology may be useful in prediction of probable resistance mutation patterns for prospective antiretroviral drug candidates prior to expensive clinical trials, and for selection of those drug candidates that are predicted to be most effective against viral variants resistant to the currently available drugs (Maggio et al. 2002). The computational mutagenesis technology may be extended to inhibitors of other viral and bacterial proteins, in which structure of the complex may be determined by X-ray crystallography or molecular modeling, and high variability of the target protein sequence is an issue for development of effective drugs. The similar approach may be applied for design and interpretation of site-directed mutagenesis studies of ligand interactions with protein targets of known 3D structure, and for design of ligands that would show selective affinities to protein targets with homologous active site structures.
Materials and methods
Wild-type HIV-1 PR–inhibitor complexes
WT complexes were built from crystal structures available in PDB. The PDB entries used were 1hxb for SQV, 1hsg for IDV, 1hxw for RTV, 1ohr for NFV, and 1hpv for APV. The crystal structure of the PR–LPV complex was obtained from Abbott Laboratories (PDB entry 1mui; Stoll et al. 2002). The chemical structures and abbreviations used for the inhibitors are shown in Figure 1 ▶. Covalent geometries of inhibitors were taken from the crystal structures and adjusted to ensure planarity of aromatic rings, rotational symmetry of particular groups, and so forth. The Gasteiger-Marsili algorithm (Gasteiger and Marsili 1980) was used to assign atomic charges to the ligands. Most of the inhibitors were considered to be electro-neutral compounds. IDV was protonated at piperazine nitrogen, which gave the best correlation with experimental binding energies in the preliminary study (Shenderovich et al. 2001a).
Molecular modeling and Monte Carlo simulations were performed using the ICM program, version 2.7 (Abagyan et al. 1994; MolSoft 1999), with the ECEPP/3 force field (Némethy et al. 1992). A regularization procedure (Maiorov and Abagyan 1998) was used to obtain relaxed structures of WT complexes with the standard ECEPP/3 amino acids and heavy atom RMSD of ≤0.5 Å from the crystal structures. Water molecules located in a 7.0 Å shell around the ligand were retained in the regularized complexes. A single “flap” water molecule (Wlodawer and Vondrasek 1998) was placed into the binding site of the PR–LPV complex. In order to find the optimal positions of ligands in the PR binding site with the force field used, Monte Carlo simulations with Minimization (Abagyan and Totrov 1994) were performed starting from the regularized crystal structures. Translational and rotational degrees of freedom, and single bond torsion angles of the ligand, as well as χ torsion angles of PR side chains located within 7.0 Å of any ligand atom were randomly perturbed during the Monte Carlo simulation steps. Movement of water molecules was allowed during the minimization. Protein–ligand interaction (binding) energies were calculated for the conformations accepted at 1000 K, and conformations with the lowest binding energies were selected as temporary models of the WT complex.
Modeling of mutant HIV-1 PR–inhibitor complexes
Modeling of mutant complexes was performed in two steps: (1) a search for an optimal conformation of each mutated side chain, and (2) optimization of the positions of the ligand and water molecules and conformations of the protein residues located near the binding site. The list of amino acid mutations was defined by alignment of the WT and mutant sequences. Individual mutations were introduced simultaneously in the two PR chains in the order of increasing distance from the mutated residues to the ligand. Initial conformation of a mutant side chain was “inherited” as much as possible from the WT side chain; that is, values of common χ torsion angles (like χ1 angled of all non-β-branched residues) were assigned to the mutant side chain. A systematic search procedure (MolSoft 1999) was applied to χ torsion angles not common for the WT and mutant side chains, which generated all combinations of three rotamers (±60° and 180°) for the torsion angles involved, minimized energy of each combination, and selected the lowest-energy conformation. The minimization involved all χ angles of the side chains located in a 5.0 Å shell (see following) around the mutant residue. After the local optimization for individual mutations, energy minimization was performed for a substructure involving the ligand, surrounding protein residues, and water molecules. The energy function used for the final minimization included ECEPP/3 van der Waals, hydrogen bonding and torsion potentials (Némethy et al. 1992), electrostatic potentials with a distance-dependent dielectric ɛ = 4.0rij, side-chain entropy, and atomic solvation energy (Abagyan and Totrov 1994). Molecular variables of the mutant complex included (1) translation and rotation variables and all torsion angles of the ligand; (2) backbone and side-chain torsion angles of PR residues that had at least one atom within a distance ≤R1 from any atom of the ligand (shell R1); (3) all torsion angles of PR residues that had at least one atom within a distance R2 from any atom of the mutated residues included in the shell R1 (shell R2); (4) translation and rotation variables of water molecules within shells R1 and R2. The radius R1 of the shell surrounding the ligand was set to 7.0 or 8.0 Å. The radii R2 of the secondary shells, which were included to relax the surroundings of mutated residues located at the periphery of the ligand-binding site, were varied between 0 and 5.0 Å.
The binding energies of PR–inhibitor complexes
Binding energies were estimated (Schapira et al. 1999, Shenderovich et al. 2001b) as
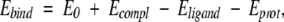 |
(1) |
where E0 is an adjustable constant, Ecompl is the energy of the complex, and Eligand and Eprot are the energies of the ligand and protein when separated. The components of the binding energy were calculated using the following energy function:
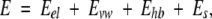 |
(2) |
where Eel is the exact-boundary electrostatics (Totrov and Abagyan 2001) that includes a Coulomb term and electrostatic solvation energy calculated with a dielectric constant ɛ = 8.0 (Schapira et al. 1999), Es is the side-chain entropy (Abagyan and Totrov 1994), and Evw and Ehb are the ECEPP/3 van der Waals and hydrogen-bonding terms.
For mutant PR–inhibitor complexes with known dissociation constants Ki, correlations between changes in the calculated binding energy
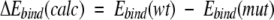 |
(3) |
and in the experimental estimate of binding free energy
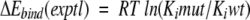 |
(4) |
were established by linear regression analysis (Shenderovich et al. 2001a). For clinical HIV-1 isolates, ratios of IC50 measured in cell-based phenotypic assays (Hertogs et al. 1998; Petropoulos et al. 2000), were used to calculate a surrogate of experimental binding energies
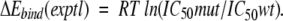 |
(5) |
For clinical HIV isolates containing a mixture of PR variants, separate ΔEbind values were calculated for all essential PR variants, and either the maximum or the best-fit individual ΔEbind(calc) value was used for correlation with experimental phenotypes.
Genotypic and phenotypic determinations
HIV-1 RNA was extracted from clinical samples submitted for RT and PR genotype determination to Quest Diagnostics Inc. or from 50 samples obtained from the California Collaborative Treatment Group 570 study (Haubrich et al. 2001) and genotyped at Quest Diagnostics Inc. The entire PR and the first 400 codons of RT were amplified by RT PCR and sequenced on an ABI 3700 capillary sequencer. Sequenced variants were aligned to an HIV-1 subtype B consensus sequence, and amino acid changes relative to the reference sequence were identified and tabulated. Genotypic resistance predictions were performed with the Quest-Diagnostics rules-based algorithm, an updated version of a published algorithm (Baxter et al. 2000). For the determination of the fold changes in IC50 values of six PIs, plasma samples were submitted for phenotypic resistance assays to ViroLogic Inc. (PhenoSense assay, http://www.ViroLogic.com) or to Tibotec-Virco (Antivirogram assay, http://www.tibotec-virco.com).
Structure-based resistance predictions
In order to define semiquantitative binding energy cutoffs for resistance predictions, the phenotypic data set for 65 clinical isolates (Virologic PhenoSense) was divided into sensitive (less than four-fold increase in phenotypic IC50) and resistant (four-fold or higher increase in phenotypic IC50) groups for each PI, and mean ΔEbind values and their standard deviations were calculated for all sensitive and resistant groups. The ΔEbind cutoff c1 defining susceptibility to each PI was set at two standard deviations above the mean of the group of samples sensitive to the respective PIs (Fig. 5 ▶). The cutoff c2 defining resistance was set at one standard deviation (1.5 S.D. for LPV) below the mean for the group of samples resistant to the respective PIs (Fig. 6 ▶). Cases of c1 ≤ ΔEbind ≤ c2 were considered equivocal. Inter-rater agreement between categorical resistance and susceptibility assignments was evaluated by the Kappa statistic (Fleiss 1981).
Acknowledgments
We gratefully acknowledge Dr. Allen McCutchan, Dr. Richard Haubrich, and their colleagues in the California Collaborative Treatment Group of the University of California, San Diego, for generous provision of access to 50 specimens from the CCTG 570 study for use in this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
3D, three-dimensional
HIV-1, human immunodeficiency virus type 1
PR, protease
PI, protease inhibitor
APV, amprenavir
IDV, indinavir
LPV, lopinavir
NFV, nelfinavir
RTV, ritonavir
SQV, saquinavir
RT, reverse transcriptase
FDA, U.S. Food and Drug Administration
RMSD, root mean square deviation
QSAR, quantitative structure-activity relationship
WT, wild type
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0301103.
References
- Abagyan, R. and Totrov, M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1002. [DOI] [PubMed] [Google Scholar]
- Abagyan, R.A., Totrov, M., and Kuznetsov, D. 1994. ICM—a new method for protein modeling and design: Applications to docking and structure prediction from the disordered native conformation. J. Comput. Chem. 15 488–506. [Google Scholar]
- Baldwin, E.T., Bhat, T.N., Liu, B., Pattabiraman, N., and Erickson, J.W. 1995. Structural basis of drug resistance for the V82A mutant of HIV-1 proteinase. Nat. Struct. Biol. 2 244–249. [DOI] [PubMed] [Google Scholar]
- Baxter, J.D., Mayers, D.L., Wentworth, D.N., Neaton, J.D., Hoover, M.L., Winters, M.A., Mannheimer, S.B., Thompson, M.A., Abrams, D.I., Brizz, B.J., et al. 2000. A randomized study of antiretroviral management based on plasma genotypic retroviral resistance testing in patients failing therapy. AIDS 14 F83–93. [DOI] [PubMed] [Google Scholar]
- Carpenter, C.C., Cooper, D.A., Fischl, M.A., Gatell, J.M., Gazzard, B.G., Hammer, S.M., Hirsch, M.S., Jacobsen, D.M., Katzenstein, D.A., Montaner, J.S., et al. 2000. Antiretroviral therapy in adults: Updated recommendations of the International AIDS Society-USA Panel. JAMA 283 381–390. [DOI] [PubMed] [Google Scholar]
- Carrillo, A., Stewart, K.D., Sham, H.L., Norbeck, D.W., Kohlbrenner, W.E., Leonard, J.M., Kempf, D.J., and Molla, A. 1998. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-387, a novel protease inhibitor. J. Virol. 72 7532–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, C., Hunt, S., Sension, M., Farthing, C., Conant, M., Jacobson, S., Nadler, J., Verbiest, W., Hertogs, K., Amesi, M., et al. 2002. A randomized trial assessing the impact of phenotypic resistance testing on antiretroviral therapy. AIDS 16 1–10. [DOI] [PubMed] [Google Scholar]
- Dam, E., Clavel, F., Calvez, V., Salmon, D., Pellegrin, J.L., Mamet, J.P., Antoun, Z., and Vauthier, J.M. 2001. Comparison of HIV-1 resistance phenotypes obtained by two different assay systems. 5th International Workshop on HIV Drug Resistance & Treatment Strategies. Scottsdale, AZ, poster #158.
- Dominy, B.N. and Brooks III, C.L. 1999. Methodology for protein ligand binding studies: Application to a model for drug resistance, the HIV/FIV protease system. Proteins 36 318–331. [DOI] [PubMed] [Google Scholar]
- Fleiss, J.L. 1981. Statistical methods for rates and proportions, 2nd ed., pp. 38–46. John Wiley, New York.
- Gasteiger, J. and Marsili, M. 1980. Iterative partial equalization of orbital electronegativities: A rapid access to atomic charges. Tetrahedron 36 3219–3228. [Google Scholar]
- Gulnik, S.V., Suvorov, L.I., Liu, B., Yu, B., Anderson, B., Mitsuya, H., and Erickson, J.W. 1995. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry 34 9282–9287. [DOI] [PubMed] [Google Scholar]
- Gulnik, S., Erickson, J.W., and Xie, D. 2000. HIV protease: Enzyme function and drug resistance. Vitam. Horm. 58 213–256. [DOI] [PubMed] [Google Scholar]
- Haubrich, R.H., Currier, J.S., Forthal, D.N., Beall, G., Kemper, C.A., Johnson, D., Dube, M.P., Hwang, J., Leedom, J.M., Tilles, J., et al. 2001. A randomized study of the utility of human immunodeficiency virus RNA measurement for the management of antiretroviral therapy. Clin. Infect. Dis. 33 1060–1068. [DOI] [PubMed] [Google Scholar]
- Hertogs, K., de Bethune, M.-P., Miller, V., Ivens, T., Schel, P., van Cauwenberge, A., van den Eynde, C., van Gerwen, V., Azin, H., van Houtte, M., et al. 1998. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob. Agents Chemother. 42 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, M.S., Brun-Vezinet, F., D’Aquila, R.T., Hammer, S.M., Johnson, V.A., Kuritzkes, D.R., Loveday, C., Mellors, J.W., Clotet, B., Conway, B., et al. 2000. Antiretroviral drug resistance testing in adult HIV-1 infection: Recommendations of an International AIDS Society-USA Panel. JAMA 283 2417–2426. [DOI] [PubMed] [Google Scholar]
- Holloway, M.K., Wai, J.M., Halgren, T.A., Fitzgerald, P.M.D., Vacca, J.R., Dorsey, B.D., Levin, R.B., Thompson, W.J., Chen, L.J., deSolms, S.J., et al. 1995. A priori prediction of activity for HIV-1 protease inhibitors employing energy minimization in the active site. J. Med. Chem. 38 305–317. [DOI] [PubMed] [Google Scholar]
- Hong, L., Zhang, X.C., Hartsuck, J.A., and Tang, J. 2000. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: Insight into the mechanisms of drug resistance. Protein Sci. 9 1898–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, N.M., Melnick, L., Prabu-Jeyabalan, M., Nalivaika, E., Yang, S.-S., Gao, Y., Zepp, C., Heefner, D.D., and Schiffer, C.A. 2002. Lack of synergy for inhibitor targeting a multi-drug-resistant HIV-1 protease. Protein Sci. 11 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabe, R.M., Bachler, L.T., Ala, P.J., Erickson-Viitanen, S., and Meek, J.L. 1998. Resistance to HIV protease inhibitors: A comparison of enzyme inhibition and antiviral potency. Biochemistry 37 8735–8742. [DOI] [PubMed] [Google Scholar]
- Kohl, N.E., Emini, E.A., Schleif, W.A., Davis, L.J., Heimbach, J.C., Dixon, R.A., Scolnick, E.M., and Sigal, I.S. 1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. 85 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio, E.T. and Ramnarayan, K. 2001. Recent developments in computational proteomics. Trends Biotechnol. 19 266–272. [DOI] [PubMed] [Google Scholar]
- Maggio, E.T., Shenderovich, M., Kagan, R., Goddette, D., and Ramnarayan, K. 2002. Structural pharmacogenomics, drug resistance and design of anti-infective super-drugs. Drug Discov. Today 7 1214–1220. [DOI] [PubMed] [Google Scholar]
- Mahalingam, B., Louis, J.M., Hung, J., Harrison, R., and Weber, I.T. 2001. Structural implications of drug-resistant mutations of HIV-1 protease: High-resolution crystal structures of the mutant protease/substrate analogue complexes. Proteins 43 455–464. [DOI] [PubMed] [Google Scholar]
- Maiorov, V. and Abagyan, R. 1998. Energy strain in three-dimensional protein structures. Fold. Des. 3 259–269. [DOI] [PubMed] [Google Scholar]
- Markland, W., Rao, B.G., Parsons, J.D., Black, J., Zuchowski, L., Tisdale, M., and Tung, R. 2000. Structural and kinetic analyses of the protease from an amprenavir-resistant human immunodeficiency virus type 1 mutant rendered resistant to saquinavir and resensitized to amprenavir. J. Virol. 74 7636–7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz, M., Mo, H., Kempf, D.J., Norbeck, D.W., Bhat, T.N., Erickson, J.W., and Ho, D.D. 1995. Selection and analysis of human immunodeficiency virus type 1 variants with increased resistance to ABT-538, a novel protease inhibitor. J. Virol. 69 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschera, B., Darby, G., Palu, G., Wright, L., Tisdale, M., Myers, R., Blair, E.D., and Furfine, E.S. 1996. Human immunodeficiency virus. Mutations in the viral protease that confer resistance to saquinavir increase the dissociation rate constant of the protease-saquinavir complex. J. Biol. Chem. 271 33231–33235. [DOI] [PubMed] [Google Scholar]
- McCarric, M.A. and Kollman, P.A. 1999. Predicting relative binding affinities of non-peptide HIV protease inhibitors with free energy perturbation calculations. J. Comput. Aided Mol. Des. 13 109–121. [DOI] [PubMed] [Google Scholar]
- Molsoft, LLC. 1999. ICM 2.7 program manual. MolSoft, San Diego, CA.
- Nair, A.C., Jayatilleke, P., Wang, X., Miertus, S., and Welsh, W.J. 2002. Computational studies on tetrahydropyrimidine-2-one HIV-1 protease inhibitors: Improving three-dimensional quantitative structure-activity relationship comparative molecular field analysis models by inclusion of calculated inhibitor- and receptor-based properties. J. Med. Chem. 45 973–983. [DOI] [PubMed] [Google Scholar]
- Némethy, G., Gibson, K.D., Palmer, K.A., Yoong, C.N., Paterlini, G., Zagari, A., Rumsey, S., and Scheraga, H.A. 1992. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J. Phys. Chem. 96 6472–6484. [Google Scholar]
- Palella Jr., F.J., Delaney, K.M., Moorman, A.C., Loveless, M.O., Fuhrer, J., Satten, G.A., Aschman, D.J., and Holmberg, S.D. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338 853–860. [DOI] [PubMed] [Google Scholar]
- Partaledis, J.A., Yamaguchi, K., Tisdale, M., Blair, E., Falcione, C., Maschera, B., Myers, R.E., Pazhanisamy, S., Futer, O., Cullinan, A.B., et al. 1995. In vitro selection and characterization of human immunodeficiency virus type 1 (HIV-1) isolates with reduced sensitivity to hydroxyethylamino sulfonamide inhibitors of HIV-1 aspartyl protease. J. Virol. 69 5228–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patick, A.K., Mo, H., Markowitz, M., Appelt, K., Wu, B., Musick, L., Kalish, V., Kaldor, S., Reich, S., Ho, D., et al. 1996. Antiviral resistance studies of AG1343, an orally bioavailable inhibitor of human immunodeficiency virus protease. Antimicrob. Agents Chemother. 40 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazhanisamy, S., Stuver, C.M., Cullinan, A.B., Margolin, N., and Rao, B.G. 1996. Kinetic characterization of human immunodeficiency virus type-1 protease-resistant variants. J. Biol. Chem. 271 17979–17985. [DOI] [PubMed] [Google Scholar]
- Perez, C., Pastor, M., Ortiz, A.R., and Gago, F. 1998. Comparative binding energy analysis of HIV-1 protease inhibitors: Incorporation of solvent effects and validation as a powerful tool in receptor-based drug design. J. Med. Chem. 41 836–852. [DOI] [PubMed] [Google Scholar]
- Petropoulos, C.J., Parkin, N.T., Limoli, K.L., Lie, Y.S., Wrin, T., Huang, W., Tiai, H., Smith, D., Winslow, G.A., Capon, D.J., et al. 2000. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 44 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qari, S.H., Respess, R., Weinstock, H., Beltrami, E.M., Hertogs, K., Larder, B.A., Petropoulos, C.J., Hellmann, N., and Heneine, W. 2002. Comparative analysis of two commercial phenotypic assays for drug susceptibility testing of human immunodeficiency virus type 1. J. Clin. Microbiol. 40 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira, M., Totrov, M., and Abagyan, R. 1999. Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recognit. 12 177–186. [DOI] [PubMed] [Google Scholar]
- Schinazi, R.F., Larder, B.A., and Mellors, J.W. 2000. Mutations in retroviral genes associated with drug resistance: 2000–2001 update. International Antiviral News 8 65–91. [Google Scholar]
- Schock, H.B., Garsky, V.M., and Kuo, L.C. 1996. Mutational anatomy of an HIV-1 protease variant conferring cross-resistance to protease inhibitors in clinical trials. Compensatory modulations of binding and activity. J. Biol. Chem. 271 31957–31967. [DOI] [PubMed] [Google Scholar]
- Shafer, R.W. 2002. Genotypic testing for human immunodeficiency virus type 1 drug resistance. Clin. Microbiol. Rev. 15 247–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenderovich, M., Ramnarayan, K., Kagan, R., and Hess, P. 2001a. Structural pharmacogenomics approach to the evaluation of drug resistant mutations in HIV-1 protease. J. Clin. Ligand Assay 24 140–144. [Google Scholar]
- Shenderovich, M.D., Tseitin, V., Fisher, C.L., and Ramnarayan, K. 2001b. Molecular modeling and binding energy calculations for drug resistant mutations in HIV-1 protease-inhibitor complexes. In Peptides: The wave of the future. Proceedings of the 2nd International and the 17th American Peptide Symposium (eds. M. Lebl and R.A Houghten), pp. 418–419. American Peptide Society, San Diego, CA.
- Stoll, V., Qin, W., Stewart, K.D., Jakob, C., Park, C., Walter, K., Simmer, R.L., Helfrich, R., Bussiere, D., Kao, J., et al. 2002. X-ray crystallographic structure of ABT-378 (Lopinavir) bound to HIV-1 protease. Bioorg. Med. Chem. 10 2803–2806. [DOI] [PubMed] [Google Scholar]
- Tomasselli, A.G. and Heinrikson, R.L. 2000. Targeting the HIV-protease in AIDS therapy: A current clinical perspective. Biochim. Biophys. Acta 1477 189–214. [DOI] [PubMed] [Google Scholar]
- Totrov, M. and Abagyan, R. 2001. Rapid boundary element solvation electrostatics calculations in folding simulations: Successful folding of a 23-residue peptide. Biopolymers 60 124–133. [DOI] [PubMed] [Google Scholar]
- Wang, W. and Kollman, P.A. 2001. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. 98 14937–14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, I. and Harrison, R.W. 1999. Molecular mechanics analysis of drug-resistant mutants of HIV protease. Protein Eng. 12 469–474. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A. and Vondrasek, J. 1998. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 27 285–327. [DOI] [PubMed] [Google Scholar]
- Xie, D., Gulnik, S., Gustchina, E., Yu, B., Shao, W., Qoronfleh, W., Nathan, A., and Erickson, J.W. 1999. Drug resistance mutations can affect dimer stability of HIV-1 protease at neutral pH. Protein Sci. 8 1702–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]