

Abstract
The PAAD/DAPIN/pyrin domain is the fourth member of the death domain superfamily, but unlike other members of this family, it is involved not only in apoptosis but also in innate immunity and several other processes. We have identified 40 PAAD domain-containing proteins by extensively searching the genomes of higher eukaryotes and viruses. Phylogenetic analyses suggest that there are five categories of PAAD domains that correlate with the domain architecture of the entire proteins. Homology models built on CARD and DD structures identified functionally important residues by studying conservation patterns on the surface of the models. Surface maps of each subfamily show different distributions of these residues, suggesting that domains from different subfamilies do not interact with each other, forming independent regulatory networks. Helix3 of PAAD is predicted to be critical for dimerization. Multiple alignment analysis and modeling suggest that it may be partly disordered, following a new paradigm for interaction proteins that are stabilized by protein–protein interactions.
Keywords: PAAD/DAPIN/Pyrin domain, phylogenetic analysis, homology modeling, function prediction
Apoptosis or programmed cell death is a major developmental process in animals and plants (Greenberg and Ausubel 1993; Jacobson et al. 1997; Aravind et al. 1999), and is important in pathogenesis (Fairbrother et al. 2001; Fiorentino et al. 2002; Stehlik et al. 2002). The proteins that signal and regulate apoptosis often have specific domains (Aravind et al. 1999), somewhat tongue-in-the-cheek often referred to as "apoptotic domains." Many such domains have been identified and extensively reviewed; one of the most comprehensive and up-to-date sources of information on this subject is the recently developed apoptosis database (Doctor et al. 2003). One of the best-characterized apoptotic domains is the death domain (DD)/death-effector domain (DED)/caspase-recruitment domain (CARD) superfamily (Huang et al. 1996; Hofmann et al. 1997; Eberstadt et al. 1998; Qin et al. 1999). This is a protein–protein interaction domain that links specific components of apoptosis’s regulatory pathways. Although the sequences of DD, DED, and CARD are very diverse, their X-ray crystallographic and nuclear magnetic resonance (NMR) structures show a similar fold of an antiparallel six-helical bundle with the Greek key topology (Liang and Fesik 1997). Therefore, it is only natural to speculate that they evolved from a common ancestor, a notion supported by their sequence similarity being recognized by distant homology and fold recognition programs (see discussion below).
Recently, a new protein domain called PAAD (Pawlowski et al. 2001), DAPIN (Staub et al. 2001), or pyrin (Bertin and DiStefano 2000; Fairbrother et al. 2001; Martinon et al. 2001) was identified by several groups as present in many proteins associated with apoptosis and other signaling pathways. Our group introduced the name PAAD: Pyrin, AIM (absent in melanoma), ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain [CARD]), and Death-domain (DD)-like. We did this to avoid confusion of the domain name with the protein named pyrin (Schaner et al. 2001), which is also called marenostrin (Booth et al. 1998), or the product of the MEFV gene (Mansfield et al. 2001). PAAD-domain proteins are so interesting because they potentially connect several important regulatory pathways, and despite their recent discovery, with over 100 manuscripts published last year (2002) alone, they have been a focus of very intensive research effort from several groups, including our own. There are several recent reviews in the field of PAAD domains and proteins with these domains (for instance, see Tschopp et al. 2003 and references therein); therefore, in the following, we provide only a short introduction to the field.
The PAAD domain was originally identified in multiple animal and viral proteins (Inohara et al. 2000; Pawlowski et al. 2001; Staub et al. 2001) using PSI-BLAST and other comparable tools. The current count exceeds 40 unique proteins, not including splicing variants. The PAAD domain is always found at the N-terminal and is followed by at least four types of domains, almost all of unknown functions. The PAAD domain in pyrin is followed by PRY and SPRY domains. In AIM2 (absent in melanoma 2), IFI (interferon inducible), and MNDA (myeloid cell nuclear differentiation antigen), the PAAD domains are followed by one or two HIN domains (Eddy 1998; Mulley 1999). The functions of the SPRY, PRY, and HIN domains are unknown. PAAD domains are also followed by NACHT domains and leucine-rich repeat domains (LRRs) in some proteins (Koonin and Aravind 2000; Tschopp et al. 2003). NACHT domains are predicted to be involved in nucleotide binding and protein oligomerization. Those proteins are known by several names: PAN (PAAD and nucleotide-binding), PYPAF (Grenier et al. 2002; Wang et al. 2002), DEFCAP (Fairbrother et al. 2001; Hlaing et al. 2001), NALP (Tschopp et al. 2003), and CATERPILLER (Harton et al. 2002)—which also serves as an example of how multiple naming conventions are a major problem in this rapidly growing field. Recent reviews shows members of this family are involved in apoptosis and inflammation (Tschopp et al. 2003). We have characterized several proteins from this group in our laboratory and shown that NAC protein is involved in cytochrome c-dependent caspase activation (Chu et al. 2001), and PAN2 mediates NF-κB suppression and is implicated in inflammatory responses (Fiorentino et al. 2002; Stehlik et al. 2002). The domain architecture in the ASC protein is different from the ones discussed so far; its PAAD domain is immediately followed by a CARD domain. ASC can activate or inhibit NF-κB depending on the cellular context (Stehlik et al. 2002). There are also proteins that are entirely composed of a PAAD domain: human PYC1 (ASC2) and four viral proteins from Myxoma, Rabbit fibroma, Yaba-like disease, and Swinepox viruses. ASC2 is currently a subject of NMR-based structural studies (Espejo et al. 2002), which at the time of this writing has not yielded a complete structure (N. Assa-Munt, pers. comm.).
Secondary structure predictions show that the PAAD domain contains six α helices; this supports results from several fold-recognition algorithms that suggests that the PAAD domain shares the fold of the death domain/CARD/DED superfamily. This would make the PAAD domain the fourth branch of the death domain/CARD/DED superfamily. Circular dichroism (Fairbrother et al. 2001) and NMR experiments (Espejo et al. 2002) support this conclusion further. Based on this, and on the position of the PAAD domain in larger proteins we hypothesized that the PAAD domain is a protein–protein interaction domain (Pawlowski et al. 2001; Fiorentino et al. 2002). This hypothesis was confirmed by several experimental studies (see below).
The DD, DED, and CARD families homotypically interact (Eberstadt et al. 1998; Qin et al. 1999; Xiao et al. 1999). Like their cousins, PAAD domains may homotypically interact: The PAAD domains are involved in ASC oligomerization (Masumoto et al. 2001; Richards et al. 2001), and the PAAD domains in MNDA self-associate (Xie et al. 1997). Therefore, PAAD domains most likely form specific homodimers analogous to the specific homodimers of the DD, DED, and CARD families. PAAD domains may guide the formation of specific complexes in the apoptotic and inflammatory pathways. The dimerization ability and the distribution of PAAD domains in various protein families makes them perfect candidates for coupling apoptotic, inflammatory, and uncharacterized pathways regulated by AIM2 and IFI.
Although we know and/or predict that PAAD domains self-associate, the experimental structure of a PAAD domain is still not available, and it is not known exactly where the dimerization interfaces are located in individual structures. It is also not known whether PAAD domains from various subgroups interact with each other or whether they show similar partner specificity as the DD, DED, and CARD families. In this article, we attempt to answer some of these questions by a combination of homology modeling, and model and phylogenetic analysis.
Materials and methods
We used the N-terminal 100 amino acids of pyrin (marenostrin) as the seed in a cascade of PSI-BLAST searches (Altschul et al. 1997). New hits were used as queries in subsequent searches (Li et al. 2000), and we verified that the selected sequences were PAAD family members by SMART (Schultz et al. 1998), PFAM (Sonnhammer et al. 1998), and FFAS (Rychlewski et al. 2000). Secondary structures were predicted with MetaServer (Bujnicki et al. 2001), and multiple alignments were prepared with T-COFFEE (Notredame et al. 2000) and then corrected by hand until consistent with the alignments to the structural templates (see below).
A profile–profile distant homology recognition methods FFAS (Rychlewski et al. 2000) was used to align PAAD family members to the DD/DED structural templates, assignment that was confirmed by other methods available at the MetaServer (Bujnicki et al. 2001). Even that such methods are often referred to as fold prediction, they are based on recognizing distant homologies, and as demonstrated on recent CASP meetings, alignments and models based on FFAS and other similar algorithms form a natural extension of homology modeling (Zemla et al. 2001).
We perform three different phylogenetic analyses to check the consistency of and explore the alternative topologies of the phylogenetic tree. We used the Neighbor Joining method and the Minimum Evolution method in 10,000 bootstrap replicates (Kumar et al. 2001), and the Bayesian approach (Huelsenbeck and Ronquist 2001; Huelsenbeck et al. 2001) for 1,000,000 generations in four independent Markov chains. When convergence was reached, a total number of 9000 trees were explored, and a consensus tree was generated with its corresponding clade confidence values. When distance (NJ tree) method was used, gaps were omitted in pairwise alignments; otherwise they were treated explicitly.
For the purposes of modeling, pairwise alignments between PAAD domains and the modeling templates were conducted using FFAS (Rychlewski et al. 2000). In distant homology recognition the alignment quality is a major concern; it was extensively studied in our group (Jaroszewski et al. 2000, 2002; Ye et al. 2003), and in this manuscript we follow the procedures described and benchmarked in these publications. In addition, the consistency between pairwise alignments between PAAD domains and the modeling templates was used to guide manual corrections in the multiple alignments of all PAAD domains (see Fig. 1 ▶). We obtained three-dimensional structure coordinate files of templates from the Protein Data Bank. The three-dimensional models were generated with JACKAL’s homology modeling method (Xiang 2001; Xiang and Honig 2002). These structures were fully minimized using the CHARM all-atoms force field to produce the final structures, which were evaluated by a PSQS (protein structure quality score). The PSQS is an energy-like measure for the quality of a protein structure, and is based on the statistical potentials of the mean interactive force between residue pairs and between single residues and solvent (P. Szczesny, unpubl.; http://www1.jcsg.org/psqs/psqs.cgi). The conserved surface patches were detected using ConSurf (Armon et al. 2001).
Figure 1.

Multiple alignment of 40 PAAD sequences. Yellow background indicates similarities between AIM and PYR/PAN group (as well as specific residues for AIM). Pink background shows differences/similarities between IFI and AIM groups. Red background shows similarities/differences between PAN5/PAN3 and PYR group, and cyan background shows similarities of PYR/AF427627 with the PYR members. Accession numbers for (1) PYR group: MEFV human gi|8928170, MEFV mouse gi|9506893, MEFV rat gi|13928876, ASC human gi|11096299, ASC pending mouse gi|12963605, ASC1 zebrafish 18858287, caspase zebrafish gi|7673640, PYC1 human gi|25024272, cryopyrin/AF427617 gi|17026378. For Fugu sequences, Genscan assembling was performed (http://genes.mit.edu/GENSCAN.html), then gi|CAAB01003190 fugu (Genscan predicted peptide 1 of 132 aa) and gi|CAAB01007457 (Genscan predicted peptide 2 of 195 aa). (2) PAN group: PAN1 human gi|8923473, PAN2 human gi|19031214, PAN3 human gi|19387136, PAN4 human gi|17483019, PAN5 human gi|17472937, PAN6 human gi|21711821, PAN7 human gi|18448933, PAN8 human gi|17461450, PAN10 human gi|21450725, PAN11 human gi|19882273. (3) AIM group: AIM2 human gi|4757734, AIM2 mouse gi|20830743, AIM2 rat gi|27678602. (4) IFI group: IFI203 mouse gi|6680355, IFI204 mouse gi|6680357, IFI205 mouse gi|20831393, MNDA human gi|730038, IFI16 human gi|5031779, IFI16 mouse gi|20830748, LOC226690 mouse gi|20830168, similar to IFI16 mouse gi|20920143, similar to IFI16 mouse gi|20830753. (5) Viral group: 18L Yaba-like gi|12085001, SPV014 Swinepox gi|18640100, GP013L rabbit fibroma virus gi|9633972, M)13L Myxoma gi|9633972. 1E41 (the top sequence) is the template selected for homology modeling. Pairwise alignments used for modeling can be obtained from the multiple alignment by reading appropriate lines: one for the target, one for the template.
Results and Discussion
Phylogenetic analyses
Over 40 instances of the PAAD domain were found in several vertebrate genomes and as part of several distinct protein families. The PAAD domain was not found in invertebrates and lower eukaryotes, so it appeared relatively recently in evolution. It can be found in several different combinations with other domains, combinations that cannot be explained by conventional gene duplication. Mechanisms such as domain accretion, shuffling, and gene conversion must have played a role in the evolution of the PAAD domain. Such mechanisms have been particularly active in proteins that are related to the immune response (Koonin and Aravind 2002).
In the case of PAAD domains, two factors must be considered to generate reliable alignments and trees. The first factor is that the sequences are extremely divergent making most pairwise alignments unreliable and affecting the quality of the input alignment. We addressed this problem by using T-COFFEE (Fig. 1 ▶), which compensates for the mis-alignments generated by pairwise approaches (Notredame et al. 2000), and by correcting the alignments on the basis of 3D models (see the modeling section of the article). The second factor is that the final alignment is short: only 80 residues. This can affect the tree’s reliability because in short alignments the bias toward few replacements could affect the final grouping. We generated trees using the Bayesian, Neighbor Joining, and Minimum Evolution methods, and found that all three gave the same topology suggesting that the overall structure of the tree is correct. For illustrative purposes only the Bayesian tree is shown (Fig. 2 ▶).
Figure 2.

Unrooted tree for PAAD sequences. This is a consensus tree after screening 9701 trees. Numbers are clade confidence values; only values higher than 80% are shown.
The result of the phylogenetic analyses is a consensus tree whose overall topology is supported by high values of confidence. The five main groups in the tree (Fig. 2 ▶) are statistically significant. The first is the viral group, which has been selected to root the tree. The second is the IFI-PAAD group, which includes all of the IFI sequences and MNDA. The third group contains the only three available sequences for AIM, which interestingly lacks paralogs in all genomes that contain it; in contrast, the other subfamilies have multiple paralogs, especially in higher organisms. The fourth group of PAN/NALP/PYCARD-PAAD is clearly separate from the fifth group, the pyrins. Both the fourth and fifth groups underwent significant expansion in recent evolutionary history, evidenced by multiple paralogs in human and mouse. The branches in the tree coincide with the domain organization, with the exception of the AIM–IFI split. This fact further supports the tree’s topology.
The most interesting case is the similarities between AIM and IFI. Both have similar domain architecture with the HIN domain and AIM’s PAAD domain is the most similar to IFI’s PAAD domain according to surface potential. However, in the tree the AIM group is the basal branch of the pyrin and PAN group. This apparent contradiction is supported by the more detailed analysis of the sequence alignment (Fig. 1 ▶): The AIM PAAD domain is more similar to the pyrin–PAN group at the sequence level. Figure 1 ▶ shows the conserved functional residues in IFI–PAAD subfamily, which no corresponding residues in AIM PAAD. In any case, the multiple alignment of all members clarifies the phylogenetic position of AIM in the tree. AIM and IFI lack the few residues after Leu28 (MEVF numbering). This insertion in the other groups (pyrin and PAN) is not functionally important, as concluded in further modeling analyses (see below). Moreover, the replacement of certain residues pushed the position of AIM closer to the PYR/PAN group, even if AIM’s structure is closer to the IFI group’s. We can also see that although AIM is the basal branch of the large pyrin–PAN group, it does not clade with the other members, being clearly separate.
There are several explanations for these apparently contradictory results between phylogenetic and functional relations between members of the PAAD family. For instance, our alignment may not reflect the evolutionary relations between the sequences because it was biased by structural similarity—it could be influenced by functional convergence of surface residues. As in all phylogenetic analyses, it is impossible to prove that a real evolutionary relation was recovered, or whether the tree obtained here is really a sequence similarity tree. Another possibility is that the AIM-IFI group duplicated, and then the PAAD of AIM rapidly changed to acquire a new function. This duplication was accepted and transmitted in consecutive gene duplications.
In the other main pyrin- or PAN-like branches, the overall topology is consistent with domain architecture, again with a few exceptions. The domain architecture in several subgroups is clearly defined via the PAAD alignment. For instance, the MEFV proteins (PAAD–PRY–SPRY domains) are in a different clade (100% value) than the other PYR members, for example, ASC proteins with PAAD–CARD domains. In the pyrin group, PAAD domain duplicated before mammalian divergence, which is reflected in the tree; the fish sequences are a basal branch of this part. A successive set of gene duplications created the current distribution of the different PYR proteins; however, there are two exceptions, namely the positions of PAN3 and PAN5 genes. Both are human proteins with PAAD and NACHT domains. They were expected to group with the other PAN members, but in the sequence alignment (Fig. 1 ▶) both are more like PAAD–pyrin. They do differ from the other PANs in their genomic locations: PAN3 and PAN5 are on chromosome 11, together with PAN8, whereas the other PAN sequences are on chromosome 19, clustered as a family of paralogs. Despite this, PAN8 still groups within the other PAN sequences. Such a distribution might result from a different mechanism for domain acquisition: shuffling or gene rearrangement instead of duplication.
Another human sequence, AF427617_1, was expected to be PYR-like based on its domain architecture, but groups together with PAN6. This continuous intermixing of sequences might reflect an exchange in gene rearrangements that increased the mammalian PAAD pool in the PYR/PAN group and has important consequences for interactions between the different regulatory pathways.
The fish sequences group with the IFI or AIM branches, suggesting that the pyrin and PAN branches separated only after fish divergence, and this separation correlates with changes in immune functions.
Why do viruses have PAAD domains? Only vital proteins and a human PYC1 (ASC2) are composed of a single PAAD domain. These proteins may mimic the PAAD domains of the IFI or AIM group; they have the same length and have similar surface residues. Viral PAAD domains could compete with host PAAD domains and block antiviral immune responses, allowing the virus to survive.
Homology modeling
Knowing the three-dimensional structure of a protein is a necessary prerequisite to understanding its molecular functions, so we modeled the PAAD domains and then analyzed the models. Several fold and distant homology recognition algorithms predicted that the PAAD domain belongs to the CARD/DD/DED superfamily. Based on this, we used the human death domain (DD) 1E41 and the human death effector domain (DED) 1A1W as our templates. The modeling of PAAD family was based on FFAS (Rychlewski et al. 2000) alignments, which were shown to reflect structural similarities much better than other types of alignments (Jaroszewski et al. 2000, 2002).
As discussed in the Materials and Methods section, the three-dimensional structure for each protein was generated using the JACKAL modeling program (Xiang 2001; Xiang and Honig 2002). The quality of the models was assessed by PSQS (P. Szczesny, unpubl. http://www1.jcsg.org/psqs/psqs.cgi), and this was used to identify one member of each subfamily for a more detailed analysis. In the pyrin subfamily, mouse MEFV was the representative, based of an average PSQS score of −0.2, a level approaching that of native protein structures (Fig. 3A ▶). The multiple alignment of the pyrin subfamily (Fig. 4 ▶) shows that there are 11 conserved hydrophobic residues: LEU9, LEU13, LEU16, PHE21, PHE24, LEU28, ILE40, LEU53, LEU57, LEU74, and LEU86. All of these residues except ILE40 are predicted to form the hydrophobic core of the structure if we define internal residues as ones whose solvent accessible area is less than 10% of the maximum solvent area (Fig. 3B ▶). All helices except helix3 have at least one of the core residues. Helix3 differs from other helices in that it has the weakest secondary structure prediction and it is located slightly away from the core. This is true for other members of the death domain superfamily. The resolved structures of DD, DED, and CARD families show that helix3 usually does not pack well with the other helices. In the crystal structure of the procaspase-9–Apaf-1 complex the negatively charged residues in helix3 are vital for dimerization (Qin et al. 1999). Helix3 of the Tube death domain significantly contributes to its interactions with Pelle (Xiao et al. 1999). Based on these observations it is tempting to speculate that helix3 is partly disordered in solution, and assumes a fully folded form only upon binding to its target.
Figure 3.

The homology model of PAAD domain. The model of MEFV_Mouse was chosen as the representation. (A) The ribbon diagram of MEFV_Mouse. (B) The conserved hydrophobic core of MEFV_Mouse. The conserved hydrophobic residues are shown in ball and stick. (A) and (B) are in the same orientation. (C) The ribbon diagram of template. For comparison, the residues that were aligned to the residues highlighted in (B) are shown.
Figure 4.
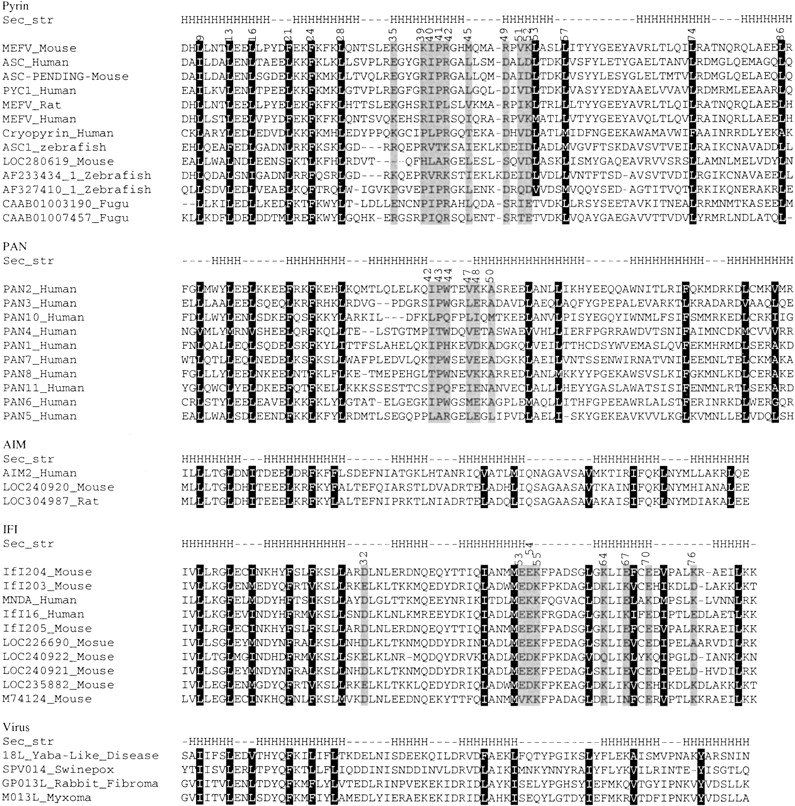
Multiple sequence alignments of the PAAD family. Black background denotes conserved external residues, gray background denotes functional important residues. The "Sec_str" line shows the secondary structure of the reference sequences in each subfamily, namely MEFV_Mouse, PAN2_Human, AIM2_Human, IFI204_Mouse and 18L_Yaba-Like_Disease. The secondary structure was identified from the homology model. The Accession numbers for sequences are the same as Figure 1 ▶.
Using the same approach, we generated the predicted structures from the PAN, AIM, IFI, and virus subfamilies. According to the PSQS, we chose the models of human PAN2, mouse IFI204, human AIM, and viral 18L to represent each subfamily (data not shown). The number of residues contributing to the hydrophobic core is 11 in the PAN subfamily and 12 in the AIM, IFI, and virus subfamilies. In these models, none of the core residues are in helix3, and this helix is clearly separated from the rest of the molecule.
Binding site prediction
We predict that the PAAD domains are involved in specific domain–domain interactions with other PAAD domains in analogy to their distant homologs: CARD, DD, and DED. We analyzed the PAAD models for the possible protein–protein binding sites.
Protein complexes are sensitive to replacements in their binding sites (Lichtarge et al. 1996; Bogan and Thorn 1998), and generally important amino acids are conserved (Zvelebil et al. 1987). Related proteins may have binding sites that are at or near the same structural region (Lichtarge et al. 1996; Russell et al. 1998). These binding sites use external residues of the protein, so these are more relevant to binding than the internal ones. We focused on the conserved external residues, which we defined as residues whose solvent accessible area is more than 30% of the maximum solvent accessibility on the surface.
The general interfaces of protein–protein interactions are mostly formed by clusters of hydrophobic residues (Young et al. 1992); however, in the death domain superfamily, the CARD–CARD (Qin et al. 1999), DD–DD (Xiao et al. 1999), and DED–DED (Eberstadt et al. 1998) interactions are electrostatic, but centered around a small and very conserved hydrophobic cluster. With this in mind, we considered both hydrophobic and charged residues on the surfaces. As mentioned in Materials and Methods, the functionally important regions of the PAAD domain were located using ConSurf (Armon et al. 2001).
Pyrin
We mapped the conserved surface residues of the pyrin group on the predicted structure of mouse pyrin (MEFV). In this family, even the most conserved residues (by ConSurf’s conservation threshold) displayed an interesting pattern of substitutions. In particular, residues LYS35, ARG42, ARG49, and LYS52 cluster together in MEFV and form a charged patch (Fig. 5A1 ▶). Another residue, LYS39, is partially conserved and located near the charged patch; LYS39 might also contribute to the binding interface in most pyrin subfamily members. Although the charged patch of mouse pyrin is positive, the multiple alignment of pyrin subfamily shows that LYS35, ARG49, and LYS52 are replaced by negatively charged residues in some members such as human ASC, mouse ASC-PENDING, and human PYC1. Those members of pyrin subfamily change the overall charge of their charged patch. Considering that the charged patches can be both positive and negative, the members of the subfamily may interact with each other via electrostatic interactions.
Figure 5.
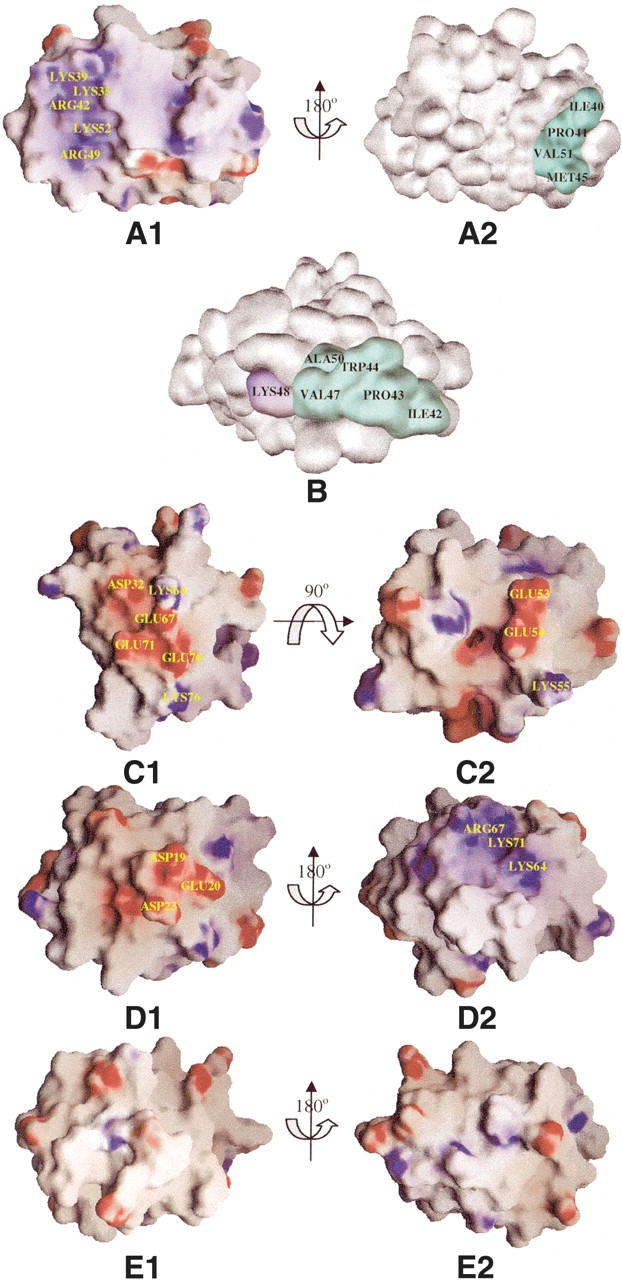
Surface representation of PAAD family. (A1, A2) The electrostatic potential of the model of the MEFV_Mouse PAAD domain, with a color scale that varies from blue to red, representing positive and negative potential, respectively. The conserved external hydrophobic residues are colored in cyan. The same color scheme is used in all subsequent figures. (A1) and (A2) are related by a 180° rotation around the indicated axis. (B) Surface representation of the model of the PAN2_Homo PAAD domain. (C1, C2) The electrostatic potential of the model of the IFI204_Mouse PAAD domain. Two views are related by a 90° rotation around the indicated axis. (D1, D2) The electrostatic potential of the model of the AIM2_Human PAAD domain. Two views are related by a 180° rotation around the indicated axis. (E1, E2) The electrostatic potential of the model of the 18L_Yaba-like_disease PAAD domain. Two views are related by a 180° rotation around the indicated axis.
At the same time, a contiguous hydrophobic patch is formed by ILE40, PRO41, MET45, and VAL51 (Fig. 5A2 ▶). This region might be protein binding site. Surface analysis shows that the two conserved patches are on the opposite sides of the protein, so this hydrophobic patch may be the interaction site with the remaining domains in pyrin.
PAN (NALP, PYCARD)
Based on the results of the ConSurf analysis, we mapped the conserved surface residues on the PAN2 PAAD model (Fig. 5B ▶), and found that most charged-conserved residues are separated. In contrast, the conserved hydrophobic residues ILE42, PERO43, VAL47, and ALA50 cluster together with the partially conserved residue TRP44 and form a large hydrophobic patch. This conserved region might be the binding interface for the PAAD domain of the PAN subfamily. At the same time, the conserved charged residue LYS48 lies close to this patch. Charged patches have a higher specificity than hydrophobic patchs, so LYS48 might enhance the specificity of the hydrophobic binding site. In conclusion, the binding site of the PAN subfamily is a hydrophobic patch intermixed with charged surface residues that are often mutated to their opposites to ensure binding specificity. This is similar to the interaction between DED and DED (Kaufmann et al. 2002).
IFI
The conserved surface residues of the IF group were mapped onto the predicted structure of mouse IFI204 (Fig. 5C ▶). Analysis of the electrostatic surface potential showed a charged region formed by ASP32, LYS64, GLU67, GLU70, GLU71, and LYS76. All of these positions in the multiple alignment are conserved except GLU71. This region is most likely the binding site for this subfamily. The multiple alignment also revealed that positions GLU67, GLU70, and LYS76 are often replaced by oppositely charged amino acids, indicating that this charged region can be an interface for dimerization within this subfamily. Detailed analysis also revealed that GLU53, GLU54, and LYS55 are strictly conserved in all IFI sequences except mouse M74124, and these three residues also cluster on the surface of the structure. This charged patch might be the second binding site of IFI subfamily. This prediction agrees with published experimental data showing that the N-terminal half of MNDA’s PAAD domain (residues 52–82) is essential for self association (Xie et al. 1997).
AIM and Viral PAADs
The approaches used to study previous subfamilies work well because the multiple alignments were composed of 10 or more sequences; however, for AIM subfamily, there are only three AIM sequences and only four PAAD domain viral proteins. This made it difficult to predict possible binding sites in both the AIM and viral subfamilies; however, the electrostatic surface potential of human AIM2 had two obviously charged regions. One is positive and is composed of LYS64, ARG67, and LYS71; the other is negative and is composed of ASP19, GLU20, and ASP23 (Fig. 5D1,D2 ▶). We found it interesting that the first region is concave while the second is convex, suggesting that AIM can homodimerize via electrostatic interactions. For viral subfamily, the electrostatic surface potential of 18L showed (Fig. 5E1,E2 ▶) that no charged residues clustered together. Therefore, the contribution of van der Waal interactions should be strong.
Our analyses are based on theoretical models. It is difficult to predict the influence of N or C termini because they are very flexible, but they might contribute to the protein–protein interactions analogous to DD–DD dimerization (Qin et al. 1999).
Conclusions
We analyzed the PAAD/pyrin/DAPIN protein–protein interaction domain in detail. Multiple alignments showed that the PAAD domain is extremely divergent. Phylogenetic analyses based on these alignments showed five well-defined groups: pyrin, IFI, AIM, PAN, and viral PAADS. The three-dimensional structures of all PAAD domains were modeled by comparative modeling, and we used these models to predict the possible binding interfaces for each family. Our models show that the PAAD domain is composed of six helices, which is in good agreement with secondary structure predictions. Helix3 is special because it does not pack well with the other helices and has the weakest helix signal in secondary structure predictions. The unusual character of helix3 was recently confirmed in the NMR experiment (Espejo et al. 2002), where one-dimensional NMR assignments corresponding to helix3 region suggested that this region is disordered, and not helical, in solution.
Each subfamily has about 10 conserved hydrophobic residues that are internal and form the hydrophobic cores of structures. We identified a series of surface patches where conserved residues form possible binding sites. In analogy to the related DD, DED, and CARD domains, electrostatic and hydrophobic interactions should be the main contributor in PAAD domain interactions. However, the identity, character, and to a smaller extent, the position of conserved surface residues, are different within the PAAD family. This strongly suggests that the PAAD domains from each subfamily interact within their subfamily, and that the regulatory networks do not crosstalk via interactions between PAAD proteins with different domain architectures. In this context, it is particularly interesting to focus on apparent inconsistencies in the phylogenetic tree, where a few proteins inconsistently cluster relative to their domain organization. For such proteins, their surface residues are distributed in a way that results in their classification in the "wrong" branch of the tree. This difference may be a result of convergent evolution. It is only natural to predict that such proteins form links between different regulatory networks.
The conservation patterns discussed here provides a detailed guidance for experimental work, such as point mutations and the study of specific interacting pairs. Several projects along these lines are in progress in our laboratory.
Acknowledgments
We thank Lukasz Jaroszewski for help with the FFAS server. Research described here was supported by NIH grant GM60049.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0359603.
References
- Altschul, S., Madden, T., Schaffer, A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind, L., Dixit, V.M., and Koonin, E.V. 1999. The domains of death: Evolution of the apoptosis machinery. Trends Biochem. Sci. 24 47–53. [DOI] [PubMed] [Google Scholar]
- Armon, A., Graur, D., and Ben-Tal, N. 2001. Consurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 307 447–463. [DOI] [PubMed] [Google Scholar]
- Bertin, J. and DiStefano, P.S. 2000. The PYRIN domain: A novel motif found in apoptosis and inflammation proteins. Cell Death Differ. 7 1273–1274. [DOI] [PubMed] [Google Scholar]
- Bogan, A.A. and Thorn, K.S. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280 1–9. [DOI] [PubMed] [Google Scholar]
- Booth, D.R., Gillmore, J.D., Booth, S.E., Pepys, M.B., and Hawkins, P.N. 1998. Pyrin/marenostrin mutations in familial Mediterranean fever. QJM 91 603–606. [DOI] [PubMed] [Google Scholar]
- Bujnicki, J.M., Elofsson, A., Fischer, D., and Rychlewski, L. 2001. Structure prediction meta serve. Bioinformatics 17 750–751. [DOI] [PubMed] [Google Scholar]
- Chu, Z.L., Pio, F., Xie, Z., Welsh, K., Krajewska, M., Krajewski, S., Godzik, A., and Reed, J.C. 2001. A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J. Biol. Chem. 276 9239–9245. [DOI] [PubMed] [Google Scholar]
- Doctor, K.S., Reed, J.C., Godzik, A., and Bourne, P.E. 2003. The apoptosis database. Cell Death Differ. 10 621–633. [DOI] [PubMed] [Google Scholar]
- Eberstadt, M., Huang, B., Chen, Z., Meadows, R.P., Ng, S.C., Zheng, L., Lenardo, M.J., and Fesik, S.W. 1998. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature 392 941–945. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models. Bioinformatics 14 755–763. [DOI] [PubMed] [Google Scholar]
- Espejo, F., Green, M., Preece, N.E., and Assa-Munt, N. 2002. NMR assignment of human ASC2, a self contained protein interaction domain involved in apoptosis and inflammation. J. Biomol. NMR 23 151–152. [DOI] [PubMed] [Google Scholar]
- Fairbrother, W.J., Gordon, N.C., Humke, E.W., O’Rourke, K.M., Starovasnik, M.A., Yin, J.P., and Dixit, V.M. 2001. The PYRIN domain: A member of the death domain-fold superfamily. Protein Sci. 10 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino, L., Stehlik, C., Oliveira, V., Ariza, M.E., Godzik, A., and Reed, J.C. 2002. A novel PAAD-containing protein that modulates NF-κ B induction by cytokines tumor necrosis factor-α and interleukin-1β. J. Biol. Chem. 277 35333–35340. [DOI] [PubMed] [Google Scholar]
- Greenberg, J.T. and Ausubel, F.M. 1993. Arabidopsis mutants compromised for the control of cellular damage during pathogenesis and aging. Plant J. 4 327–341. [DOI] [PubMed] [Google Scholar]
- Grenier, J.M., Wang, L., Manji, G.A., Huang, W.J., Al-Garawi, A., Kelly, R., Carlson, A., Merriam, S., Lora, J.M., Briskin, M., et al. 2002. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-κB and caspase-1. FEBS Lett. 530 73–78. [DOI] [PubMed] [Google Scholar]
- Harton, J.A., Linhoff, M.W., Zhang, J., and Ting, J.P. 2002. CATERPILLER: A large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 169 4088–4093. [DOI] [PubMed] [Google Scholar]
- Hlaing, T., Guo, R.F., Dilley, K.A., Loussia, J.M., Morrish, T.A., Shi, M.M., Vincenz, C., and Ward, P.A. 2001. Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J. Biol. Chem. 276 9230–9238. [DOI] [PubMed] [Google Scholar]
- Hofmann, K., Bucher, P., and Tschopp, J. 1997. The CARD domain: A new apoptotic signaling motif. Trends Biochem. Sci. 22 155–156. [DOI] [PubMed] [Google Scholar]
- Huang, B., Eberstadt, M., Olejniczak, E.T., Meadows, R.P., and Fesik, S.W. 1996. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384 638–641. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck, J.P. and Ronquist, F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17 754–755. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck, J.P., Ronquist, F., Nielsen, R., and Bollback, J.P. 2001. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294 2310–2314. [DOI] [PubMed] [Google Scholar]
- Inohara, N., Kosekin, T., Lin, J., del Peso, L., Lucas, P.C., Chen, F.F., Ogura, Y., and Nunez, G. 2000. An induced proximity model for NF-B activation in the Nod1/RICK and RIP signaling pathways. J. Biol. Chem. 275 27823–27831. [DOI] [PubMed] [Google Scholar]
- Jacobson, M.D., Weil, M., and Raff, M.C. 1997. Programmed cell death in animal development. Cell 88 347–354. [DOI] [PubMed] [Google Scholar]
- Jaroszewski, L., Rychlewski, L. and Godzik, A. 2000. Improving the quality of twilight-zone alignments Protein Sci. 91476–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroszewski, L., Li, W., and Godzik, A. 2002. In search for more accurate alignments in the twilight zone. Protein Sci. 11 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, M., Bozic, D., Briand, C., Bodmer, J., Zerbe, O., Kolh, A., Tschopp, J., and Grutter, M.G. 2002. Identification of a basic surface area of the FADD death effector domain critical for apoptotic signaling. FEBS Lett. 527 250–254. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V. and Aravind, L. 2000. The NACHT family—A new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem. Sci. 25 223–224. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Origin and evolution of eukaryotic apoptosis: The bacterial connection. Cell Death Differ. 9 394–404. [DOI] [PubMed] [Google Scholar]
- Kumar, S., Tamura, K., Jakobsen, I.B., and Nei, M. 2001. MEGA2: Molecular evolutionary genetics analysis software. Bioinformatics 17 1244–1245. [DOI] [PubMed] [Google Scholar]
- Li, W., Pio, F., Pawlowski, K., and Godzik, A. 2000. Saturated BLAST: An automated multiple intermediate sequence search used to detect distant homology. Bioinformatics 16 1105–1110. [DOI] [PubMed] [Google Scholar]
- Liang, H. and Fesik, S.W. 1997. Three-dimensional structures of proteins involved in programmed cell death. J. Mol. Biol. 274 291–302. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996. An evolutionary trace method defines binding surfacs common to protein families. J. Mol. Biol. 257 342–358. [DOI] [PubMed] [Google Scholar]
- Mansfield, E., Chae, J.J., Komarow, H.D., Brotz, T.M., Frucht, D.M., Aksentijevich, I., and Kastner, D.L. 2001. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 98 851–859. [DOI] [PubMed] [Google Scholar]
- Martinon, F., Hofmann, K., and Tschopp, J. 2001. The pyrin domain: A possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr. Biol. 11 R118–R120. [DOI] [PubMed] [Google Scholar]
- Masumoto, J., Taniguchi, S., and Sagara, J. 2001. Pyrin N-terminal homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem. Biophys. Res. Commun. 280 652–655. [DOI] [PubMed] [Google Scholar]
- Mulley, J.C. 1999. The genetic basis for periodic fever. Am. J. Hum. Genet 64 939–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302 205–217. [DOI] [PubMed] [Google Scholar]
- Pawlowski, K., Pio, F., Chu, Z., Reed, J.C., and Godzik, A. 2001. PAAD—A new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem. Sci. 26 85–87. [DOI] [PubMed] [Google Scholar]
- Qin, H., Srinivasula, S.M., Wu, G., Fernandes-Alnemri, T., Alnemri, E.S., and Shi, Y. 1999. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399 549–557. [DOI] [PubMed] [Google Scholar]
- Richards, N., Schaner, P., Diaz, A., Stuckey, J., Shelden, E., Wadhwa, A., and Gumucio, D.L. 2001. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 276 39320–39329. [DOI] [PubMed] [Google Scholar]
- Russell, R.B., Sasieni, P.D., and Sternberg, M.J. 1998. Supersites with superfolds. Binding site similarity in the absence of homology. J. Mol. Biol. 282 903–918. [DOI] [PubMed] [Google Scholar]
- Rychlewski, L., Jaroszewski, L., Li, W., and Godzik, A. 2000. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 9 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaner, P., Richards, N., Wadhwa, A., Aksentijevich, I., Kastner, D., Tucker, P., and Gumucio, D. 2001. Episodic evolution of pyrin in primates: Human mutations recapitulate ancestral amino acid states. Nat. Genet. 27 318–321. [DOI] [PubMed] [Google Scholar]
- Schultz, J., Milpetz, F., Bork, P., and Ponting, P.C. 1998. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. 95 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer, L.L.E., Eddy, S.R., and Durbin, R. 1998. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 28 405–420. [DOI] [PubMed] [Google Scholar]
- Staub, E., Dahl, E., and Rosenthal, A. 2001. The DAPIN family: A novel domain links apoptotic and interferon response proteins. Trends Biochem. Sci. 26 83–85. [DOI] [PubMed] [Google Scholar]
- Stehlik, C., Fiorentino, L., Dorfleutner, A., Bruey, J.M., Ariza, E.M., Sagara, J., and Reed, J.C. 2002. The PAAD/PYRIN-family protein ASC is a dual regulator of a conserved step in nuclear factor κB activation pathways. J. Exp. Med. 196 1605–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp, J., Martinon, F., and Burns, K. 2003. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 4 95–104. [DOI] [PubMed] [Google Scholar]
- Wang, L., Manji, G.A., Grenier, J.M., Al-Garawi, A., Merriam, S., Lora, J.M., Geddes, B.J., Briskin, M., DiStefano, P.S., and Bertin, J. 2002. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-κ B and caspase-1-dependent cytokine processing. J. Biol. Chem. 277 29874–29880. [DOI] [PubMed] [Google Scholar]
- Xiang, Z. 2001. Extending the accuracy limit of side-chain prediction. J. Mol. Biol. 311 421–430. [DOI] [PubMed] [Google Scholar]
- Xiang, Z. and Honig, B. 2002. Evaluating configurational free energies: The colony energy concept and its application to the problem of protein loop prediction. Proc. Natl. Acad. Sci. 99 7432–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, T., Towb, P., Wasserman, S.A., and Sprang, S.R. 1999. Three-dimensional structure of a complex between the death domains of Pelle and Tube. Cell 99 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, J., Briggs, J.A., and Briggs, R.C. 1997. MNDA dimerizes through a complex motif involving an N-terminal basic region. FEBS Lett. 408 151. [DOI] [PubMed] [Google Scholar]
- Ye, Y., Jaroszewski, L., Li, W., and Godzik, A. 2003. A segment alignment approach to protein comparison. Bioinformatics 19 742–749. [DOI] [PubMed] [Google Scholar]
- Young, L., Jernigan, R.L., and Covell, D.G. 1994. A role for surface hydrophobicity in protein–protein recognition. Protein Sci. 3 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemla, A., Venclovas, Č., Moult, J., and Fidelis, K. 2001. Processing and evaluation of predictions in CASP4. Proteins Suppl 5 13–21. [DOI] [PubMed] [Google Scholar]
- Zvelebil, M.J., Barton, G.J., Taylor, W.R., and Sternberg, M.J. 1987. Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J. Mol. Biol. 195 957–961. [DOI] [PubMed] [Google Scholar]