

Abstract
Malate synthase, an enzyme of the glyoxylate pathway, catalyzes the condensation and subsequent hydrolysis of acetyl-coenzyme A (acetyl-CoA) and glyoxylate to form malate and CoA. In the present study, we present the 1.95 Å–resolution crystal structure of Escherichia coli malate synthase isoform G in complex with magnesium, pyruvate, and acetyl-CoA, and we compare it with previously determined structures of substrate and product complexes. The results reveal how the enzyme recognizes and activates the substrate acetyl-CoA, as well as conformational changes associated with substrate binding, which may be important for catalysis. On the basis of these results and mutagenesis of active site residues, Asp 631 and Arg 338 are proposed to act in concert to form the enolate anion of acetyl-CoA in the rate-limiting step. The highly conserved Cys 617, which is immediately adjacent to the presumed catalytic base Asp 631, appears to be oxidized to cysteine-sulfenic acid. This can explain earlier observations of the susceptibility of the enzyme to inactivation and aggregation upon X-ray irradiation and indicates that cysteine oxidation may play a role in redox regulation of malate synthase activity in vivo. There is mounting evidence that enzymes of the glyoxylate pathway are virulence factors in several pathogenic organisms, notably Mycobacterium tuberculosis and Candida albicans. The results described in this study add insight into the mechanism of catalysis and may be useful for the design of inhibitory compounds as possible antimicrobial agents.
Keywords: Malate synthase, citrate synthase, protein crystallography, hydrogen abstraction, cysteine-sulfenic acid
In the glyoxylate cycle, isocitrate is cleaved by isocitrate lyase to form glyoxylate and succinate, whereas malate synthase catalyzes the Claisen condensation of glyoxylate and acetyl-coenzyme A (acetyl-CoA) to form malate. In this manner, two oxidative steps of the Krebs cycle are bypassed, retaining carbon that would otherwise be lost as CO2 (Kornberg and Krebs 1957). The glyoxylate cycle thus permits plants, some worms, and various unicellular organisms (Cioni et al. 1981), including Archaea (Oren and Gurevich 1995; Uhrigshardt et al. 2002), to use two-carbon compounds for biosynthetic purposes.
Recently, glyoxylate shunt enzymes have been implicated as virulence factors in several different pathogens, for example, Mycobacterium tuberculosis (Graham and Clark-Curtiss 1999; Honer zu Bentrup et al. 1999; McKinney et al. 2000), Candida albicans (Lorenz and Fink 2001), Rhodococcus fascians (Vereecke et al. 2002), and Pseudomonas aeruginosa (Ha and Jin 1999). These organisms respectively cause tuberculosis, fungal infections in humans, leafy gall formation in plants, and, in the case of P. aeruginosa, opportunistic infections in burn wounds or immunocompromised patients. In M. tuberculosis and C. albicans, it has been reported that genes encoding glyoxylate shunt enzymes are up-regulated in response to phagocytosis, indicating that this may be important for survival in the environment within the phagolysosome (Honer zu Bentrup et al. 1999; McKinney et al. 2000; Lorenz and Fink 2001). In R. fascians and P. aeruginosa, glyoxylate and other aldo- and keto-acids are expected to be present at elevated levels, in which case induction of the glyoxylate shunt genes may function to prevent toxic buildup of these compounds (Ha and Jin 1999; Vereecke et al. 2002). The glyoxylate cycle has not been found in mammals; thus, these enzymes form excellent targets for drug design.
Malate synthases described to date fall into two major families, isoforms A and G. The ∼80-kD monomeric malate synthase isoform G (MSG) has been found only in bacteria, whereas the oligomeric malate synthase isoform A (MSA, ∼65 kD per subunit) occurs in plants and several other organisms. Recently, two examples of malate synthase have been discovered in Archaea. Both are oligomeric and may belong to at least one new isoform (Serrano et al. 1998; Serrano and Bonete 2001; Uhrigshardt et al. 2002).
MSG is a magnesium-dependent (Dixon et al. 1960) enzyme based on the TIM-barrel fold. It contains an insertion that forms a separate α/β domain and an additional C-terminal helical plug (Howard et al. 2000; Smith et al. 2003). The TIM barrel and C-terminal plug are presumed to be essential for catalysis as the cleft between them forms the active site. The α/β domain, which has no known function, appears to be absent from MSA (Howard et al. 2000).
Crystal structures of MSG in complex with glyoxylate have been determined for enzymes from E. coli (Howard et al. 2000) and M. tuberculosis (Smith et al. 2003), giving a clear picture of how this substrate is activated upon binding to the enzyme. Bound glyoxylate is polarized by hydrogen bonds to main- and side-chains on the enzyme, as well as salt links to bound Mg2+, activating the carbonyl carbon of glyoxylate for nucleophilic attack. Smith et al. (2003) have also described the structure of a complex with malate and CoA, permitting some hypotheses to be formed regarding how the proposed reaction intermediate malyl-CoA might be hydrolyzed to form products.
The mode of acetyl-CoA binding and the means of activating this substrate remained to be determined. In this study, we describe how the enzyme binds acetyl-CoA and the competitive inhibitor pyruvate. Comparison to previously determined structures reveals conformational changes of an active-site loop that may be important for catalysis. A highly conserved active site residue, Cys 617, appears to be oxidized to cysteine-sulfenic acid. Oxidation of Cys 617 can explain earlier observations of the susceptibility of the enzyme to inactivation and aggregation upon X-ray irradiation (Zipper and Durchschlag 1981) and indicates that this residue might play a role in redox regulation of enzyme activity in vivo.
Results
Overall structure
Malate synthase crystallized in space group P212121 with unit cell dimensions of a = 73.9 Å, b = 107.4 Å, and c = 205.0 Å and with two molecules in the asymmetric unit. The final model includes two molecules of pyruvate, magnesium, and acetyl-CoA; four molecules of polyethylene glycol; and 583 water molecules. The crystallographic R-factor is 0.197 for all data, and the model has acceptable stereochemistry. Data collection and final refinement statistics are presented in Tables 1 and 2, respectively. The overall fold of malate synthase is as described previously (Howard et al. 2000; Smith et al. 2003). The enzyme contains a central TIM barrel buttressed on one side by an N-terminal α-helical clasp, on the other side by an α/β domain formed by two insertions into the TIM barrel sequence and terminating with a C-terminal five-helix plug (Fig. 1 ▶). For the A molecule, electron density permitted residues 4–150, 159–302, and 308–722 to be modeled but was weak for residues 151–158 and 303–307. Because of weak electron density, 31 side-chains were truncated to some extent. In the B molecule, residues 8–151, 156–302, and 309–722 could be modeled, although 45 side-chains were truncated. Electron density is present but weak for residues 152–155 and 303–308. As defined by PROCHECK (Laskowski et al. 1993), there are no residues with disallowed backbone conformational angles and only four (Ser A8, Tyr B219, Glu B272, and Ala B585) in "generously allowed" regions of the ϕ/ξ diagram. Electron density is clear for bound acetyl-CoA (Fig. 2A ▶) and pyruvate (Fig. 2B ▶), although breaks in density occur on either side of the sulfur of acetyl-CoA and between the ribose ring and 5′ phosphates. Electron density peaks >4 σ were observed within atomic bonding distance to the sulfur atoms in Cys 617 in both A and B molecules, as well as on Cys 688 molecule B. These three residues were successfully modeled as cysteine-sulfenic acids as evidenced by the lack of significant peaks in Fo-Fc difference electron density maps at these locations.
Table 1.
Data collection statistics
| Total reflections | 591,192 |
| Unique reflections | 108,075 |
| Spacegroup | P212121 |
| Cell dimensions (Å) | 73.9, 107.4, 205.0 |
| Resolution (Å) | 39.22–1.95 |
| Highest resolution shell (Å) | 2.06–1.95 |
| Completeness (%) | 90.3 (90.3) |
| Multiplicity | 3.3 (3.3) |
| Average I/σ | 10.4 (3.4) |
| Rmerge | 0.051 (0.212) |
Values in parentheses indicate the statistics for the highest resolution shell.
Table 2.
Crystallographic refinement statistics
| Resolution (Å) | 39.22–1.95 |
| Number of protein moleculesa | 2 |
| Number of protein atomsa | 10,773 |
| Number of solvent atomsa,b | 616 |
| Number of substrate atomsc | 116 |
| Crystallographic Rfactor/Rfreed | 19.4/29.4 |
| Average B-factors (Å2) | 30.7 |
| Protein B-factors (Å2) | 30.4 |
| Substrate B-factors (Å2)d | 53.6 |
| Solvent B-factors (Å2)c | 32.4 |
| Root mean square deviations: | |
| Bond lengths (Å) | 0.022 |
| Bond angles (degrees) | 2.7 |
| B-factor correlations (Å2) | 5.9 |
a Per asymmetric unit.
b Five hundred eighty-three water molecules and 33 polyethylene glycol atoms.
c Substrate defined as magnesium, pyruvate, and acetyl-coenzyme A.
d R = ∑∥Fobs| − |Fcalc∥/∑|Fobs|. Ten percent of the data were reserved for calculation of Rfree. In the deposited coordinates, all reflections were used in the final cycles of crystallographic refinement, with Rfactor = 19.7.
Figure 1.

Ribbon diagram of malate synthase. Domains are indicated by color: N-terminal α-helical clasp (blue), extended surface loop linker (turquoise), TIM barrel (red), α/β domain (yellow), and C-terminal plug (purple). Magnesium, pyruvate, and acetyl-coenzyme A are shown in green ball-and-stick form.
Figure 2.
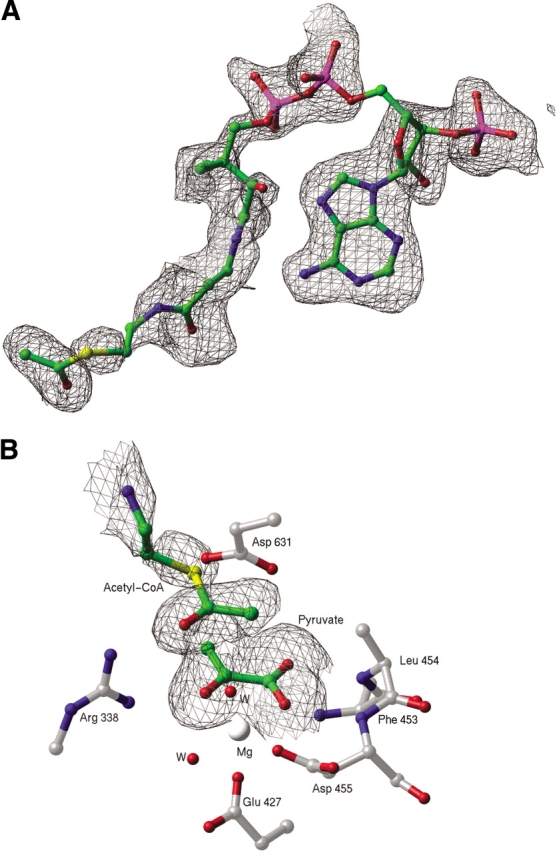
(A) Portion of an Fo-Fc electron density omit map, contoured at 2 σ. The refined model of acetyl-coenzyme A (acetyl-CoA), which was omitted for the purpose of map calculation, is superimposed and shown as a ball-and-stick model. (B) Portion of an Fo-Fc electron density omit map, contoured at 2 σ, showing bound pyruvate and the acetyl group of acetyl-CoA.
In a recent NMR study, it was found that Asn 305 of E. coli MSG forms an isoaspartyl linkage with Gly 306 (Tugarinov et al. 2002). Unfortunately, this loop is disordered in all crystal forms obtained to date, so we were unable to observe the conformation and environment of this unusual backbone component. As indicated by Tugarinov et al., backbone flexibility may be important for this posttranslational modification to occur.
The A and B molecules in the asymmetric unit were compared by using the programs EDPDB (Zhang and Matthews 1995), DynDom (Hayward and Berendsen 1998), and match3d. The overall root mean square deviation (RMSD) for Cα atoms is 0.41 Å. Small but significant conformational differences were found to cluster in three distinct areas: residues 150–250, 562–567, and 583–591. The elevated RMSD values of residues 150–250, which comprise the larger of the two segments of the α/β domain, can be accounted for by a small rigid body rotation of 3.8° and 0.1 Å translation of this domain with respect to the overall transformation relating the two molecules. Residues 562–567 and 583–591 are located in a connector between the TIM barrel and C-terminal plug and have higher than average temperature factors, which indicates flexibility. The observed positional differences are likely due to crystal contacts, which between the A and B molecules are different in these three regions.
Binding of acetyl-CoA
The binding site of acetyl-CoA is a remarkably deep (∼15 Å) and narrow pocket (Fig. 2C ▶ in Smith et al. 2003) located between the TIM barrel and the C-terminal plug (Fig. 1 ▶). Although the phosphate groups are partially exposed to solvent, the adenine ring and pantethenic acid tail are bent into a "J" shape inserted into the pocket such that the acetyl group closely approaches bound pyruvate. The adenine ring binds to a hydrophobic patch, wedged between TIM barrel residues Tyr 126 and Pro 538 (Fig. 3A ▶). Hydrogen bonds from the N6 position of the adenine ring are made to the backbone of TIM barrel residues Val 118 and Pro 536. The compact portion of the "J" configuration is characterized by an intramolecular hydrogen bond, which forms between the N7 of the adenine ring and a hydroxyl group on the pantetheine tail. These interactions are summarized in Figure 3B ▶.
Figure 3.
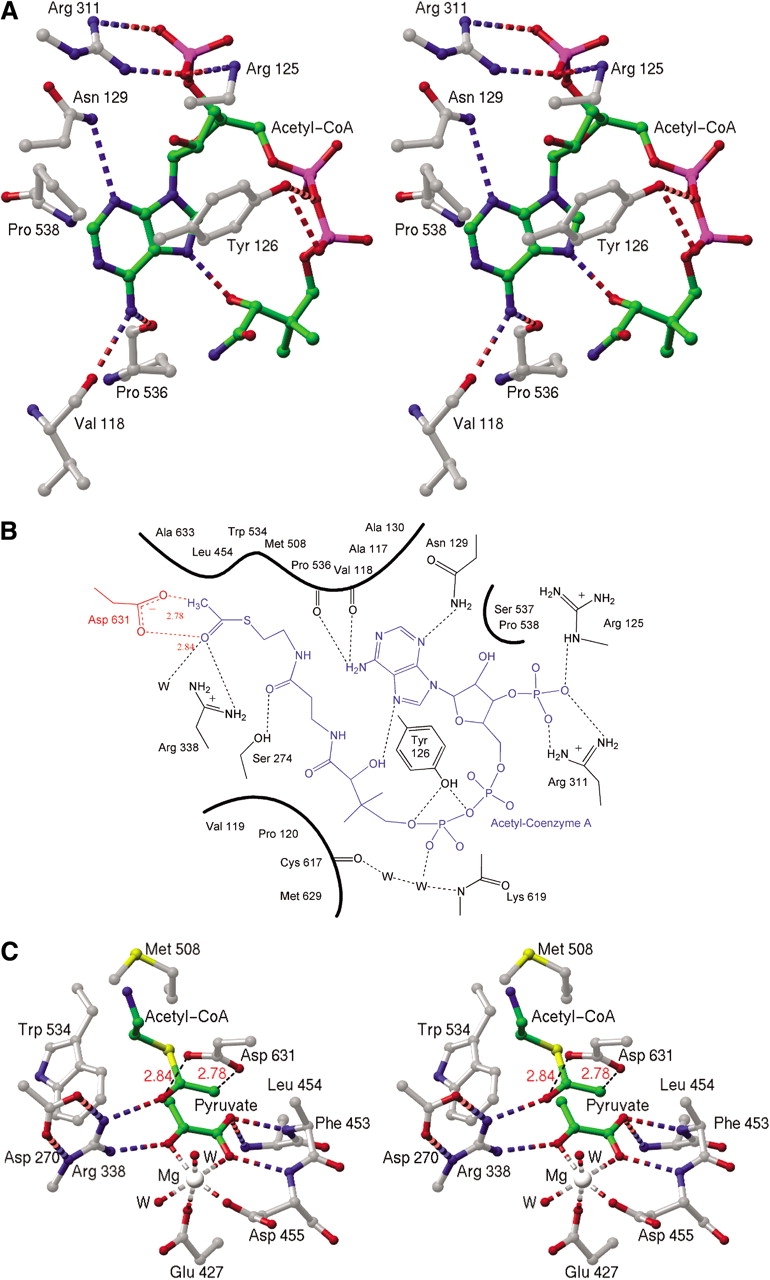
(A) Stereo view of the adenosine portion of the acetyl-coenzyme A (acetyl-CoA) binding site. Hydrogen bonds are shown as dashed cylinders. Green spheres represent carbon atoms of acetyl-CoA, and carbon atoms of the protein are shown in gray. (B) Schematic diagram of the acetyl-CoA binding site. For simplicity, the three-dimensional structure has been projected onto a plane. Acetyl-CoA is shown in blue, waters are shown as black W’s, and protein bonds are black except for the catalytic Asp 631, shown in red. Heavily shaded curves indicate hydrophobic interactions. Hydrogen bonds are shown as dashed lines, except for unfavorable contacts between Asp 631 and the terminal atoms of acetyl-CoA, which are represented with dashed red lines. (C) Stereo view of active site. As in A, but with unfavorable interaction as black dashed lines.
Active site
The active site, which is buried in MSG between the TIM barrel and the C-terminal plug (Fig. 1 ▶), is approached on one side via the previously described entrance tunnel forming the acetyl-CoA binding site. The side-chains of residues Phe 453 and Leu 454 block the opposite side of the active site. Pyruvate, which is bound essentially as described for glyoxylate (Howard et al. 2000), makes hydrogen bonds to the backbone (residues 453–455) and salt bridges to the side-chain of conserved Arg 338 (Fig. 3C ▶). The methyl group of pyruvate has hydrophobic interactions with the sulfur and carbonyl carbon of the acetyl terminus of acetyl-CoA; another hydrophobic interaction is with Trp 534. The methyl group of pyruvate also has unfavorable contacts with two structurally conserved water molecules located ∼3 Å away. The magnesium has nearly perfect octahedral coordination with interactions with pyruvate, Glu 427, Asp 455, and two water molecules. The surroundings are otherwise hydrophobic with Phe 453, Leu 454, Met 508, and Trp 534 located nearby.
Of particular interest are the interactions of the enzyme with the acetyl group of acetyl-CoA. Hydrogen bonds and salt bridges, which are presumably critical for catalysis, are formed between the carbonyl oxygen of the acetyl group and the side-chains of conserved residues. Arg 338 interacts directly with this atom, whereas Asp 273 interacts via a water molecule bound to the active site magnesium ion. It is quite striking that the proposed catalytic base Asp 631 makes very unfavorable (∼2.8 Å) contacts with the terminal methyl group and carbonyl oxygen of acetyl-CoA (Fig. 3B,C), indicating that these groups are forced together by the enzyme to facilitate proton abstraction (see Discussion).
Mutational analysis of active site residues
To determine the importance of certain active site residues, the mutations D631N, R338K, and C617S were constructed and the enzymes purified. Activity measurements and kinetic analyses were carried out on wild-type and mutant enzymes, and the results are presented in Table 3. Two of the mutants had substantially reduced activity. D631N had no detectable activity, consistent with its proposed role as the active site base, whereas the R338K mutant had a specific activity of 2.39 ± 0.07 μmole malate formed/min/mg protein, which is only 6.6% of the wild-type activity. Interestingly, in the C617S mutant, the Km for acetyl-CoA is 46 ± 3 μM, more than five times greater than that of wild type, although its specific activity was comparable.
Table 3.
Enzyme kinetic parameters
| Enzyme | Wild type | R338K | C617S | D631Nb |
| Specific activitya | 36.1 ± 0.5 | 2.39 ± 0.07 | 31.8 ± 0.5 | 0 |
| Wild-type activity (%) | 100 | 6.62 | 88.1 | 0 |
| kcat | 48.1 ± 0.6 s−1 | 3.19 ± 0.09 s−1 | 42.4 ± 0.7 s−1 | 0 |
| Km, glyoxylate | 21 ± 2 μM | 27 ± 5 μM | 50 ± 4 μM | ND |
| Km, acetyl-CoA | 9.0 ± 1.1 μM | 110 ± 20 μM | 46 ± 3 μM | ND |
| KI Pyruvate | 1.0 ± 0.2 mM | 1.6 ± 0.5 mM | 1.00 ± 0.09 mM | ND |
a Specific activity in terms of μmole malate formed/min/mg protein at 37°C.
b No activity was detected for the D631N mutant under any conditions assayed.
Discussion
Binding site of acetyl-CoA
Cofactors such as ATP (Saraste et al. 1990), FAD (Dym and Eisenberg 2001), and NAD(P) (Wierenga et al. 1986; Carugo and Argos 1997) are well known to interact with their cognate enzymes via recognizable structural and/or amino acid sequence motifs. However, despite the ubiquitous appearance of CoA derivatives in enzymatic reactions, there appears to be no strictly conserved sequence or structural motif for CoA binding (Engel and Wierenga 1996). Therefore, it is of interest to closely examine new examples of CoA binding sites. Denessiouk et al. (2001) proposed a sequence-independent adenine binding motif, first identified in citrate synthase (Remington et al. 1982), which may be found in ATP, CoA, NAD(P), and FAD-binding proteins. The motif uses a three-residue loop to form hydrogen bonds between the protein backbone to the adenine ring, whereas a fourth residue may provide hydrophobic interactions. Malate synthase does not seem to use this motif for recognition of CoA. However, as commonly found in other CoA-dependent enzymes, the recognition of the adenine ring is accomplished primarily through hydrogen bonds to the polypeptide backbone and hydrophobic interactions with a small number of side-chains (Fig. 3A,B).
Although the residues that interact with acetyl-CoA are well conserved in the known MSG sequences, the actual mode of CoA binding is more highly conserved than one might expect. The M. tuberculosis MSG (TBMSG; Smith et al. 2003) structures reveal that the E. coli and M. tuberculosis enzymes have essentially identical backbone conformations despite only 56% sequence identity. There is only a single amino acid difference in the CoA-binding sites of the two enzymes. Tyr 126 in E. coli MSG (ECMSG) is replaced by phenylalanine in TBMSG, which is unable to form the hydrogen bond with the 5′ phosphates observed for Tyr 126 in ECMSG. The conformation of the CoA moiety is essentially the same in the two structures, although the first isopeptide group from the acetyl terminus of acetyl-CoA is flipped ∼180° in TBMSG. The corresponding carbonyl oxygen is clearly defined by the electron density map (Fig. 2A ▶). Given the lower resolution (2.7 Å) of the TBMSG study, this conformational difference may derive from a difference in interpretation of the electron density maps. There are also minor positional differences at the 5′ phosphates, which is to be expected, as in ECMSG these are able to interact with the side-chain of Tyr 126 and as a result are slightly closer to the protein surface.
Although sequence identity is high within individual isoforms of malate synthase, between isoforms A and G sequence identity drops to ∼15% (Serrano and Bonete 2001). This combined with the size disparity between the two classes and the absence of a representative MSA structure prevents us from speculating on how this isoform interacts with acetyl-CoA.
CoA-binding modes among mechanistically or structurally related enzymes
Methylmalonyl-CoA mutase is the only other TIM barrel–containing protein for which the structure in complex with CoA or an analog is available (Mancia et al. 1996). This enzyme is a 150-kD heterodimer that catalyzes the conversion of succinyl-CoA to methylmalonyl-CoA. Although sequence homology between methylmalonyl-CoA mutase and malate synthase is not significantly above the background, comparison of the atomic models reveals that in both cases, the adenine rings make hydrogen bonds to the protein backbone and bound water molecules and are wedged between two hydrophobic residues. However, despite the similarities in overall folds of the two enzymes, the position and conformation of the bound CoA moiety in each enzyme is remarkably different. Methylmalonyl-CoA mutase binds the partial substrate desulpho-CoA inside the TIM barrel along its axis, which is allowed by the somewhat unusual feature of the interior of the barrel being lined with small, hydrophilic side-chains as opposed to bulky hydrophobic groups (Brandon and Tooze 1999). In addition, the conformation of CoA is linear rather than bent, as in MSG. This underscores the disparity in CoA recognition among different enzyme families, indicating that recognition of CoA by protein enzymes evolved independently many times.
Although citrate synthase and malate synthase catalyze essentially the same reaction, CoA binds differently to each enzyme. This is not surprising because these enzymes are unrelated in either amino acid sequence or three-dimensional structure (Howard et al. 2000). Citrate synthase binds acetyl-CoA via an "adenine recognition loop" and hydrophobic interactions with the adenine ring as previously described (Remington et al. 1982; Denessiouk et al. 2001). In both MSG and citrate synthase, the CoA conformation is compact and characterized by an intramolecular hydrogen bond between the adenine ring and the pantethenic acid tail. However, the configuration of CoA as bound to citrate synthase is "S" shaped due to an additional bend between the isopeptide linkages. This may be attributed to the different relationships between the catalytic and the adenine-binding sites.
Enzymatic activation of acetyl-CoA
The enzymatic abstraction of a proton from an acidic carbon is the rate-limiting step in reactions catalyzed by malate synthase (Lenz and Eggerer 1976; Clark et al. 1988); citrate synthase (Lenz et al. 1971); and other enzymes, for example, triose phosphate isomerase (for a review, see Remington 1992) and in nonenzymic base-catalyzed aldol condensation (Bove et al. 1959). Further evidence of a stepwise reaction in malate synthase is that α-ketoacids closely resembling glyoxylate, such as the competitive inhibitor pyruvate, stimulate the rate of enolization but do not proceed to condensation (Eggerer and Klette 1967). A central question concerns that nature of the reactive intermediate formed, which has been proposed to be either the enolate anion or the neutral enol of acetyl-CoA.
There is some experimental evidence in support of an enolate intermediate for malate synthase. Fluoroacetyl-CoA is a substrate for both citrate and malate synthase. In the latter case, (2R, 3R) and (2R, 3S) 3-fluoromalate are produced in a 3:4 ratio (Keck et al. 1980; Marletta et al. 1981). This implies that malate synthase does not strongly distinguish between the pro-R and pro-S hydrogens in the rate-limiting step. Theoretical calculations provide evidence that the Z and E enolates are approximately isoenergetic, so that an enzyme should produce these intermediates with equal likelihood. On the other hand, the Z enol is calculated to be more stable than the E enol (O’Hagan and Rzepa 1994), in which case an enzyme should efficiently produce the former. On the basis of these considerations, formation of the Z enol intermediate has been taken by O’Hagan and Rzepa (1994) to explain the observation that citrate synthase forms only the (2R,3R) stereoisomer of fluorocitrate (Fanshier et al. 1964).
Howard et al. (2000) proposed that in malate synthase Asp 631 acts as the base, abstracting a proton to form the enol(ate) of acetyl-CoA, which in turn is stabilized by the presence of positively charged Arg 338. This proposal is strongly supported by the observation that in the ternary complex, the carboxylate of Asp 631 makes an unfavorable 2.8 Å contact with the terminal methyl group of acetyl-CoA, similar to that reported for citrate synthase (Karpusas et al. 1990, 1991; Usher et al. 1994). Proton abstraction by Asp 631 would take place on the opposite side of the methyl group from the substrate glyoxylate (Fig. 3C ▶), in accord with the observed stereochemical inversion of proton configuration on the methyl group of acetyl-CoA (Cornforth et al. 1969; Luthy et al. 1969). In turn, Arg 338 is ideally positioned to interact simultaneously with the carbonyl oxygens of both substrates (Fig. 3B,C).
This indicates that Asp 631 acts to deprotonate the methyl group of acetyl-CoA, whereas Arg 338 acts to stabilize the intermediate that is formed. The central question is whether the intermediate is protonated by Arg 338. The pKa for arginine in solution is 12.48 (Dawson et al. 1986), whereas the pKaE for the neutral enol is estimated by analogy to ketones to be 10 to 11 (Chiang and Kresge 1991). Arg 338 must be charged as it forms a salt bridge with Asp 270 (Fig. 3C ▶). Therefore, the pKa of Arg 338 on the enzyme is expected to be substantially higher than the solution value, making protonation of the enolate by Arg 338 highly unlikely. The available evidence favors the proposal that in the malate synthase reaction, the enolate of acetyl-CoA (Fig. 4 ▶) is the reactive intermediate.
Figure 4.
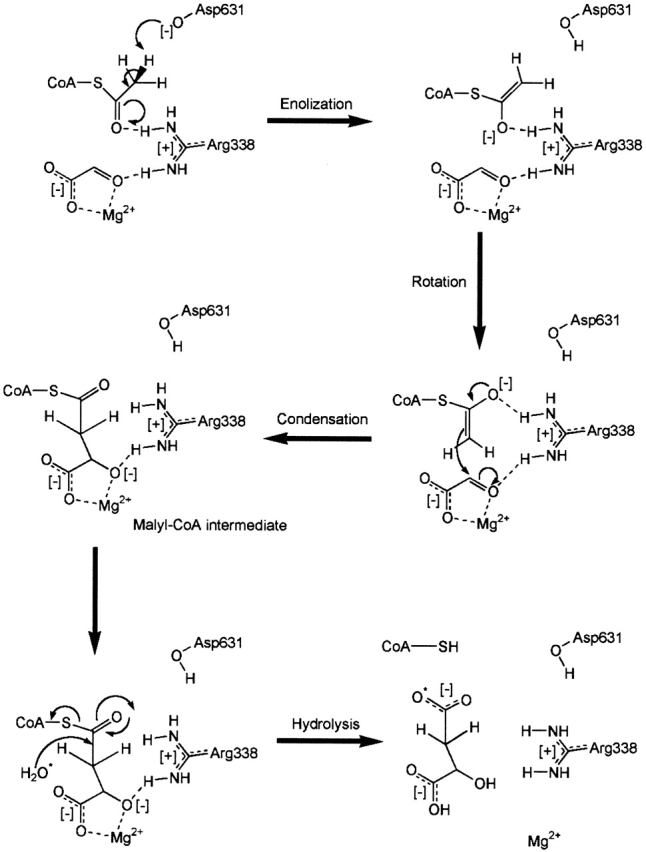
Proposed catalytic mechanism of malate synthase G, adapted from Howard et al. (2000).
Preliminary mutational analysis and kinetic studies of ECMSG were performed in order to test these hypotheses, and the results are presented in Table 3. Asp 631 is absolutely essential, and replacement with Asn results in complete loss of catalytic function, in keeping with the inability of Asn to act as a proton acceptor. On the other hand, Arg 338 is not essential. The relatively conservative replacement of Arg 338 with lysine reduced catalysis to 6.6% of wild-type activity. The Km for glyoxylate is essentially unchanged by the R338K substitution, but Km for acetyl-CoA is increased by over an order of magnitude. We interpret this to mean that Lys 338 interacts strongly with glyoxylate and fulfills the role of Arg 338 in this regard, but interacts less well with acetyl-CoA, resulting in weaker binding. The positive charge is probably very important for enzymatic rate enhancement, but proton transfer from position 338 to the intermediate does not seem to be required for catalysis. Goh et al. (2001) report similar results with Streptomyces coelicolor MSA. The mutations D453A and R171L (analogous to the ECMSG Asp 631 and Arg 338, respectively) resulted in negligible catalytic activity.
A second major question in the reaction mechanism of malate synthase is how the malyl-CoA intermediate is cleaved to produce the products malate and CoA. In the glyxoylate-bound, as well as the pyruvate and acetyl-CoA–bound structures, the water molecules involved with coordinating the magnesium form hydrogen bonds with either Asp 274 or Glu 273 (273 and 272 in ECMSG). In the product complex of TBMSG, the 2-hydroxyl of malate replaces the water coordinating the magnesium ion and hydrogen bonding Asp 274. Given the high conservation and proximity of Asp 274 and Glu 273 to this water molecule, these two residues were proposed to activate this water toward hydrolysis of the malyl-CoA intermediate (Smith et al. 2003). Although this hypothesis is intriguing, some issues need to be addressed. First, the magnesium ion is proposed to be coordinated by different oxygen atoms in malate compared with glyoxylate, requiring reorientation of the substrate at some point in the catalytic cycle. Second, the proposed magnesium coordination in the product complex is distorted square pyramidal, which is not the preferred configuration. In all other MSG structures, the magnesium is octahedrally coordinated, and such coordination is the most preferred configuration found in the Cambridge Structural Database. Removal of a water from an octahedrally coordinated magnesium is energetically unfavorable (Markham et al. 2002).
Domain motions associated with acetyl-CoA binding
Measurements from small-angle X-ray scattering (SAXS) experiments were consistent with a decrease in the radius of gyration of yeast malate synthase upon substrate binding (Zipper and Durchschlag 1977). Circular dichroism (CD) studies on malate synthase from Zea mays provided evidence for conformational change upon glyoxylate binding (Beeckmans 1994). Similar studies with S. cerevisiae malate synthase (Schmid et al. 1974) evidenced a conformational change upon both glyoxylate and acetyl-CoA binding. On the basis of this and other considerations, Howard et al. (2000) proposed that the C-terminal domain might move to allow substrate entry and product release.
Three different studies, including this one, appear to have laid this proposal to rest. We found no evidence that complex formation with pyruvate and acetyl-CoA results in significant shifts in domain position, in agreement with results reported by Smith et al. (2003) for substrate and product complexes with the M. tuberculosis enzyme. NMR solution studies of ECMSG (Tugarinov and Kay 2003) are also in agreement with the crystallographic results, which seems to eliminate the objection that crystal contacts might inhibit domain motion.
Loop movement in the acetyl-CoA binding site
Upon binding of acetyl-CoA, an antiparallel loop of β-sheet (residues 616–632) swings up to 2.5 Å (Fig. 5 ▶) away from its position in the MSG:glyoxylate complex, and the side-chain of highly conserved Met 629 folds up against the loop (Fig. 5 ▶), making van der Waals contacts with the acetyl group of acetyl-CoA. Changes in 15N and 1HN chemical shifts are observed in the 616–632 loop region in response to both pyruvate and acetyl-CoA binding (Tugarinov and Kay 2003), as well as in the nearby 300–310 loop. Loop 300–310 is located near the 3′ phosphate of acetyl-CoA, but does not directly interact with the substrate itself. It is largely disordered in glyoxylate-bound structures (Howard et al. 2000; Smith et al. 2003), but several more residues become visible in structures containing CoA or acetyl-CoA. The formation of a salt bridge between the conserved Arg 311 and the 3′ phosphate may account for this increased order.
Figure 5.

Stereo view comparison of loop 616–632 between structure with (yellow) and without (blue) bound acetyl-coenzyme A.
Residues analogous to ECMSG loop 616–632 are displaced by a much smaller degree in the TBMSG stuctures (Smith et al. 2003) and the side-chain of Met 631 (analogous to ECMSG Met 629) is found in a conformation intermediate to that found in the two ECMSG structures (Fig. 5 ▶). The loop movement seen in ECMSG may be due in part to the formation of a cysteine-sulfenic acid on Cys 617. If Cys 617 is oxidized to cysteine-sulfenic acid, then Met 629 cannot be in the intermediate conformation seen in the TBMSG structures, as it would have unfavorable contact distances with the oxygen of the cysteine-sulfenic acid (Fig. 5 ▶; see below).
Possible formation of cysteine-sulfenic acid
Much attention has recently been directed toward the partial oxidation products of cysteine, such as cysteic acid, as a potential means of regulating enzyme activity, protein–protein interactions, or protein–nucleic acid interactions (for a review, see Claiborne et. al. 1999). There is some evidence that malate synthase is sensitive to environmental conditions that affect the oxidation state of the enzyme. Malate synthase is prone to aggregation and inactivation by X-ray irradiation, and both are partially reversible by addition of DTT (Durchschlag and Zipper 1988). Substrates and substrate analogs are potent inhibitors of aggregation and/or inactivation (Durchschlag et al. 1981; Durchschlag and Zipper 1988).
There is crystallographic evidence for formation of cysteine-sulfenic acid in the ECMSG crystals. Large peaks in Fo-Fc difference electron density maps were observed adjacent to the sulfur atom of Cys 617 and were successfully modeled as cysteine-sulfenic acid in both molecules of the asymmetric unit (Fig. 6 ▶). Previous work was reevaluated, and a similar electron density peak was found adjacent to Cys 617 in the structure of ECMSG:glyxoylate complex (Howard et al. 2000). The possibility of cysteine oxidation was not discussed in the TBMSG structures (Smith et al. 2003), and the analogous Cys 619 was modeled as cysteine. Inspection of the TBMSG atomic models (PDB ID code: 1N8I, 1N8W) reveals that presence of cysteine-sulfenic acid seems unlikely for reasons of steric interference by Met 631 (analogous to ECMSG Met 629)
Figure 6.

Stereo view of a portion of an Fo-Fcomit map contoured at 2.5 σ. The map shows additional electron density associated with Cys 617, interpreted to result from partial oxidation of Cys 617 to cysteine sulfenic acid. Asp 631 and Cys 617 were omitted for the purpose of phase calculation. Portions of the final refined model, including the terminal portion of acetyl-coenzyme A are superimposed. Ribbon and tube segments represent portions of the protein backbone.
The two most likely causes of cysteine oxidation are the presence of agents such as molecular oxygen in the crystallization solutions and radiation-induced damage. Both crystal structures were determined by using data collected at synchrotron sources, but ECMSG took months to crystallize versus weeks for TBMSG (Smith et al. 2003). Therefore, oxidation of Cys 617 by molecular oxygen seems the most likely scenario.
Given the close proximity of Cys 617 to both the catalytic base Asp 631 and acetyl-CoA, it is plausible that oxidation of the highly conserved cysteine 617 may be part of a redox regulatory mechanism. As an initial experiment to investigate this possibility, we constructed and prepared the C617S mutant, as it cannot undergo an analogous oxidative event and as it is occasionally observed in MSA sequences. C617S has 88% of wild-type–specific activity. However, the Km for glyoxylate increases by a factor of two (to 50 μM), whereas the acetyl-CoA Km increases by a factor of five (to 46 μM). kcat is slightly lowered and was found to be 42 s−1 versus 48 s−1 for wild type (Table 3). Serine at position 617 in ECMSG appears to be disadvantageous for enzymatic activity at low substrate concentrations. Given the larger size of cysteine-sulfenic acid, the effect of oxidation of Cys 617 could be substantial. Further investigation of the possibility of redox regulation of malate synthase activity is in progress.
Materials and methods
Protein preparation and crystallization
C-terminal hexahistidine-tagged ECMSG was prepared for crystallization as described previously (Howard et al. 2000), except that the Sephadex column was found to be unnecessary. Crystals were grown at room temperature in hanging drops containing 7 μL of protein solution containing 27.7 mg/mL ECMSG, 8.3 mM MgCl2, 8.3 mM Tris (pH 7.9), 4.2 mM DTT, 50 mM pyruvate, and 4.2 mM acetyl-CoA added to 7 μL of well solution containing 50 mM HEPES (pH 7.0), 150 mM sodium acetate, 5 mM MgCl2, and 16% PEG-8000 (Hampton). Resulting crystals were used to seed hanging drops under identical conditions. Crystals forming from this were, in turn, used for streak-seeding sitting drops at room temperature containing 7.5 μL protein solution containing the same buffer as before, but with 20 mg/mL protein and 7.5 μL of well solution containing 50 mM HEPES (pH 7.0), 150 mM sodium acetate, 5 mM MgCl2, and 23.5% PEG-8000 (Hampton). Crystals grew to ∼20 μm on a side, which were crushed and streak-seeded into room temperature hanging drops containing 7 μL protein solution and 7 μL well solution. Protein solution contained 28 mg/mL protein, 5.8 mM Tris (pH 7.9), 5.8 mM MgCl2, 50 mM pyruvate, 5 mM acetyl-CoA, and 3.4 mM DTT. Well solution contained 15% PEG-8000 (Hampton), 50 mM HEPES (pH 7.0), and 150 mM sodium acetate. Crystals grew to ∼0.08 mm across and up to 3 mm long after ∼3 months and were suitable for data collection.
Diffraction data collection and structure refinement
Diffraction data were collected from a single flash-frozen crystal at beamline 7–1 at the Stanford Synchrotron Radiation Laboratory (SSRL). Data were indexed and reduced by using the program MOSFLM (Leslie 1996), and intensities were scaled by using SCALA (CCP4 program suite). Molecular replacement solutions were found by using EPMR (Kissinger et al. 1999), using the structure of ECMSG with magnesium and glyoxylate (PDB ID code: 1D8C; Howard et al. 2000) as a search model. Model building was done by using O (Jones et al. 1991), and refinement was carried out with the program TNT (Tronrud et al. 1987) by using 90% of the data save for the last 20 rounds of refinement when all the data were used from 39.22 to 1.95 Å. Solvent molecules were placed where evidenced by large positive difference density features with appropriate proximity to hydrogen bond partners.
Mutagenesis, protein preparation, and kinetics
ECMSG mutants were prepared using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), using the ECMSG clone (Howard et al. 2000) as a template. All mutations were verified by DNA sequencing. Single colonies were picked, and 10 mL of culture grown in Luria-Burtani (LB) broth overnight was used to inoculate 1L fresh LB. Cultures were grown to an OD600 of 0.7 to 1.8 before induction with 0.125 mg/mL (final concentration) IPTG for 3 to 6 h. Cultures were immediately harvested by centrifugation and pellets were stored at −80°C overnight. Pellets were resuspended in 10 mL lysis buffer (50 mM HEPES at pH 7.9, 0.3 M NaCl, 10% glycerol, 10 mM MgCl2, and 3 mM β-mercaptoethanol) and lysed for 20 min on ice with lysozyme (Sigma) and DNase I (Roche). Protein was purified by using Ni-NTA Agarose (Qiagen), eluting with 50 mM HEPES (pH 7.9), 0.3 M NaCl, 100 mM imidazole, 10 mM MgCl2, and 3 mM β-mercaptoethanol. Protein was then dialyzed for 24 h with two buffer changes against 2 L of 50 mM HEPES (pH 7.9), 10 mM MgCl2, and 2 mM DTT and was then buffer-exchanged on a PD-10 Sepharose column (Pharmacia) into 25 mM potassium phosphate (pH 8.0), 10 mM MgCl2, 2 mM DTT, and 2 mM K2Mg EDTA. Protein purity was assayed by using precast 20% homogeneous SDS-PAGE (Amersham Biosciences).
The reaction of acetyl-CoA and glyoxylate was quantified by measuring the loss of absorbance at 232 nm (ɛ232acetyl CoA = 4500 M−1cm−1) due to cleavage of the thioester bond of acetyl-CoA (Eggerer and Klette 1967; Durchschlag et al. 1981). The reaction mixture—containing 0.2 mM acetyl-CoA, 0.5 mM glyoxylate, 50 mM potassium phosphate (pH 8.0), 10 mM MgCl2, and 2 mM K2Mg EDTA—was kept at 37°C in a volume of 500 μL, and the reaction initiated by addition of 10 μL of enzyme. Protein concentrations were ∼1 μg/mL final concentration for wild type and C617S, ∼10 μg/mL for R338K and up to 25 μg/mL for D631N, as determined by measuring absorbance at 280 nm. Molar extinction coefficients were determined by using ExPASy (Appel et al. 1994). At 37°C but not at 25°C (Beeckmans 1994), HEPES reacts rapidly with glyoxylate, which necessitated the change to a potassium phosphate/EDTA buffer. Kinetic parameters for substrates and the inhibitor pyruvate were estimated for wild-type and mutant proteins using the above conditions but with varying pyruvate and/or substrate concentrations. Least-squares fits were determined with the program Enzfitter (Biosoft). Figures were prepared by using Ribbons (Carson 1997) and Chemdraw.
Acknowledgments
This work was supported in part by NSF grant MCB 0111053 (to S.J.R.) and NIH Graduate Training Grant in Molecular Biology and Biophysics T32GM07759 (to D.M.A.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03174303.
References
- Appel, R.D., Baroch, A., and Hochstrasser, D.F. 1994. A new generation of information retrieval tools for biologists: The example of the ExPASy WWW server. Trends. Biochem. Sci. 19 258–261. [DOI] [PubMed] [Google Scholar]
- Beeckmans, S., Khan, A.S., Kanarek, L., and van Driessche, E. 1994. Ligand binding onto maize (Zea mays) malate synthase: A structural study. Biochem. J. 303 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove, J., Martin, R.O., Ingraham, L.L., and Stumpf, P.K. 1959. Studies of the mechanism of action of the condensing enzyme. J. Biol. Chem. 234 999–1003. [PubMed] [Google Scholar]
- Brandon, C. and Tooze, J. 1999. Introduction to protein structure. Garland Publishing, New York.
- Carson, M. 1997. Ribbons. Methods Enzymol. 277 493–505. [PubMed] [Google Scholar]
- Carugo, O. and Argos, P. 1997. NADP-dependent enzymes, I: Conserved stereochemistry of cofactor binding. Proteins 28 10–28. [DOI] [PubMed] [Google Scholar]
- Chiang, Y. and Kresge, A.J. 1991. Enols and other reactive species. Science 253 395–400. [DOI] [PubMed] [Google Scholar]
- Cioni, M., Pinzauti, G., and Vanni, P. 1981. Comparative biochemistry of the glyoxylate cycle. Comp. Biochem. Physiol. 70B 1–26. [Google Scholar]
- Claiborne, A., Yeh, J.I., Mallett, T.C., Luba, J., Crane Jr., E., Charrier, V., and Parsonage, D. 1999. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 38 15407–15416. [DOI] [PubMed] [Google Scholar]
- Clark, J.D., O"Keefe, S.J., and Knowles, J.R. 1988. Malate synthase: Proof of a stepwise Claisen condensation using the double-isotope fractionation test. Biochemistry 27 5961–5971. [DOI] [PubMed] [Google Scholar]
- Cornforth, J.W., Redmond, J.W., Eggerer, H., Buckel, W., and Gutschow, C. 1969. Asymmetric methyl groups, and the mechanism of malate synthase. Nature 221 1212–1213. [DOI] [PubMed] [Google Scholar]
- Dawson, R.M.C., Elliot, D.C., Elliot, W.H., and Jones, K.M. 1986. Data for biochemical research, 3rd ed. (eds. R.M.C. Dawson et al.), pp. 1–31. Oxford University Press, New York.
- Denessiouk, K.A., Rantanen, V.V., and Johnson, M.S. 2001. Adenine recognition: A motif present in ATP-, CoA-, NAD-, NADP-, and FAD-dependent proteins. Proteins 44 282–291. [DOI] [PubMed] [Google Scholar]
- Dixon, G.H., Kornberg, H.L., and Lund, P. 1960. Purification and properties of malate synthase. Biochim. Biophys. Acta 41 217–233. [DOI] [PubMed] [Google Scholar]
- Durchschlag, H. and Zipper, P. 1988. Primary and post-irradiation inactivation of the sulfhydryl enzyme malate synthase: Correlation of protective effects of additives. FEBS Lett. 237 208–212. [DOI] [PubMed] [Google Scholar]
- Durchschlag, H., Biedermann, G., and Eggerer, H. 1981. Large-scale purification and some properties of malate synthase from baker’s yeast. Eur. J. Biochem. 114 255–262. [DOI] [PubMed] [Google Scholar]
- Dym, O. and Eisenberg, D. 2001. Sequence-structure analysis of FAD-containing proteins. Protein Sci. 10 1712–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggerer, H. and Klette, A. 1967. The principle of catalysis of malate synthase. Eur. J. Biochem. 1 447–475. [DOI] [PubMed] [Google Scholar]
- Engel, C. and Wierenga, R. 1996. The diverse world of coenzyme A binding proteins. Curr. Opin. Struct. Biol. 6 790–797. [DOI] [PubMed] [Google Scholar]
- Fanshier, D.W., Gottwald, L.K., and Kun, E. 1964. Studies on specific enzyme inhibitors, VI: Characterization and mechanism of action of the enzyme-inhibitory isomer of monofluorocitrate. J. Biol. Chem. 239 425–434. [PubMed] [Google Scholar]
- Goh, L.L., Loke, P., and Sim, T.S. 2001. Replacement of arginine-171 and aspartate-453 in Streptomyces coelicolor malate synthase A by site-directed mutagenesis inactivates the enzyme. Appl. Microbiol. Biotech. 57 363–367. [DOI] [PubMed] [Google Scholar]
- Graham, J.E. and Clark-Curtiss, J.E. 1999. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. 96 11554–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, U. and Jin, S. 1999. Expression of the soxR Gene of Pseudomonas aeruginosa is inducible during infection of burn wounds in mice and is required to cause efficient bacteremia. Infect. Immun. 67 5324–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, S. and Berendsen, H.J. 1998. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 30 144–154. [PubMed] [Google Scholar]
- Honer zu Bentrup, K., Miczak, A., Swenson, D.L., and Russell, D.G. 1999. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J. Bacteriol. 181 7161–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, B.R., Endrizzi, J.A., and Remington, S.J. 2000. The crystal structure of Escherichia coli malate synthase G complexed with magnesium and glyoxylate at 2.0 Å resolution: Mechanistic implications. Biochemistry 39 3156–3168. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta. Crystallogr. A 47 110. [DOI] [PubMed] [Google Scholar]
- Karpusas, M., Branchaud, B., and Remington, S.J. 1990. Proposed mechanism for the condensation reaction of citrate synthase: 1.9 Å structure of the ternary complex with oxaloacetate and carboxymethyl coenzyme A. Biochemistry 29 2213–2219. [PubMed] [Google Scholar]
- Karpusas, M., Holland, D., and Remington, S.J. 1991. 1.9 Å structures of ternary complexes of citrate synthase with d- and l-malate: Mechanistic implications. Biochemistry 30 6024–6031. [DOI] [PubMed] [Google Scholar]
- Keck, R., Haas, H., and Retey, J. 1980. Synthesis of stereospecific deuterated fluoracetic acid and its behaviour in enzymic aldol-type condensations. FEBS Lett. 114 287–290. [DOI] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D 55 484–491. [DOI] [PubMed] [Google Scholar]
- Kornberg, H.L. and Krebs, H.A. 1957. Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature 179 988–991. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structure. J. Appl. Crystallogr. 26 283. [Google Scholar]
- Lenz, H. and Eggerer, H. 1976. Enzymic generation of chiral acetates: A quantitative evaluation of their configurational assay. Eur. J. Biochem. 65 237–246. [DOI] [PubMed] [Google Scholar]
- Lenz, H., Buckel, W., Wunderwald, P., Biedermann, G., Buschmeier, V., Eggerer, H., Cornforth, J.W., Redmond, J.W., and Mallaby, R. 1971. Stereochemistry of si-citrate synthase ant ATP-citrate-lyase reactions. Eur. J. Biochem. 24 207–215. [DOI] [PubMed] [Google Scholar]
- Leslie, A.G.W. 1996. Molecular data processing. In Crystallographic computing (eds. D. Moras, et al.), pp. 50–61. Oxford University Press, Oxford, UK.
- Lorenz, M.C. and Fink, G.R. 2001. The glyoxylate cycle is required for fungal virulence. Nature 412 83–86. [DOI] [PubMed] [Google Scholar]
- Luthy, J., Retey, J., and Arigoni, D. 1969. Preparation and detection of chiral methyl groups. Nature 221 1213–1215. [DOI] [PubMed] [Google Scholar]
- Mancia, F., Keep, N.H., Nakagawa, A., Leadlay, P.F., McSweeney, S., Rasmussen, B., Bosecke, P., Diat, O., and Evans, P.R. 1996. How coenzyme B12 radicals are generated: The crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 4 339–350. [DOI] [PubMed] [Google Scholar]
- Markham, G.D., Glusker, J.P., and Bock, C.W. 2002. The arrangement of first- and second-sphere water molecules in divalent magnesium complexes: Results from molecular orbital and density functional theory and from structural crystallography. J. Phys. Chem. B 106 5118–5134. [DOI] [PubMed] [Google Scholar]
- Marletta, M.A., Srere, P.A., and Walsh, C. 1981. Sterochemical outcome of processing of fluorinated substrates by ATP citrate lyase and malate synthase. Biochemistry 20 3719–3723. [DOI] [PubMed] [Google Scholar]
- McKinney, J.D., Honer zu Bentrup, K., Munoz-Elias, E.J., Miczak, A., Chen, B., Chan, W.T., Swenson, D., Sacchettini, J.C., Jacobs, W.R.J., and Russell, D.G. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406 683–685. [DOI] [PubMed] [Google Scholar]
- O’Hagan, D. and Rzepa, H.S. 1994. Stereoelectronic effects of fluorine in enzyme chemistry. Stereospecific control of citrate synthase mediated synthesis of (2R,3R) 3-fluorocitrate by the relative stabilities of the intermediate fluoroenolates. J. Chem. Soc. Chem. Commun. 2029–2030.
- Oren, A. and Gurevich, P. 1995. Isocitrate lyase activity in halophilic archaea. FEMS Microbiol. Lett. 130 91–95. [Google Scholar]
- Remington, S., Wiegand, G., and Huber, R. 1982. Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 Å resolution. J. Mol. Biol. 158 111–152. [DOI] [PubMed] [Google Scholar]
- Remington, S.J. 1992. Mechanisms of citrate synthase and related enzymes (triose phosphate isomerase and mandelate racemase). Curr. Biol. 2 730–735. [Google Scholar]
- Saraste, M., Sibbald, P.R., and Wittinghofer, A. 1990. The P-loop: A common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 15 430–434. [DOI] [PubMed] [Google Scholar]
- Schmid, G., Durchschlag, H., Biedermann, G., Eggerer, H., and Jaenicke, R. 1974. Molecular structure of malate synthase and structural changes upon ligand binding to the enzyme. Biochem. Biophys. Res. Commun. 58 419–426. [DOI] [PubMed] [Google Scholar]
- Serrano, J.A. and Bonete, M.J. 2001. Sequencing, phylogenetic and transcriptional analysis of the glyoxylate bypass operon (ace) in the halophilic archaeon Haloferax volcanii. Biochim. Biophys. Acta 1520 154–162. [DOI] [PubMed] [Google Scholar]
- Serrano, J.A., Camacho, M., and Bonete, M.J. 1998. Operation of glyoxylate cycle in halophilic archaea: Presence of malate synthase and isocitrate lyase in Haloferax volcanii. FEBS Lett. 434 13–16. [DOI] [PubMed] [Google Scholar]
- Smith, C.V., Huang, C.-C., Miczak, A., Russell, D.G., Sacchettini, J.C., and Honer zu Bentrup, K. 2003. Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis. J. Biol. Chem. 278 1735–1743. [DOI] [PubMed] [Google Scholar]
- Tronrud, D.E., Ten Eyck, L.F., and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Cryst. A43 489–503. [Google Scholar]
- Tugarinov, V. and Kay, L.E. 2003. Quantitative NMR studies of high molecular weight proteins: Application to domain orientation and ligand binding in the 723 residue enzyme malate synthase G. J. Mol. Biol. 327 1121–1133. [DOI] [PubMed] [Google Scholar]
- Tugarinov, V., Muhandiram, R., Ayed, A., and Kay, L.E. 2002. Four-dimensional NMR spectroscopy of a 723-residue protein: Chemical shift assignments and secondary structure of malate synthase g. J Am. Chem. Soc. 124 10025–10035. [DOI] [PubMed] [Google Scholar]
- Uhrigshardt, H., Walden, M., John, H., Petersen, A., and Anemuller, S. 2002. Evidence for an operative glyoxylate cycle in the thermoacidophilic crenarchaeon Sulfolobus acidocaldarius. FEBS Lett. 513 223–229. [DOI] [PubMed] [Google Scholar]
- Usher, K.C., Remington, S.J., Martin, D.P., and Drueckhammer, D.G. 1994. A very short hydrogen bond provides only moderate stabilization of an enzyme-inhibitor complex of citrate synthase. Biochemistry 33 7753–7759. [DOI] [PubMed] [Google Scholar]
- Vereecke, D., Cornelis, K., Temmerman, W., Jaziri, M., Van Montagu, M., Holsters, M., and Goethals, K. 2002. Chromosomal locus that affects pathogenicity of Rhodococcus fascians. J. Bacteriol. 184 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga, R.K., Terpstra, P., and Hol, W.G. 1986. Prediction of the occurrence of the ADP-binding βαβ-fold in proteins, using an amino acid sequence fingerprint. J. Mol. Biol. 187 101–107. [DOI] [PubMed] [Google Scholar]
- Zhang, X.J. and Matthews, B.W. 1995. EDPDB: A multifunctional tool for protein structure analysis. J. Appl. Crystallogr. 28 624–630. [Google Scholar]
- Zipper, P. and Durchschlag, H. 1977. Small-angle X-ray studies on malate synthase from baker’s yeast. Biochem. Biophys. Res. Commun. 75 394–400. [DOI] [PubMed] [Google Scholar]
- ———. 1981. Small angle X-ray scattering studies on the X-ray–induced aggregation of malate synthase II: Inactivation and aggregation experiments. Monatshefte für Chemie 112 1–23. [Google Scholar]