

Abstract
Phospholipases C (PLCs) reversibly associate with membranes to hydrolyze phosphatidylinositol-4, 5-bisphosphate (PI[4,5]P2) and comprise four main classes: β, γ, δ, and ɛ. Most eukaryotic PLCs contain a single, N-terminal pleckstrin homology (PH) domain, which is thought to play an important role in membrane targeting. The structure of a single PLC PH domain, that from PLCδ1, has been determined; this PH domain binds PI(4,5)P2 with high affinity and stereospecificity and has served as a paradigm for PH domain functionality. However, experimental studies demonstrate that PH domains from different PLC classes exhibit diverse modes of membrane interaction, reflecting the dissimilarity in their amino acid sequences. To elucidate the structural basis for their differential membrane-binding specificities, we modeled the three-dimensional structures of all mammalian PLC PH domains by using bioinformatic tools and calculated their biophysical properties by using continuum electrostatic approaches. Our computational analysis accounts for a large body of experimental data, provides predictions for those PH domains with unknown functions, and indicates functional roles for regions other than the canonical lipid-binding site identified in the PLCδ1-PH structure. In particular, our calculations predict that (1) members from each of the four PLC classes exhibit strikingly different electrostatic profiles than those ordinarily observed for PH domains in general, (2) nonspecific electrostatic interactions contribute to the membrane localization of PLCδ-, PLCγ-, and PLCβ-PH domains, and (3) phosphorylation regulates the interaction of PLCβ-PH with its effectors through electrostatic repulsion. Our molecular models for PH domains from all of the PLC classes clearly demonstrate how a common structural fold can serve as a scaffold for a wide range of surface features and biophysical properties that support distinctive functional roles.
Keywords: Phospholipase C, pleckstrin homology domain, bioinformatics, continuum electrostatics, membrane association, computational biology
Pleckstrin homology (PH) domains are small protein modules (∼130 residues) that are present in many proteins involved in cellular processes in which membrane association plays an integral role, for example, signal transduction, vesicular trafficking, and cytoskeletal rearrangements (Lemmon and Ferguson 2000; Hurley and Meyer 2001). A number of PH domains have been implicated in membrane targeting through specific interactions with particular phosphoinositides, but in most cases, the structural basis for these interactions has not been elucidated. Indeed, function for the majority of PH domains remains to be established. To date, structures for 14 PH domains have been solved. In each case, the structural core consists of a β-sandwich of two nearly orthogonal β-sheets. One end of the β-sandwich is capped by a long C-terminal α-helix, and the other end contains three "variable loops," which are highly diverse in both length and amino acid composition (Lemmon and Ferguson 2000). Despite having the same overall structural fold, PH domains have sequences that share quite low similarity, reflecting the diversity in membrane-binding behavior observed throughout the family (Rebecchi and Scarlata 1998).
Most of the PH domains of known structure are electrostatically polarized, with a highly basic surface corresponding to the position of the three variable loops; structural and biochemical studies implicate this region as the lipid-binding site. Comprehensive model building of the PH domain family indicates that this electrostatic polarization is a feature common to many PH domains (Blomberg and Nilges 1997; Blomberg et al. 1999; Lemmon and Ferguson 2000). Recently, however, a class of PH domains from Caenorhabditis elegans has been characterized that exhibits a highly negative electrostatic profile (Blomberg et al. 2000), consistent with the notion that PH domains may perform functions other than directly mediating membrane association (Blomberg et al. 1999; Rhee 2001; Philip et al. 2002). Given the large size and extreme diversity in sequence and function of the PH domain family, it is not practical to characterize in detail the family as a whole. Therefore, we further subclassify PH domains according to the protein families in which they occur and, in the present study, focus on the set of PH domains found in one such family, the phosphoinositide-specific phospholipases C (PLCs). However, as shown in the literature and further indicated by the work presented here, even the PH domains from this single protein family exhibit a rich array of biophysical properties and functions.
PLCs constitute a large family of closely related enzymes that reversibly associate with membranes to carry out hydrolysis of its membrane resident phosphoinositide substrate, phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2; Rhee 2001). PLCs can be organized into four classes—β, γ, δ, and ɛ—each containing various isoforms (Rebecchi and Pentyala 2000; Rhee 2001). In addition, another protein similar to PLCs, PLC-L or p130, has recently been identified (Kanematsu et al. 1996; Takeuchi et al. 2000). All of the PLC isozymes are soluble multidomain proteins comprised of a core catalytic X/Y domain and various combinations of regulatory domains, for example, PH, C2, SH2, and SH3 domains. The regulatory domains serve to target PLCs to the vicinity of their substrates, activators, and/or effectors through protein–lipid or protein–protein interactions (Rhee 2001). Most eukaryotic PLCs contain a single PH domain located in their N-terminal region. The structure of the PLCδ1 PH domain was determined experimentally and has been instrumental in explaining how its interaction with PI(4,5)P2 targets PLCδ1 to membrane surfaces (Ferguson et al. 1995). Other structural studies indicate that the PLCδ1 PH domain is attached to the rest of the enzyme by a flexible linker region and may, thus, serve a membrane tethering role (Essen et al. 1996). Furthermore, PH domains from a number of PLC classes have been shown to facilitate the interaction of the enzyme with membrane surfaces (Falasca et al. 1998; Pawelczyk and Matecki 1999; Wang et al. 1999; Razzini et al. 2000; Matsuda et al. 2001; Varnai et al. 2002). Moreover, experimental studies provide evidence that the membrane-binding properties of PH domains from different PLC classes are quite distinct: The PLC-δ PH domains bind specifically and with high affinity to membranes containing PI(4,5)P2 (Rebecchi et al. 1992; Pawelczyk and Lowenstein 1993; Ferguson et al. 1995; Garcia et al. 1995), and the PLC-γ PH domains target membranes containing phosphatidylinositol 3,4,5-triphosphate (PI[3,4,5]P3; Falasca et al. 1998; Pawelczyk and Matecki 1999). In contrast, the PLC-β PH domains bind nonspecifically to membranes of a wide range of lipid compositions (Runnels et al. 1996; Wang et al. 1999). However, in some cases, PLC PH domains have also been shown to mediate protein–protein interactions, which indicates that they perform functions beyond that of a simple membrane tether (Wang et al. 1999; Thodeti et al. 2000; Chang et al. 2002; Philip et al. 2002). The goal of the present study is to use computational models to rationalize the observed functional differences for the PLC PH domains and to provide predictions when function is unknown.
Recent studies using homology modeling and calculations of electrostatic properties of both experimentally determined structures and computational models have been successful in describing the membrane-binding behaviors of a number of other lipid-interacting signal transduction domains, namely, C2, FYVE, and PX domains. For example, finite difference Poisson-Boltzmann (FDPB) calculations explained how calcium binding can drive the association of some C2 domains to negatively charged membranes and others to neutral zwitterionic membranes (Murray and Honig 2002). In addition, the biophysical properties of homology models for the C2 domains from PLCδ isoforms and 5-lipoxygenase were shown to correlate with the calcium-dependent lipid-binding preferences of the C2 domains in both biochemical and cellular assays (Ananthanarayanan et al. 2002; Kulkarni et al. 2002). Electrostatic calculations also provide a model for how the binding of both FYVE and PX domains to poly-phosphoinositides facilitates the membrane penetration of these domains: The multivalent, negatively charged poly-phosphoinositides neutralize strong regions of positive potential surrounding hydrophobic residues that are adjacent to the lipid-binding sites, thus decreasing the unfavorable desolvation that occurs upon inserting these residues into the membrane interface (Stahelin et al. 2002; Diraviyam et al. 2003; Stahelin et al. 2003). These studies illustrate the utility of computational approaches in describing, at the molecular level, the regulation of lipid-interacting protein domains.
As illustrated by the multiple sequence alignment in Figure 1 ▶, the sequences of the PLC PH domains share a limited number of conserved residues, many of which correspond to hydrophobic residues that contribute to the structural core of the domains. This lack of sequence conservation is illustrated more dramatically by the all-on-all pairwise sequence comparisons for these domains (see Electronic Supplemental Material). It is clear that sequence similarity is relatively high within classes, but is much less statistically significant for sequences from different classes. In agreement with experiments, this diversity in sequence indicates the existence of class-specific properties and regulatory roles. By considering sequence information alone, it is possible to explain lipid-binding specificity for only the δ2, δ3, and δ4 PH domains (see Results). However, this characterization relies on the knowledge obtained from the structure of the δ1 PH domain complexed with Ins(1,4,5)P3. Furthermore, although the lipid-binding specificity of the γ PH domains is known, it has previously been noted that it is difficult to extract a sequence motif indicative of PI(3,4,5)P3 binding due to a number of alternative ways in which PH domains have been shown to bind this lipid (Baraldi et al. 1999; Ferguson et al. 2000; Lietzke et al. 2000). Moreover, sequence information alone cannot account for nonspecific contributions to membrane-binding, nor can it distinguish those PH domains that do not target membranes. Thus, it would be of great utility to examine how biophysical properties are arrayed on the surfaces of the PH domains. To this end, we modeled the three-dimensional structures of the PLC PH domains by using a number of different computational tools, including fold recognition, sequence-to-profile alignments, and homology modeling. We then calculated the biophysical properties of our models by using continuum electrostatic approaches (Honig and Nicholls 1995). The results of this analysis are robust with respect to alternative models for a given sequence. This is significant because although the core of the PH domains is expected to be well conserved structurally, there is uncertainty in modeling the surface loops, which are quite variable across the PLC classes. Importantly, we found that many members of the PLC-PH family do not exhibit the electrostatic polarity characteristic of the majority of PH domains (Blomberg and Nilges 1997; Blomberg et al. 1999; Lemmon and Ferguson 2000), something that could not be predicted on the basis of sequence alone. In addition, the analysis of our models shows that their biophysical and structural features correlate well with observed functional behaviors and provides the basis for experimentally testable hypotheses for those domains with unknown functions.
Figure 1.

Multiple sequence alignment of the N-terminal PLC PH domains. The sequences were aligned with the program Pattern-Induced (local) Multiple Alignment (PIMA) using the default parameters (Smith and Smith 1992). Columns in the alignment are shaded from light gray to dark gray to black to denote increasing numbers of conserved residues at those positions. White lettering indicates residues that are conserved across all or nearly all sequences. Residues of the PLCδ1 PH domain that directly contact Ins(1,4,5)P3 are highlighted in orange. Note that the alignments with respect to PLCδ1-PH are not necessarily the alignments used in homology modeling (see Materials and Methods). The Swiss-Prot, PIR, and NCBI accession numbers and residues delimiters for the sequences in the alignment are as follows: beta1_bov, P10894(16–139); beta2_hum, Q00722(11–135); beta3_mus, P51432(16–140); beta4_rat, Q9QW07(11–134); epsilon_hum, AAG28341(544–599); gamma1a_bov, P08487(18–144); gamma2a_hum, P16885(11–133); p130_rat, NP_445908(105–224); delta2_bov, S14113(10–127); delta3_hum, AC002117; delta4_rat, Q62711(10–126); and delta1_rat, P10688(12–131). For more detail, see Electronic Supplemental Material.
The focus of our computational analysis on a subset of PH domains, those from the PLC isoforms, constitutes a family-specific strategy for functional annotation in the absence of experimentally determined structures. Consistent with this notion, out of 14 possible PH domain structures, the structure of PLCδ1-PH was identified as the best structural representative for all of the PLC PH domain sequences we examined, and homology models constructed based on the alignment of the PLC PH domain sequences to this structural template all scored well according to structure evaluation analysis. This indicates that our subclassification is reasonable. Our approach provides detailed models for the molecular basis of the interactions of the PLC PH domains with membranes, as well as with other proteins, through the detection of biophysical similarities and differences both within and across the PLC classes. The examination of the PH domains from each PLC class, in turn, results in a more complete picture of their role in the regulation of PLC isozymes than is possible through the analysis of the domains taken either individually or more broadly in the context of the entire PH domain family.
Results
The analysis of the structure and function of PH domains is a challenging task due to the diversity in their sequences and membrane-binding modes. However, by integrating information available on sequence, structure, and function with homology model building and the calculation of electrostatic potentials, we derive computational models that can account for experimental observations and lead to useful biological predictions that can be tested experimentally. All of our models score well according to the structure evaluation programs (see Materials and Methods), indicating that the conclusions based on them are reliable. The sequences and coordinate files representing our models for all mammalian PLC PH domains, as well as other supplementary information, for example, GRASP images and Verify3D profiles, are available at our Web site (http://maat.med.cornell.edu/ph_PVsuppl.html).
PLC-δ
Although four distinct PLCδ isoforms (δ1, δ2, δ3, and δ4) have been identified, the mechanisms that activate and regulate these enzymes remain unclear. The sensitivity of PLCδ isozymes to Ca2+ is greater than that of the other classes. Hence, increases in the intracellular Ca2+ concentration alone may be sufficient to trigger activation of PLCδ (Rhee 2001). Studies of the role of the PH domain in regulating PLCδ activity have focused on PLCδ1 due to the availability of experimentally determined structures for both its PH domain and catalytic core (Ferguson et al. 1995; Essen et al. 1996). Experiments show that both PLCδ1 and PLCδ1-PH bind with high affinity and stereospecificity to membranes containing PI(4,5)P2 (Rebecchi et al. 1992; Pawelczyk and Lowenstein 1993; Garcia et al. 1995). It is, therefore, thought that the PH domain localizes or tethers the enzyme to membranes containing its substrate (Essen et al. 1996). Ligand-binding studies demonstrate that Ins(1,4,5)P3, a product of the hydrolysis of PI(4,5)P2 by PLCδ1, competes effectively with PI(4,5)P2 for binding to PLCδ1-PH and may, thus, interfere with membrane attachment and serve as a negative feedback regulator of catalysis (Cifuentes et al. 1994; Lemmon et al. 1995; Hirose et al. 1999).
Figure 2A ▶ shows the backbone structure of PLCδ1-PH along with electrostatic equipotential contours, which illustrate the electrostatic polarity characteristic of many PH domains (Lemmon and Ferguson 2000). The PLCδ1-PH structure revealed that the 4- and 5-phosphoryl groups of Ins(1,4,5)P3 interact with the side-chains of specific amino acids present in the variable loops of the domain (Fig. 1 ▶, orange highlights), which accounts for the preference for PI(4,5)P2 over other poly-phosphoinositides. Although the binding of δ1-PH to PI(4,5)P2 has been well characterized, not much is known about the phosphoinositide or membrane-binding properties of the other isoforms. As illustrated in Figure 2 ▶, A–D, the structure and our models for the PLCδ PH domains share similar biophysical features: All have a deep PI(4,5)P2-binding pocket that is highly positively charged. Consistent with this, the sequences of the PH domains from the δ2, δ3, and δ4 isozymes share a relatively high level of identity with the δ1-PH sequence (33%–38% identity). Based on the multiple sequence alignment of the PLCδ PH domains (Fig. 1 ▶), many of the residues identified in the PLCδ1-PH structure as making direct contacts with Ins(1,4,5)P3 are conserved in the other isoforms. Therefore, it is expected that all δ PH domains will bind PI(4,5)P2, although the affinity may vary among the different isoforms because not all Ins(1,4,5)P3-binding residues are strictly conserved across the multiple alignment. For example, nine of the 10 key ligand-binding residues observed in the PLCδ1-PH/Ins(1,4,5)P3 complex (highlighted in Fig. 1 ▶: K30, K32, W36, R38, R40, E54, S55, R56, K57, and T107) are either identical or similar at the corresponding positions in the δ3-PH sequence. Based on the conservation of amino acids at the δ1-PH ligand-binding positions, we predict that the δ PH domains have decreasing affinity for PI(4,5)P2 in the order δ1 ∼ δ3 > δ4 > δ2.
Figure 2.

Electrostatic properties of the PLCδ1 PH structure and homology models for the δ2, δ3, and δ4 PH domains. In all panels, the electrostatic potentials were calculated in 0.1 M KCl and contoured at +1 kT/e (blue) and –1 kT/e (red) by using the program GRASP (Nicholls et al. 1991). The structure and models are represented as Cα backbone traces. (A) PLCδ1-PH structure (PDB identifier: 1mai). (B–D) Homology models for the δ2, δ3, and δ4 PH domains. (E–H) Same as in A through D, except that Ins(1,4,5)P3 (yellow spheres) is docked in the lipid-binding loops. The net charge assigned to Ins(1,4,5)P3 for the electrostatic potential calculations is −5, the net charge it would have as the headgroup of intact PI(4,5)P2.
Experiments indicate that interactions in addition to those directly mediated by PI(4,5)P2 may be important for membrane association. For example, mutating basic residues on the variable loops that are not involved in direct interactions with PI(4,5)P2 moderately weakens the binding to membranes containing PI(4,5)P2; this implies the existence of secondary, lower-affinity membrane association sites within the PH domain (Yagisawa et al. 1998). These secondary sites may mediate interactions with anionic phospholipids in the membrane and contribute to membrane association through nonspecific electrostatic interactions. A role for electrostatic interactions in the membrane association of PLCδ1-PH is also indicated by observations that the presence of monovalent acidic phospholipids in PI(4,5)P2-containing vesicles increases the binding of the isolated PH domain by ∼10-fold and that PLC-δ1 activity is stimulated by interaction with the monovalent acidic lipid phosphatidic acid (Rebecchi et al. 1992; Garcia et al. 1995; Henry et al. 1995). Interestingly, the δ3 PH domain is predicted to lack the electrostatic polarity of δ1-PH and is, instead, engulfed in a strong, contiguous positive potential profile (Fig. 2C ▶), which indicates that δ3-PH should interact strongly and nonspecifically with negatively charged membrane surfaces (see below). This is consistent with the observation that PLCδ3 interacts with membranes containing PI(4,5)P2 or phosphatidic acid in a PH domain–dependent manner (Pawelczyk and Matecki 1999).
Our calculations based on the FDPB method (see Materials and Methods) predict that nonspecific electrostatic interactions between the positively charged regions of the δ-PH domains and acidic phospholipids in the membrane contribute significantly to the membrane localization of these PH domains. Figure 3 ▶ illustrates the electrostatic component of the membrane-binding free energy for the PLCδ1-PH structure and the PLCδ3-PH homology model as a function of distance between the surfaces of the domains and membrane. As depicted by the free energy curves, the electrostatic attraction to a membrane containing 33 mole % acidic lipid; that is, 2 : 1 phosphatidylcholine (PC)/ phosphatidylserine (PS; PC, z = 0; PS, z = −1), in 0.1 M KCl is long-range and, thus, could facilitate the diffusion of the PH domains to membranes containing acidic phospholipids. As seen in Figure 3A ▶, the predicted minimum electrostatic free energy of interaction between PLCδ1-PH and a 2 : 1 PC/PS membrane is quite strong (–4 kcal/mole), and that for PLCδ3-PH even stronger (−9 kcal/mole). The minimum electrostatic free energy of interaction with a membrane of lower negative surface charge density (5 : 1 PC/PS; Fig. 3B ▶) is correspondingly weaker. These calculations indicate that nonspecific electrostatic interactions alone may be sufficient to localize these PH domains, especially PLCδ3-PH, to membranes containing acidic phospholipids, and that the interaction with membrane regions containing higher negative surface charge density is significantly favored. The nonspecific accumulation of a PH domain at the membrane surface may then facilitate the specific 1 : 1 interaction with membrane-embedded PI(4,5)P2.
Figure 3.

Electrostatic free energy curves for the membrane interaction of the δ1 and δ3 PH domains. The electrostatic free energy of interaction (in kcal/mole) with 2 : 1 PC/PS (A) and 5 : 1 PC/PS (B) membranes in 0.1 M KCl as a function of the distance between the van der Waals surfaces of the PH domain and membrane. The electrostatic free energies were calculated using the finite difference Poisson-Boltzmann (FDPB) method as described in Materials and Methods (Gallagher and Sharp 1998).
Our FDPB calculations also provide an explanation for why δ1-PH prefers soluble Ins(1,4,5)P3 to membrane embedded PI(4,5)P2 (Lemmon et al. 1995). As described above, the nonspecific association of the δ PH domains with membrane surfaces involves two main electrostatic components: (1) a "coulombic" attraction between the positively charged PH domain and acidic lipids in the membrane and (2) a short-range desolvation penalty due to the loss of favorable interactions between the aqueous solvent and the charged and polar groups on both the PH domain and membrane when they are in close apposition. These two components are illustrated in Figure 3 ▶ by the electrostatic free energy curves for the δ1 and δ3 PH domains. Far from the membrane, the PH domains experience an electrostatic attraction toward the negatively charged membrane surface, but at short distances, the desolvation penalty dominates and the electrostatic interaction becomes much less favorable and, in some cases, repulsive (Fig. 3B ▶). Thus, the minimum electrostatic free energy of interaction with the membrane is attained when the surfaces of the PH domains and membrane are separated by a distance of about the thickness of a layer of water. This orientation maximizes the coulombic attraction and minimizes the desolvation repulsion (Ben-Tal et al. 1996). When the PH domains bind PI(4,5)P2, they are anchored at the membrane surface and are no longer necessarily free to assume an orientation that minimizes the nonspecific electrostatic contribution to the membrane association. Importantly, as illustrated in Figure 2 ▶, E through H, the PI(4,5)P2 headgroup dramatically alters the electrostatic profile of the PH domains. In particular, the electrostatic potential in the membrane-binding region of δ1-PH becomes predominantly negative. The PI(4,5)P2-bound PH domain would, thus, experience highly repulsive electrostatic interactions with the surrounding membrane environment due both to unfavorable coulombic interactions with negatively charged lipids and the desolvation penalty that occurs close to the membrane surface. These nonspecific interactions would oppose the energetically favorable specific interactions with PI(4,5)P2.
To get an idea of the magnitude of the nonspecific repulsion, we performed FDPB calculations of the membrane interaction of δ1-PH when it is bound to Ins(1,4,5)P3. Table 1 compares the electrostatic free energy of interaction of both the Ins(1,4,5)P3-free and Ins(1,4,5)P3-bound forms with a 2 : 1 PC/PS membrane in 0.1 M KCl for small separations between the domain and membrane. In all cases, the interaction of the Ins(1,4,5)P3-bound form is highly repulsive. These calculations indicate that when the bulk membrane contains acidic phospholipid, the interaction of δ1-PH with soluble Ins(1,4,5)P3 is energetically more favorable than the interaction with membrane-embedded PI(4,5)P2. In the former case, the PH domain retains the favorable specific interactions with the lipid headgroup without the repulsive electrostatic interactions due to the surrounding membrane environment that are experienced in the latter case.
Table 1.
Comparison of the electrostatic free energy of interaction of the PLCδ1 PH domain in the absence and presence of bound Ins(1,4,5)P3 with a 2 : 1 PC/PS membrane in 0.1 M KCl
| Ra (Å) | δGel (kcal/mole), δ1-PH, no Ins(1,4,5)P3 | δGel (kcal/mole), δ1-PH with bound Ins(1,4,5)P3 |
| 0 | 0.36 | 11.3 |
| 0.5 | −1.3 | 8.7 |
| 1.0 | −2.3 | 6.3 |
| 1.5 | −3.6 | 4.0 |
| 2.0 | −3.9 | 2.7 |
a R is the distance between the van der Waals surfaces of the PH domain and membrane. The electrostatic free energies were calculated by using the finite difference Poisson-Boltzmann (FDPB) method as described in Materials and Methods (Gallagher and Sharp 1998).
PLC-L/p130
p130 was originally identified as a catalytically incompetent PLC with lipid-binding properties similar to those of PLCδ1 (Kanematsu et al. 1992; Yoshida et al. 1994). Although it has the same domain organization as PLCδ1, that is, PH, EF-hand, catalytic X/Y, and C2 domains, it exhibits distinct functional characteristics. Even though p130-PH is capable of binding PI(4,5)P2 in membranes, both the PH domain and intact protein are found principally in the cytoplasm of cells (Takeuchi et al. 2000; Várnai et al. 2002). This is in sharp contrast to PLCδ1 and the δ1-PH domain, which are both predominantly localized to the plasma membrane. p130 has been implicated in the down-regulation of Ins(1,4,5)P3-mediated Ca2+ signaling (Kanematsu et al. 1996; Takeuchi et al. 2000). Studies show that the PH domain, which binds with high affinity to Ins(1,4,5)P3, is responsible for this functionality (Takeuchi et al. 2000): The sequestration of cellular Ins(1,4,5)P3 by the p130 PH domain prevents the activation of Ins(1,4,5)P3 receptors and the subsequent increase in intracellular calcium.
We compared the biophysical properties of our homology model for p130-PH with those of the PLCδ1-PH structure. As described above, the membrane targeting of PLCδ1-PH is mediated by specific interactions with PI(4,5)P2 as well as nonspecific electrostatic interactions between basic residues surrounding the lipid-binding pocket and acidic lipids in the bulk membrane environment. In addition, one of the membrane-binding loops of PLCδ1-PH contains a pair of hydrophobic residues (Val and Met) that are well positioned to penetrate the membrane interface when the PH domain binds PI(4,5)P2 (Fig. 4 ▶, green). The corresponding elements in p130-PH combine to produce a PH domain that we predict should interact significantly more weakly with PI(4,5)P2-containing membranes than does PLCδ1-PH. First, as shown in Figure 5 ▶, the potential profile surrounding the membrane-binding loops of p130-PH is significantly less positive than that of δ1-PH and contains a region of negative potential. Therefore, the favorable nonspecific electrostatic free energy between p130-PH and the membrane is expected to be diminished with respect to δ1-PH. Second, although the sequences are quite similar, p130-PH is missing some of the residues implicated in mediating interactions with the 1- and 5-phosphates of PI(4,5)P2 (Fig. 4 ▶). Third, the residue corresponding to the valine in PLCδ1-PH is aspartic acid in p130-PH (Fig. 4 ▶). In contrast, all of the δ PH domains contain a nonpolar residue in this position (Fig. 1 ▶). The aspartate contributes to the negative potential profile of p130-PH (Fig. 5 ▶) and is adjacent to basic residues that are expected to directly contact the PI(4,5)P2 headgroup (Fig. 4 ▶). Therefore, if the domain was bound to PI(4,5)P2, the aspartate would be positioned close to the membrane surface and would, thus, result in electrostatic repulsion from the negatively charged membrane surface due both to unfavorable charge-charge interactions and desolvation. Given its proximity to potential PI(4,5)P2-binding residues, it may also disrupt specific interactions with the lipid headgroup.
Figure 4.

Sequence alignment of p130-PH and PLCδ1-PH. Conserved residues are represented as white letters on a black background. Residues of the PLCδ1 PH domain that make direct contacts with Ins(1,4,5)P3 are highlighted in orange. Hydrophobic residues (Val, Met) that may contribute to the membrane partitioning of PLCδ1-PH are highlighted in green.
Figure 5.

Electrostatic properties of the homology model of p130 PH domain. Electrostatic equipotential profiles and Cα backbones of PLCδ1-PH (A) and p130-PH (B) were calculated and visualized in GRASP (Nicholls et al. 1991). The blue and red meshes represent the +1 kT/e and −1 kT/e equipotential contours, respectively, for 0.1 M KCl. V58 and M59 in PLCδ1-PH, and D61 and L62 in p130-PH are represented by yellow spheres. Ins(1,4,5)P3 (green rods) is depicted in the PLCδ1-PH structure to illustrate the domain orientation with respect to the membrane surface but was not included in the electrostatic potential calculations.
PLC-β
The PLCβ class is comprised of four isoforms: β1, β2, β3, and β4, and is distinguished from the other PLC classes by the presence of a long C-terminal coiled-coil sequence (Singer et al. 2002) that is implicated in membrane association (Kim et al. 1996) and in activation by heterotrimeric G-protein subunits (Rhee 2001). Experiments indicate that the β class exhibits a broad subcellular distribution. PLCβ has been shown to be present in the nucleus (Martelli et al. 1992) and associated with both the soluble and particulate fractions from cells (Smrcka and Sternweis 1993). Biochemical measurements indicate that PLCβ1 and PLCβ2 bind strongly and nonspecifically to phospholipid (PC, PS) membranes and that the interaction is independent of the presence of poly-phosphoinositides (Romoser et al. 1996; Wang et al. 1999). In addition, complementary experiments with the isolated PH domains indicate that they have similar membrane-binding properties as their intact enzymes (Wang et al. 1999). However, a recent study indicates that PLCβ1-PH specifically recognizes PI(3)P (Razzini et al. 2000).
Figure 6 ▶ depicts our alignment of the sequences and secondary structure elements of the β3 and δ1 PH domains. The correspondence in the locations of the predicted secondary structure elements of β3-PH with those from the δ1-PH structure indicates that the alignment is suitable for homology modeling despite the low sequence similarity. Similar results were obtained for the PH domains from other β isoforms (see Electronic Supplemental Material).
Figure 6.

Sequence alignment of PLCβ3-PH and PLCδ1-PH and the correspondence of observed and predicted secondary structure elements. The top two rows represent the sequence alignment between the PH domains of PLCδ1 (Delta1) and PLCβ3 (Beta3) that was used in modeling the structure of β3-PH (PLCδ1-PH served as the structural template). The highlighted residues in the PLCδ1-PH sequence represent the location of secondary structure elements (gray, α-helix; black, β-strand). The bottom five rows depict the output of various secondary structure prediction programs (for details, see Materials and Methods) for the PLCβ3-PH sequence. Letters highlighted in black/gray and colored white denote the secondary structure predictions for β3-PH (H, α-helix; E, β-strand); C indicates random coil.
As illustrated in Figure 7 ▶, the electrostatic profiles of our β PH domain models are strikingly different from those of other PH domains. In contrast to the observations of Razzini et al. (2000), our models predict that these domains are unable to interact specifically with phosphoinositides. In all cases, the canonical lipid-binding site is predominantly negatively charged (Fig. 7 ▶). Moreover, our models predict that regions other than this site may mediate membrane partitioning. As depicted in Figure 7 ▶, all of the models have a significant patch of surface-exposed hydrophobic residues (Leu, Phe, Tyr, Trp) located mainly on β-strands 3 and 4. Leu, Phe, Tyr, and Trp residues have been shown, in the context of model peptides, to partition favorably into the interface of phospholipid membranes (Wimley and White 1996). In addition, the models for β2- and β3-PH have significant patches of surface-exposed basic residues (Fig. 7 ▶, blue meshes). Figure 8 ▶ depicts the electrostatic free energy of interaction of our model for the PLCβ2 PH domain with a 2 : 1 PC/PS membrane in 0.1 M KCl as a function of the distance between the van der Waals surfaces of the domain and membrane. Nonspecific electrostatic interactions are predicted to contribute significantly to the membrane-binding free energy, that is, ∼−5 kcal/mole; this is similar to what is predicted for the PLCδ1-PH domain (Fig. 3 ▶). The nonpolar residues may partition hydrophobically into the interface of both electrically neutral and negatively charged membranes, whereas the basic residues may mediate electrostatic interactions with acidic phospholipids. Thus, our models support the observation that the β PH domains bind nonspecifically to phospholipid membranes (Wang et al. 1999).
Figure 7.

Electrostatic properties of the homology models for the PLCβ PH domains. Electrostatic equipotential profiles for the PLCβ PH domain models were calculated and visualized in GRASP (Nicholls et al. 1991): β1-PH (A), β2-PH (B), β3-PH (C), and β4-PH (D). The loops that correspond to the PI(4,5)P2-binding loops in the PLCδ1-PH structure are located at the bottom of the images. All β PH domains are in an orientation similar to that as the PLCδ PH domains in Figure 2 ▶, except for β2-PH (B), which is rotated 180 degrees about the vertical axis to highlight its large basic surface patch. Clusters of surface-exposed hydrophobic residues (Leu, Phe, Tyr, Trp) are colored green. The blue and red meshes represent, respectively, the +1 kT/e and −1 kT/e equipotential contours for 0.1 M KCl.
Figure 8.

Electrostatic free energy curve for the membrane interaction of the β2 PH domain. The electrostatic free energy of interaction (in kcal/mole) with a 2 : 1 PC/PS membrane in 0.1 M KCl as a function of the distance between the van der Waals surfaces of the PH domain and membrane. The free energies were calculated by using the finite difference Poisson-Boltzmann (FDPB) method as described in Materials and Methods (Gallagher and Sharp 1998).
The basic surface patches found on our PLCβ-PH models may also mediate the observed interaction with Gβγ-subunits of heterotrimeric G proteins (Wang et al. 1999, 2000; Barr et al. 2000). Our previous computational studies indicate that Gβγ is oriented at the membrane surface such that the basic surface patch surrounding the site of prenylation on the γ-subunit interacts with acidic lipid headgroups (Murray et al. 2001). Gβγ has a net charge of −12 and is dramatically electrostatically polarized. The basic surface patch on Gβγ that is implicated in membrane binding is localized to the region surrounding the prenyl group. The rest of the protein is highly negatively charged, indicating that Gβγ and PLCβ PH domains interact through the complementarity of their electrostatic surfaces (positive on the PH domain, negative on Gβγ). Indeed, there is a serine residue in PLCβ3-PH whose phosphorylation is implicated in disrupting the interaction with Gβγ (Xia et al. 2001). Our model for PLCβ3-PH predicts that this serine is located in the center of a basic surface patch that could serve as a potential Gβγ-binding site. As shown in Figure 9 ▶, phosphorylation of this serine significantly reduces the positive potential in this region of the PH domain and may, thus, disrupt the interaction with Gβγ through an electrostatic repulsion mechanism.
Figure 9.

The effect of phosphorylation of Ser26 on the electrostatic properties of PLCβ3-PH. The PH domain is oriented differently than in Figure 7 ▶ so that the surface containing Ser26 is facing the viewer. (A, B) Electrostatic equipotential contours are shown for PLCβ3-PH in the unphosphorylated (A) and phosphorylated (B) states. The Cα backbone is colored green at the position of Ser26. The blue and red meshes represent, respectively, the +1 kT/e and −1 kT/e equipotential contours. (C, D) The electrostatic potentials are mapped to the molecular surface of PLCβ3-PH in the unphosphorylated (C) and phosphorylated (D) states. The surface potentials are color-graded from −4 kT/e (red) to +4 kT/e (blue). All potentials were calculated with 0.1 M KCl and visualized in GRASP (Nicholls et al. 1991).
PLC-γ
The γ class of PLCs consists of two isoforms, PLCγ1 and PLCγ2, both of which are activated by polypeptide growth factor stimulation of receptor and nonreceptor protein tyrosine kinases (Cockcroft and Thomas 1992; Rhee and Bae 1997). Both isoforms have two PH domains, one in their N terminus and one occurring in the linker sequence between the two halves of the catalytic domain. Experimental studies indicate that the N-terminal PLCγ PH domains are targeted to the plasma membrane and bind specifically to PI(3,4,5)P3 in a PI3 kinase–dependent manner (Falasca et al. 1998; Matsuda et al. 2001).
The PLCγ-PH sequences are slightly more similar to the δ1-PH sequence than are the β-PH sequences. Our PLCγ PH domain models exhibit the characteristic electrostatic polarization typical of PH domains that bind poly-phosphoinositides (e.g., cf. Fig. 10 ▶ and Fig. 2A ▶). As predicted for the δ PH domains, nonspecific electrostatic interactions between basic residues in the lipid-binding loops and acidic phospholipids in the membrane are expected to contribute to the membrane localization of the γ PH domains.
Figure 10.

Electrostatic properties of the homology models of the N-terminal PLCγ PH isoforms. The Cα backbone traces of the models for γ1-PH (A, C) and γ2-PH (B, D) without (A, B) and with (C, D) bound Ins(1,3,4,5)P4 (yellow spheres) are depicted. The electrostatic potentials were calculated and visualized in GRASP (Nicholls et al. 1991). The blue and red meshes represent, respectively, the +1 kT/e and −1 kT/e equipotential contours for 0.1 M KCl.
Figure 11 ▶ depicts the multiple structure-based sequence alignment for the PLCγ1 and γ2 PH domain models with PI(3,4,5)P3-binding PH domains of known structure. Because the sequences in this group share such low similarity, superposition of the structures with our homology models is the most reliable way of deriving a multiple sequence alignment. From the experimentally determined structures, it is known that residues in the β1/β2 loop are important for dictating PI(3,4,5)P3 specificity and that residues in the β3/β4 and β6/β7 loops also contribute, but to varying degrees (Falasca et al. 1998; Baraldi et al. 1999; Cullen and Chardin 2000; Ferguson et al. 2000; Lemmon and Ferguson 2000; Lietzke et al. 2000; Thomas et al. 2001). For PLCγ1-PH, experiments determined that the β3/β4 loop plays a key role in mediating the interaction with PI(3,4,5)P3 (Falasca et al. 1998). As pointed out by others (Baraldi et al. 1999; Ferguson et al. 2000; Lietzke et al. 2000) and as illustrated by the alignment in Figure 11 ▶, the residues in the lipid-binding loops are not well conserved across this group of PH domains and do not provide a simple consensus sequence for PI(3,4,5)P3 specificity.
Figure 11.

Multiple structure-based sequence alignment among the PLCγ-PH models and structures of PI(3,4,5)P3-binding PH domains. Protein sequences were aligned based on structural superposition by using the multiple structure alignment module of PrISM (Yang and Honig 1999). 1bwn indicates PH domain of Bruton’s tyrosine kinase (Btk) with the E → K mutation (denoted by arrow); 1b55, wild-type Btk PH domain; 1fao, PH domain of DAPP1/Phish; 1fgy, PH domain of Grp1; Gamma1a, homology model of the N-terminal PLCγ1 PH domain; and Gamma2a, homology model for the N-terminal PLCγ2 PH domain. Residues in the experimentally determined structures that are implicated in forming direct contacts with PI(3,4,5)P3 are colored orange. Black bars denote the positions of the variable lipid-interacting loops.
However, based on the multiple structure–based sequence alignment (Fig. 11 ▶), we are able to identify several residues in the PLCγ-PH sequences that together may define a PI(3,4,5)P3-binding function. In our models, there is a conserved basic residue in the β3/β4 loop that has been shown to be important for membrane-binding activity in other PI(3,4,5)P3-binding PH domains. For example, a Glu-to-Lys mutation in Btk-PH (Fig. 11 ▶, arrow) has been reported to contribute to the binding to PI(3,4,5)P3-containing membranes by increasing nonspecifically the membrane affinity of the PH domain (Li et al. 1995). In addition, a lysine at this position in DAPP1-PH forms direct contacts with the PI(3,4,5)P3 headgroup (Ferguson et al. 2000). Therefore, the basic residue at the same position in the PLCγ PH domains may also form specific interactions with the PI(3,4,5)P3 headgroup. Moreover, the alignment in Figure 11 ▶ indicates that the PLCγ PH domains have other functionally important residues at conserved positions: (1) β-Strand 2 contains basic and aromatic residues at its N and C termini, respectively, that are highly conserved across the PI(3,4,5)P3-binding PH domains, but not the PLCδ PH domains, which bind PI(4,5)P2; and (2) a number of basic residues occur in the β1/β2 and β6/β7 loops, which have been shown in other PI(3,4,5)P3-binding PH domains to contribute to lipid specificity. Overall, the characteristic electrostatic polarity of our models (Fig. 10 ▶) and the conservation of residues that interact with PI(3,4,5)P3 in other PH domains of known structure (Fig. 11 ▶) indicate a similar function for the PLCγ PH domains and provide a molecular basis for the experimentally observed interaction with PI(3,4,5)P3 (Falasca et al. 1998; Matsuda et al. 2001).
PLCγ1 and γ2 also possess a purported second PH domain that is located within a sequence insert between the two halves of the catalytic domain (X and Y). Our analysis predicts that in both cases, the sequence corresponding to the additional PH domain is split into two parts by a span of sequence that corresponds to the one SH3 and two SH2 domains that are characteristic of the PLCγ class. Experimental studies (Chang et al. 2002) indicate that the N-terminal portion of this "split PH domain" binds specifically to phosphoinositides, including PI(4)P and PI(4,5)P2. Other studies implicate the domain in protein–protein interactions (Thodeti et al. 2000; Chang et al. 2002).
While attempting to identify the segments of sequence that correspond to these split PH domains, we found significant ambiguities in the sequence assignments provided by different databases and domain detection programs (Schultz et al. 1998; Boekmann et al. 2003; Wheeler et al. 2003). We, therefore, used a combination of fold recognition and secondary structure prediction (see Materials and Methods) to identify the sequence segments corresponding to the two halves of each split PH domain. Once assembled into contiguous pieces (excluding the sequences representing the SH domains), these sequences are predicted to have all of the characteristic secondary structure elements of the PH domain fold (Fig. 12 ▶). In addition, these "patched" sequences pick up PH domain structures with very high statistical significance when submitted to fold recognition programs (data not shown). Therefore, these sequences have the potential to assemble into PH domains. Indeed, the homology models representing these split PH domains score very high according to structure validation analysis (see Materials and Methods; Electronic Supplemental Material). Furthermore, a recent study demonstrated that a functional PH domain assembles from two separate sequence segments (both attached to coiled-coil forming regions) which correspond to the N- and C-terminal halves of PLCδ1-PH, establishing, in principle, the possibility of the formation of a stable PH domain structure from noncontiguous sequence segments when they are tethered together (Sugimoto et al. 2003). However, each of the PLCγ split PH domains has an insert in the β3/β4 loop, which corresponds to the SH3 and SH2 domains, which we have not attempted to include in our models. The impact of these large insertions on the structural organization and function of the split PH domains is unknown.
Figure 12.

Sequence assignment of the split PLCγ PH domains. The top portion of the figure shows the two alternative domain architectures assigned to PLCγ by the SMART database (Schultz et al. 1998). This arrangement indicates that the location of the second PH domain overlaps with that of the SH3 domain. We predict that the two halves of the domain are located in the regions between (1) the X portion of the catalytic domain and the first SH2 domain (gray arrows) and (2) the SH3 domain and the Y portion of the catalytic domain (black arrows). The sequence segments that are combined to comprise our prediction for the sequence of the PLCγ1 PH domain are highlighted in purple.
The electrostatic profiles of our homology models for the split PH domains are overall negative with minor positive regions (data not shown). Therefore, contrary to previous studies (Chang et al. 2002), our models indicate that these domains do not bind poly-phosphoinositides. However, the experimental studies were based on a construct that corresponds to only the N-terminal portion of the domain rather than the complete domain, which we have modeled here. Hence, the experimental characterization may not be an accurate representation of lipid-binding function if the two portions of the split domain, identified here, do indeed come together and fold up into a complete PH domain structure.
PLC-ɛ
PLCɛ is a recently discovered member of the PLC family and has been shown to be regulated by the small GTPase H-Ras and α-subunits (Gα12) of heterotrimeric G proteins (Kelley et al. 2001; Rhee 2001; Wing 2001). PLCɛ was originally distinguished by the absence of an N-terminal PH domain, but in agreement with the recent work of Wing et al. (2001), we have detected a potential PH domain by aligning the PLCɛ sequence with the PH domain sequences from other PLC classes by using multiple sequence and secondary structure alignments. Figure 13 ▶ depicts the alignment between the sequences of the PLCδ1 and ɛ PH domains. Although the details in the sequence assignments differ, our alignment and that of Wing et al. (2001) both predict a long insert between the β3- and β4-strands. In addition, Wing et al. (2001) predict that the C-terminal α-helix of ɛ-PH extends significantly beyond that of δ1-PH and that this "molten helix" may be important for mediating interactions with Gβγ subunits as seen for other PH domains (Fushman et al. 1998; Carman et al. 2000). The β3/β4-loop typically contributes to lipid binding in those PH domains that are known to associate with poly-phosphoinositides. In our model, the structure of the insertion is not well defined due to the absence of a suitable structural template. However, because the insertion is located in a loop region, it most likely does not affect the core structure of the PH domain.
Figure 13.

Sequence alignment of PLCɛ-PH and PLCδ1-PH. The top two rows represent the sequence alignment between the PH domains of PLCδ1 (Delta1) and PLCɛ (Epsilon) that was used in modeling the structure of ɛ-PH (PLCδ1-PH served as the structural template). The highlighted residues in the PLCδ1-PH sequence represent the location of secondary structure elements (gray, α-helix; black, β-strand). The bottom five rows depict the output of various secondary structure prediction programs (for details, see Materials and Methods) for the PLCɛ-PH sequence. Letters highlighted in black/gray and colored white denote the secondary structure predictions for ɛ-PH (H, α-helix; E, β-strand); C indicates random coil. The arrow denotes the serine residue predicted by Prosite (Falquet et al. 2002) to be a protein kinase C phosphorylation site.
As shown in Figure 13 ▶, the predicted secondary structure elements of PLCɛ-PH are well aligned to the core structural elements of PLCδ1-PH. The large insertion is predicted to have a short α-helical segment flanked by regions with random coil conformation, although its structure and orientation with respect to the PH domain core is uncertain based on our analysis. As illustrated in Figure 14 ▶, the electrostatic profile of the PLCɛ-PH domain model is predominantly negative with a positive region in the vicinity of the insert. The β3/β4 insertion could contribute to membrane association because it contains 11 hydrophobic residues, several of which (five Leu and one Phe) have been shown, in the context of model peptides, to partition favorably into the membrane interface (Wimley and White 1996), as well as three Lys residues that may interact electrostatically with acidic lipids. Hence, like other long unstructured sequences in proteins such as MARCKS, Gap43, and phospholipase D (Wang et al. 2002), the long β3/β4-loop may interact with membrane surfaces through nonspecific electrostatic and hydrophobic interactions. Such membrane-adsorbed sequences have been shown to sequester PI(4,5)P2 in phospholipid vesicles (Wang et al. 2002). Hence, this sequence may function to localize the enzyme in the vicinity of its substrate. The insert contains a serine at position 903 of the PLCɛ sequence (Fig. 13 ▶, arrow), which Prosite (Falquet et al. 2002) predicts with high probability to be a protein kinase C phosphorylation site. This indicates a potential mechanism whereby membrane association is regulated: Phosphorylation would introduce negative charge into a region containing basic and hydrophobic residues, produce an electrostatic repulsion from the membrane surface, and, thus, weaken the membrane association of the insert sequence. Although the PLCɛ PH domain may contribute to nonspecific membrane association, it is clear from Figure 14 ▶ that its electrostatic profile, like those for the β and split γ PH domains, deviates significantly from the electrostatic polarization of most PH domains previously studied (Lemmon and Ferguson 2000; Hurley and Meyer 2001).
Figure 14.

Electrostatic properties of the homology model of the PLCɛ PH domain. The Cα backbone trace for the model of the PLCɛ PH domain, based on the alignment in Figure 13 ▶, is depicted. The backbone is colored cyan in the region of the β3/β4-loop insert (see Fig. 13 ▶). The electrostatic potentials were calculated and visualized in GRASP (Nicholls et al. 1991). The blue and red meshes represent, respectively, the +1 kT/e and +1 kT/e equipotential contours for 0.1 M KCl.
Discussion
Although PH domains share a common structural fold, they are highly diverse in their sequences, biophysical properties and functions, which makes it particularly challenging to derive general "rules" that characterize their targeting and regulatory roles. Computational studies that examine broadly the entire PH domain family highlight conserved characteristics, but provide a coarse-grained, incomplete, functional annotation (Blomberg and Nilges 1997; Blomberg et al. 1999). In contrast, computational studies that focus on a single PH domain provide significantly more detailed information on the molecular basis of function, but lack context with regard to related PH domains (Rong et al. 2001). Here, we have chosen to focus on a subset of PH domains that belongs to the same protein family, the phosphoinositide-specific PLCs. This type of examination allows for detailed molecular-level descriptions of the individual PH domains and provides an additional dimension to functional annotation through comparisons among related PH domains.
Despite low conservation in the sequences of the PH domains from the different PLC classes (Fig. 1 ▶), the most suitable structural template for modeling them was consistently identified as the PH domain from PLC-δ1 (Protein Databank [PDB] identifier: 1mai; Ferguson et al. 1995). A major finding of our study is that even though we used a single PH domain of well-defined structure and function as the template for homology modeling, the biophysical properties of our derived models are quite distinct but correlate well with the observed characteristics of the respective PH domains. (See Materials and Methods for a discussion of the reliability of our models and conclusions.) This supports the notion that the structural core of the PH domain serves as a stable scaffold that can support a wide range of functions (Lemmon and Ferguson 2000). Moreover, this study brings to light PH domains with electrostatic profiles that differ markedly from the polarized profile commonly associated with PH domains: (1) Unique among the PH domains that bind poly-phosphoinositides, the PLCδ3 PH domain is predicted to have a dramatically positive electrostatic potential profile (Fig. 2C ▶); (2) the electrostatic profiles of the C-terminal "split" PH domains from PLCγ are predicted to be highly negative and, thus, are similar to the PH domain from a C. elegans muscle protein, UNC-89, the structure of which was recently solved (Blomberg et al. 2000); (3) negatively charged, positively charged, and hydrophobic surface patches are predicted to exist in various combinations for the PH domains from the different PLCβ isoforms (Fig. 7 ▶) and PLCɛ (Fig. 14 ▶). The atypical patterns of electrostatic potential and hydrophobicity of these models implicate regions of the PH domain surface other than the canonical membrane-binding site in important functional interactions.
Taken as a whole, the PLC PH domains exhibit a rich variety of biophysical properties and functions that are well described by our computational models. In each case, electrostatics is predicted to play a major role in dictating function. However, the different family members use electrostatic interactions in quite distinct ways. First, the region surrounding the PI(4,5)P2-binding site in the PLCδ PH domains is highly positively charged (Fig. 2 ▶). Many of the basic residues that contribute to this lobe of positive potential are involved in coordinating the PI(4,5)P2 headgroup (Fig. 1 ▶, orange highlights). In addition, these positively charged regions are predicted to contribute significantly to the membrane localization of the δ PH domains by mediating nonspecific electrostatic interactions with acidic phospholipids (Fig. 3 ▶). The electrostatic profiles of the δ PH domains are altered upon binding PI(4,5)P2 (Fig. 2 ▶). Specifically, the δ1 PH domain becomes overall negatively charged. Our calculations indicate that this change underlies the preference of PLCδ1-PH for soluble Ins(1,4,5)P3 over membrane-embedded PI(4,5)P2 by introducing electrostatic repulsions with the surrounding membrane environment (Table 1). Second, the comparison of the PH domains from PLCδ1 and p130 (or PLC-L; Fig. 4 ▶) highlights features of the latter, in particular, an Asp residue in one of the Ins(1,4,5)P3-binding loops, that may account for the strong preference of this PH domain for soluble Ins(1,4,5)P3 over PI(4,5)P2 in membranes (Varnai et al. 2002). Based on our model, this aspartate is predicted to disfavor binding to PIP(4,5)P2 by causing an electrostatic repulsion from the membrane surface. Third, Figure 7 ▶ illustrates the unique electrostatic and hydrophobic surface properties of the PLCβ PH domains. In each model, the region surrounding the canonical lipid-binding loops is highly negatively charged, arguing against a phosphoinositide-binding function for these PH domains (Razzini et al. 2000). The basic and hydrophobic surface features (Figs. 7, 8 ▶ ▶) may mediate the nonspecific membrane binding observed in biochemical studies of the isolated PH domains (Wang et al. 1999). In addition, our models suggest that the β2 and β3 PH domains interact with β2 subunits from heterotrimeric G proteins (Wang et al. 1999; Barr et al. 2000; Wang et al. 2000) though electrostatic complementarity (positive charge on the PH domain and negative on Gβ). Indeed, the recent structure of the complex of the G protein-coupled receptor kinase 2 (GRK2) and Gβ1γ2 (reveals that the GRK2 PH domain interacts with an acidic region on the surface of Gβ1γ2. (Lodowski et al. 2003). Furthermore, our models provide a mechanism for the regulation of the interaction of PLCβ3-PH with βγ-subunits from heterotrimeric G proteins (Xia et al. 2001): The phosphorylation of a serine in the middle of a basic surface patch on β3-PH is predicted to disrupt the electrostatic complementarity between Gβγ and the PH domain (Fig. 9 ▶). Fourth, our models for the N-terminal PLCγ PH domains exhibit the polarized electrostatic potential characteristic of phosphoinositide-binding PH domains (Fig. 10 ▶). The positive lobe should mediate nonspecific electrostatic interactions with the membrane surface as predicted for the PLCδ PH domains. In addition, the lipid-binding loops contain residues that coordinate the headgroup of PI(3,4,5)P3 in PH domains of known structure, reflecting the observed lipid specificity (Fig. 11 ▶). Moreover, we have defined the sequences for a second C-terminal PH domain for the γ class (Fig. 12 ▶). The models for these PH domains have a highly negatively charged electrostatic profile, indicating functionality other than membrane targeting. Finally, the sequence corresponding to the large β3/β4-loop in our PLCɛ PH domain model has basic and hydrophobic residues that could potentially mediate the membrane association of this PH domain (Fig. 14 ▶). Overall, our models predict that the PLC PH domains use electrostatic interactions in both specific and nonspecific ways to mediate and regulate protein–lipid and protein–protein interactions.
In addition to being able to account for the experimentally observed behaviors of a number of the PLC PH domains, our models have utility in that they provide the basis for directed experimental tests of function. For example, Figure 2 ▶, E through H, illustrates how the electrostatic profiles of the PLCδ PH domains change upon binding the headgroup of PI(4,5)P2. The δ1 PH domain becomes overall negatively charged, whereas the δ3 PH domain remains highly positively charged. The electrostatic profiles indicate that the PI(4,5)P2-bound, membrane-associated form of δ1-PH is targeted laterally to different regions of the plasma membrane than the δ3 isoform. Recent studies show that PI(4,5)P2 is laterally sequestered by membrane-adsorbed basic peptides and that the sequestration is driven by nonspecific electrostatic attraction between the multivalent anionic lipid and the positively charged peptide (Wang et al. 2002). In addition, PI(4,5)P2 co-localizes in membrane ruffles and nascent phagosomes with the protein MARCKS (myristoylated alanine-rich C kinase substrate; Honda et al. 1999; Botelho et al. 2000; Laux et al. 2000; Tall et al. 2000). MARCKS exists at high concentrations in cells and associates with membrane surfaces, in part, through favorable electrostatic interactions between its highly positively charged effector domain (MARCKS[151–175], net charge +13) and acidic phospholipids in the membrane (Kim et al. 1991; Seykora et al. 1996; Swierczynski and Blackshear 1996). The PI(4,5)P2-bound δ1 PH domain (Fig. 2E ▶) should be laterally directed, through favorable electrostatic interactions, to regions enriched in the MARCKS protein, with a membrane-adsorbed effector domain that constitutes a positively charged basin of attraction for negatively charged molecules. Indeed, experiments show that the interaction of a peptide based on MARCKS(151–175) with PI(4,5)P2 does not displace PI(4,5)P2-bound δ1-PH from the membrane (A. Gambhir and S. McLaughlin, pers. comm.). A possible interpretation is that both PI(4,5)P2 and the PI(4,5)P2-bound PH domain are laterally sequestered by membrane-adsorbed MARCKS(151–175) because both species are negatively charged. Similarly, the PI(3,4,5)P3-bound γ1 PH domain (Fig. 10C ▶), which also acquires a strong negative character, should be localized to these regions as well. Although highly speculative, this electrostatic sequestration provides a mechanism whereby PLCδ1 and PLCγ1 (when PI[3,4,5]P3 is present) are localized to regions of membrane that are enriched in their substrate, PI(4,5)P2 (Wang et al. 2002). Conversely, PLCδ3-PH should be excluded from these regions due to its overall basic character (Fig. 2G ▶). The visualization of green fluorescent protein constructs of the δ1, γ1, and δ3 PH domains expressed in cells would provide a test of these predictions.
Our model for the p130 (or PLC-L) PH domain and its comparison with the PH domain from PLCδ1 indicate a number of point mutations (R129W, Y131R, and D152V) that could alter the function of p130-PH so that it may bind to PI(4,5)P2 in membranes rather than just the soluble headgroup, Ins(1,4,5)P3, as observed (Varnai et al. 2002). In addition, our model for PLCβ3-PH indicates that mutating S26 to an acidic residue should result in a PH domain unable to interact with Gβγ heterodimers. Moreover, our models for the PLCγ N-terminal PH domains implicate a number of residues responsible for PI(3,4,5)P3 binding that may be mutated to determine their role in PI3 kinase–dependent membrane recruitment. Finally, the sequence of the unstructured insertion in the elongated β3/β4-loop of PLCɛ-PH may be removed or mutated to determine whether it mediates the association of PLCɛ to membranes containing acidic lipids.
The present study is another in a line of recent computational investigations of the association of signal transduction domains with membrane surfaces. The previous work and the work presented here not only illustrate the success of homology modeling and the calculation of biophysical properties in providing models that account for observed membrane targeting behaviors but also highlight how different lipid-interacting domains, that is, PH, C2, FYVE, and PX domains, share common mechanisms to control membrane association. Nonspecific electrostatic interactions were shown to be a general feature of many C2 domains, including those that are calcium independent (Ananthanarayanan et al. 2002; Kulkarni et al. 2002; Murray and Honig 2002). Similarly, PH domains from all of the PLC classes are predicted to use electrostatic interactions to facilitate their membrane association. FYVE and PX domains, which interact specifically with poly-phosphoinositides, also experience favorable nonspecific electrostatic interactions with acidic phospholipids in membranes (Stahelin et al. 2002, 2003; Diraviyam et al. 2003). Moreover, as shown for the PLCδ and γ PH domains (Figs. 2, 10 ▶ ▶), the electrostatic properties of FYVE and PX domains change dramatically upon binding their phosphoinositide ligands. Similarly, the electrostatic properties of C2 domains are altered, but here the ligand is calcium rather than a lipid. Therefore, for PH, FYVE, and PX domains, the binding of poly-phosphoinositides neutralizes positive charge in the lipid-binding loops, whereas for C2 domains, the binding of calcium ions neutralizes negative charge in the calcium-binding loops. In all cases, membrane localization signals are unmasked upon ligand binding: (1) The PLCδ1 PH domain may co-localize with PI(4,5)P2 and MARCKS in plasma membrane domains because of the overall negative character it acquires; (2) electrostatic potential is reduced around the ligand-binding sites on FYVE, PX and a number of C2 domains so that nonpolar residues in these regions are able to penetrate hydrophobically into the membrane interface; and (3) positive potential due to basic residues in the calcium-binding loops on some C2 domains dominates in the calcium-bound state and drives the binding to membranes containing acidic phospholipids. In essence, each of these domains, upon binding ligand, undergoes a type of "switch" that alters their electrostatic properties in ways that are important for their membrane targeting function.
In summary, the present study demonstrates how three-dimensional models allow us to define structural and biophysical features that are important for function and, thus, add significant value over the analysis of sequence information alone. Including all of the PLC isoforms in the present study was instrumental in discovering both class-specific and isoform-specific features. The family-based analysis described here for PLC PH domains is a powerful approach to functional annotation and should be equally applicable to PH domains from other protein families, for example, serine/threonine protein kinases belonging to the Akt/Rac subfamily and guanine nucleotide exchange factors.
Materials and methods
Sequences modeled
The sequences of the mammalian PLC PH domains were retrieved from publicly available sequence databases: Swiss-Prot/TrEMBL (Boeckmann et al. 2003), PIR (Wu et al. 2002), and NCBI (Wheeler et al. 2003), except that for PLCδ3-PH, which was obtained from Pawelcyzk and Matecki (1999). The boundaries of the PH domain sequences were assigned based on the consensus of the output of a number of secondary structure prediction programs: PSIpred (McGuffin et al. 2000), PHDsec (Rost 1996), SAM-T99 (Karplus and Hu 2001), and SSPro2 (Baldi et al. 1999). This ensured that the sequences encompass all of the secondary structure elements characteristic of PH domains as discerned from structure determinations. Sequence information related to the PLC PH domains modeled in the present study is delineated in the legend to Figure 1 ▶.
Modeling methodology
Because PH domains share low sequence similarity, routine homology modeling alone is not expected to provide adequate results. To generate high-quality models for the PLC PH domains, we implemented a scheme based on the use of multiple approaches at each step: (1) choice of a suitable structural template, (2) alignment of the template and target sequences, (3) model building, and (4) model evaluation and refinement.
Structural template
The PH domain of PLCδ1 was used as the structural template for all of the N-terminal PLC PH domain sequences modeled (PDB identifier: 1mai; Ferguson et al. 1995), whereas the PH domain from Dapp1/Phish was used as the structural template for the C-terminal "split" PH domains of PLCγ (PDB identifier: 1fb8; Ferguson et al. 2000). Both were identified as the best structural templates for the respective sequences based on: (1) sequence searches against the PDB by using the Smith-Waterman algorithm (Smith and Watermann 1981) and BLAST (Altschul et al. 1990), (2) profile alignments as implemented by PSI-BLAST (Altschul et al. 1997) and reverse PSI-BLAST (Marchler-Baur et al. 2002), and (3) fold recognition as implemented in 3D-PSSM (Kelley et al. 2000), 123D+ (Alexandrov et al. 1996), and FUGUE (Shi et al. 2001). The fold recognition results were highly significant: The hits obtained from 123D+, FUGUE and 3D-PSSM were scored, respectively, with e values between 10−8 and 10−4, z scores between 7 and 23, and z scores between 10 and 23.
Sequence alignment
The alignment of the template and target sequences is the most important step in generating a high-quality model, as the accuracy of the modeled structure has been shown to be greatly dependent on the quality of the alignment (Venclovas et al. 2001). Therefore, we used a number of different approaches to generate the sequence alignment so as to combine information from sequence, sequence profiles, secondary structure profiles, and fold recognition. The different programs that were used in generating these alignments include (1) PrISM (Yang and Honig 1999) for both local (Smith-Waterman) and global (Needleman-Wunsch) pairwise alignments; (2) ClustalW (Thompson et al. 1994), TCOFFEE (Notredame et al. 2000), and Gtop Reverse Psi Blast (Kawabata et al. 2002) for multiple sequence alignment; (3) Jigsaw-3D (Bates and Sternberg 1999) for automatic alignment and model building; and (4) 123D+ (Alexandrov et al. 1996), 3D-PSSM (Kelley et al. 2000), and FUGUE (Shi et al. 2001) for fold recognition. In addition, all alignments were assessed and manually edited based on the correspondence of the positions of the secondary structure elements of the template with the predicted consensus secondary structure assignments for the target sequence (as described above). In addition, alignment editing was performed iteratively with model evaluation (described below). The program Verify3D scores each of the residues in a structural model, inputted as a PDB file, according to biochemical criteria extracted from the analysis of high resolution structures (Luthy et al. 1992). The scores are plotted as a function of residue number, which provides a "fitness profile." We reconsidered an alignment between target and template sequences in regions corresponding to low-scoring portions of the Verify3D plot. Generally, manual editing of the alignment in these regions improved the profile of a model.
The multiple sequence alignment of the PLC PH domain family depicted in Figure 1 ▶ was constructed by using the program PIMA (Smith and Smith 1992) in order to highlight overall features of the family that are either conserved across all classes or specific to a single class. The actual alignments between each sequence and the modeling template, PLCδ1-PH, differ slightly from the pairwise alignments that can be extracted from the family alignment in Figure 1 ▶ due to manual editing, as described above. The alignments used in constructing our homology models are available as part of the Electronic Supplemental Material.
Model building
Jigsaw-3D (Bates and Sternberg 1999) was used to construct homology models when there was high sequence identity between the target and template sequences. PrISM (Yang and Honig 1999Yang and Honig 2000) was used when the sequence alignments were generated with its alignment modules. Modeller (Sali and Blundell 1993) and Nest (Z. Xiang and B. Honig, unpubl.) were used when an alignment generated from another program or a manually edited alignment was used. Loop and side-chain prediction, when needed, were implemented by using the programs Loopy (Xiang et al. 2002) and Scap (Xiang and Honig 2001), respectively.
The head group of PI(4,5)P2, Ins(1,4,5)P3, was docked onto the homology models for the PLCδ2, δ3, and δ4 PH domains by structurally aligning the models with the experimentally determined structure of the PLCδ1-PH/Ins(1,4,5)P3 complex (PDB identifier: 1mai) by using the program CE (Shindyalov and Bourne 1998). The structure alignment was used to transfer the coordinates of Ins(1,4,5)P3 from the structure to the models. Similarly, the head group of PI(3,4,5)P3, Ins(1,3,4,5)P4, was docked onto the homology models for the PLCγ1 and γ2 PH domains by structurally aligning the models with the experimentally determined structure of the Bruton’s tyrosine kinase PH domain (PDB identifier: 1b55).
Model evaluation
The quality of the models was assessed by using the structure verification program Verify3D (Luthy et al. 1992), which tests the compatibility of a protein structure with its amino acid sequence. Verify3D constructs a profile for the three-dimensional model in which each residue position is characterized by its environmental score. These scores were derived from a statistical analysis of high-resolution protein structures from the PDB. The Verify3D profile is graphically represented by the numerical scores as a function of the residue number in the structure or model. For high-resolution, experimentally determined structures, the Verify3D scores are positive and consistently high (>0.2), indicating that they provide a reliable means to assess the quality of a protein structure. We have observed in other work (data not shown) that homology models constructed based on alignments to templates of decreasing sequence similarity have correspondingly degraded Verify3D scores, indicating that Verify3D can assess the quality of modeled structures, as well as discriminate among potential models for a single sequence. Because scores are calculated for each residue, the Verify3D profiles were used to identify unreliable regions that had been modeled improperly; these were subsequently improved by manually editing the alignment between query and template sequences.
The Verify3D profiles are exceptionally good for the β3, δ1, δ2, δ3, δ4, N- and C-terminal γ1, C-terminal γ2, and p130 PH domain models. The β1, β2, β4, N-terminal γ2, and ɛ PH domains have profiles that are consistently positive but have relatively lower absolute scores (see Electronic Supplemental Material). We further evaluated our models with the program ProsaII, which calculates energy profiles for a structural model by using a molecular mechanics force field (Sippl 1993). The results of this analysis correlate well with the Verify3D results.
Sensitivity analysis of homology models
In all cases, residues corresponding to the hydrophobic core of the PH domain fold were reliably identified. On the other hand, as expected, loop regions were much more variably defined and modeled. However, we used secondary structure profiles (Figs. 6, 13 ▶ ▶) to refine the alignment between target and template in these regions. Still, it was sometimes the case that the alignment had significant gaps and insertions in the vicinity of the loops. The use of the loop modeling program, Loopy, often improved the quality of a model. But more definitively, we examined the robustness of biophysical properties across alternative models for a given sequence. Because in many cases the sequence identity between target and template is low, it was possible to generate a number of alternative alignments for each target and produce a series of structural models that scored equally well according to Verify3D and ProsaII. In addition, we used PH domain structures other than that of PLCδ1-PH as structural templates in constructing alternative models. The biophysical features quantified and described in this paper—PI(4,5)P2-binding residues for the δ class, the surface location of Ser26 in PLCβ3-PH, and charged or hydrophobic surface patches—were constant across the different models for a given sequence and, in particular, were insensitive to changes in loop structure and side-chain rotamers.
Analysis of the models
The models were analyzed according to their sequence, structural, and biophysical properties. To compare the lipid-binding residues in the models for the PLCγ PH domains with those from other PI(3,4,5)P3-binding PH domains, structure superpositions, and structure-based sequence alignments were made with the program PrISM. The analysis of biophysical properties including the electrostatics, hydrophobicity, and shape of each model was conducted by using the surface property analysis tools in the program GRASP (Nicholls et al. 1991).
The electrostatic free energy component of the membrane interaction of the PLCδ1 and δ3 and PLCβ2 PH domains was obtained from a modified version of the DelPhi program (Gallagher and Sharp 1998) that solves the nonlinear Poisson Boltzmann equation for protein–membrane systems (Ben-Tal et al. 1996). DelPhi produces FDPB solutions for a system in which the solvent is described in terms of a bulk dielectric constant and concentrations of mobile ions, whereas solutes (here, PH domains and phospholipid membranes) are described in terms of the coordinates of the individual atoms, as well as their atomic radii and partial charges. The FDPB method has previously been shown to yield excellent agreement with experimental measurements of the binding of peptides and proteins to charged membranes (Ben-Tal 1996, 1997; Murray et al. 1998, 2001; Burden et al. 1999; Provitera et al. 2000; Murray and Honig 2002).
In the calculations described in this work, the PH domains and phospholipid bilayers are represented in atomic detail, whereas the solvent is modeled as a homogeneous dielectric medium. Each atom of a protein/bilayer system is assigned a radius and partial charge that is located at its nucleus; the protein/membrane model is then mapped onto a three-dimensional lattice of l3 points, each of which represents a small region of the protein, membrane, or solvent. The charges and radii used for the amino acids were taken from a CHARMM22 parameter set (Brooks et al. 1983), and those used for the lipids are the ones described by Peitzsch et al. (1995) and were used in previous studies (Ben-Tal et al. 1996, 1997; Murray et al. 1998). The partial charges for Ins(1,4,5)P3 were taken from similar functional groups from the CHARMM22 parameter set so that the net charge on the lipid headgroup was −5 or −6, which corresponds to the net charge the headgroup would have in the context of PI(4,5)P2 or in the context of soluble Ins(1,4,5)P3, respectively. Ins(1,3,4,5)P4 was similarly modeled. Regions inside the molecular surfaces of the protein and membrane are assigned a dielectric constant of two to account for electronic polarizability, and those outside are assigned a dielectric constant of 80 (Sharp and Honig 1990). An ion exclusion layer is added to the solutes and extends 2 Å beyond the molecular surfaces. The nonlinear Poisson-Boltzmann equation is solved in the finite difference approximation, and the numerical calculation of the potential is iterated to convergence, which is defined as the point at which the potential changes <10−4 kT/e between successive iterations. Electrostatic free energies are obtained from the calculated potentials (Sharp and Honig 1990), and the electrostatic free energy of interaction is determined as the difference between the electrostatic free energy of a PH domain in a specific orientation with respect to the membrane surface, Gel(P × M), and the electrostatic free energies of the PH domain, Gel(P), and membrane, Gel(M), infinitely far apart, that is, taken separately:
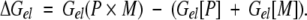 |
A sequence of focusing runs (Gilson et al. 1987) of increasing resolution was used to calculate the electrostatic potentials (e.g., 0.375, 0.75, 1.5, and 3.0 grid/Å). In the initial calculation, the PH domain/membrane model encompassed a small percentage of the lattice (∼10%), and the potentials at the boundary points of the lattice are approximately zero; this procedure ensures that the system is electroneutral. Lattice sizes of 2733 and 3693 were used, and the calculations were performed to final resolutions of 3 grid/Å and 4 grid/Å, respectively. The precision in the electrostatic free energies of interaction, determined as the difference between the results obtained at the two highest resolution scales, is <0.2 kcal/mole for all calculations.
Phospholipid bilayers were built as described previously (Ben-Tal et al. 1996). Each lipid leaflet contains 192 lipids distributed uniformly in a hexagonal lattice. Each lipid headgroup occupies an area of 68 Å2 in the plane of the membrane, and the lipid head group regions from the two opposing membrane leaflets encompass about one-half the thickness of the bilayer. It is assumed that the lipids change neither structure nor position upon interaction with the protein. These approximations are not unreasonable for the PLCδ PH domains studied here because they are highly charged and do not penetrate the membrane interface to a large degree. Previous work has shown that the membrane partitioning of basic peptides that reside outside the polar envelope of the membrane is independent of whether the membrane is in the liquid crystalline or gel phase, indicating that the use of static bilayer models is appropriate for calculating the binding of peripheral proteins (Ben-Tal et al. 1996). Hydrogen atoms were added to the heavy atoms in the PH domain structures with the program CHARMM (Brooks et al. 1983). The structures with hydrogens were subjected to conjugate gradient minimization with a harmonic restraint force of 50 kcal/mole/Å2 applied to the heavy atoms located at the original crystallographic coordinates.
In this article, we have not calculated the full membrane partition coefficient as described in previous work, which requires treating many different orientations of the protein with respect to the membrane (Murray et al. 1997). Rather, we have considered the orientation of minimum electrostatic free energy (Ben-Tal et al. 1997). Previous work has established that for peripheral association, both the relative binding free energies and the electrostatic contribution to the absolute binding free energies are well described by consideration of this orientation alone (Ben-Tal et al. 1996, 1997; Murray et al. 1998; Arbuzova et al. 2000). Therefore, the membrane-associated orientations of the PLCδ and β PH domains, for which data were presented in this study, are the orientations of minimum electrostatic free energy. These were determined by comparing the electrostatic free energies of interaction of different orientations sampled about an orientation obtained by visual inspection of the electrostatic potential profiles in GRASP.
Electronic supplemental material
The sequences and coordinate files representing our models for all mammalian PLC PH domains as well as other supplementary information, for example, GRASP images and Verify3D profiles, are available at our Web site (http://maat.med.cornell.edu/ph_PVsuppl.html).
Acknowledgments
This work was supported by NIH grant GM66147.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0358803.
References
- Alexandrov, N.N., Nussinov, R., and Zimmer, R.M. 1996. Fast protein fold recognition via sequence to structure alignment and contact capacity potentials. Pac. Symp. Biocomput. 53–72. [PubMed]
- Altschul, S.F., Gish, W., Miller, W., Meyers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthanarayanan, B., Das, S., Rhee, S.G., Murray, D., and Cho, W. 2002. Membrane targeting of C2 domains of phospholipase C-δ isoforms. J. Biol. Chem. 277 3568–3575. [DOI] [PubMed] [Google Scholar]
- Arbuzova, A., Wang, L., Wang, J., Hangyas-Mihalyne, G., Murray, D., Honig, B., and McLaughlin, S. 2000. Membrane binding of peptides containing both basic and aromatic residues: Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry 39 10330–10339. [DOI] [PubMed] [Google Scholar]
- Baldi, P., Brunak, S., Frasconi, P., Soda, G., and Pollastri, G. 1999. Exploiting the past and the future in protein secondary structure prediction. Bioinformatics 15 937–946. [DOI] [PubMed] [Google Scholar]
- Baraldi, E., Carugo, K.D., Hyvonen, M., Surdo, P.L., Riley, A.M., Potter, B.V., O’Brien, R., Ladbury, J.E., and Saraste, M. 1999. Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure Fold Des. 7 449–460. [DOI] [PubMed] [Google Scholar]
- Barr, A.J., Ali, H., Haribabu, B., Snyderman, R., and Smrcka, A.V. 2000. Identification of a region at the N-terminus of phospholipase C-β3 that interacts with G protein βγ subunits. Biochemistry 39 1800–1806. [DOI] [PubMed] [Google Scholar]
- Bates, P.A. and Sternberg, M.J. 1999. Model building by comparison at CASP3: Using expert knowledge and computer automation. Proteins 3 47–54. [DOI] [PubMed] [Google Scholar]
- Ben-Tal, N., Honig, B., Peitzsch, R.M., Denisov, G., and McLaughlin, S. 1996. Binding of small basic peptides to membranes containing acidic lipids: Theoretical models and experimental results. Biophys. J. 71 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tal, N., Honig, B., Miller, C., and McLaughlin, S. 1997. Electrostatic binding of proteins to membranes: Theoretical predictions and experimental results with charybdotoxin and phospholipid vesicles. Biophys. J. 73 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg, N. and Nilges, M. 1997. Functional diversity of PH domains: An exhaustive modeling study. Fold Des. 2 343–355. [DOI] [PubMed] [Google Scholar]
- Blomberg, N., Gabdoulline, R.R., Nilges, M., and Wade, R.C. 1999. Classification of protein sequences by homology modeling and quantitative analysis of electrostatic similarity. Proteins 37 379–387. [DOI] [PubMed] [Google Scholar]
- Blomberg, N., Baraldi, E., Sattler, M., Saraste, M., and Nilges, M. 2000. Structure of a PH domain from the C. elegans muscle protein UNC-89 suggests a novel function. Structure Fold Des. 8 1079–1087. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.C., Estreicher, A., Gasteiger, E., Martin, M.J., Michoud, K., O’Donovan, C., Phan, I., et al. 2003. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho, R.J., Teruel, M., Dierckman, R., Anderson, R., Wells, A., York, J.D., Meyer, T., and Grinstein, S. 2000. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell. Biol. 151 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4 187–217. [Google Scholar]
- Burden, L.M., Rao, V.D., Murray, D., Ghirlando, R., Doughman, S.D., Anderson, R.A., and Hurley, J.H. 1999. The flattened face of type II β phosphatidylinositol phosphate kinase binds acidic phospholipid membranes. Biochemistry 38 15141–15149. [DOI] [PubMed] [Google Scholar]
- Carman, C.V., Barak, L.S., Chen, C., Liu-Chen, L.Y., Onorato, J.J., Kennedy, S.P., Caron, M.G., and Benovic, J.L. 2000. Mutational analysis of Gβγ and phospholipid interaction with G protein–coupled receptor kinase 2. J. Biol. Chem. 275 10443–10452. [DOI] [PubMed] [Google Scholar]
- Chang, J.S., Seok, H., Kwon, T.K., Min, S., Ahn, B.H., Lee, Y.H., Suh, J.W., Kim, J.W., Iwashita, S., Omori, A., et al. 2002. Interaction of elongation factor-1α and pleckstrin homology domain of phospholipase C-γ1 with activating its activity. J. Biol. Chem. 277 19697–19702. [DOI] [PubMed] [Google Scholar]
- Cifuentes, M.E., Delaney, T., and Rebecchi, M.J. 1994. D-myo-inositol 1,4,5-trisphosphate inhibits binding of phospholipase C-δ1 to bilayer membranes. J. Biol. Chem. 269 1945–1948. [PubMed] [Google Scholar]
- Cockcroft, S. and Thomas, G.M. 1992. Inositol-lipid-specific phospholipase C isoenzymes and their differential regulation by receptors. Biochem. J. 288 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen, P.J. and Chardin, P. 2000. Membrane targeting: What a difference a G makes. Curr. Biol. 10 R876–R878. [DOI] [PubMed] [Google Scholar]
- Diraviyam, K., Stahelin, R.V., Cho, W., and Murray, D. 2003. Computer modeling of the membrane interaction of FYVE domains. J. Mol. Biol. 328 721–736. [DOI] [PubMed] [Google Scholar]
- Essen, L.O., Perisic, O., Cheung, R., Katan, M., and Williams, R.L. 1996. Crystal structure of a mammalian phosphoinositide-specific phospholipase C δ. Nature 380 595–602. [DOI] [PubMed] [Google Scholar]
- Falasca, M., Logan, S.K., Lehto, V.P., Baccante, G., Lemmon, M.A., and Schlessinger, J. 1998. Activation of phospholipase Cγ by PI 3-kinase–induced PH domain-mediated membrane targeting. EMBO J. 17 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falquet, L., Pagni, M., Bucher, P., Hulo, N., Sigrist, C.J.A., Hofmann, K., and Bairoch, A. 2002. The PROSITE database: Its status in 2002. Nucleic Acids Res. 30 235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, K.M., Lemmon, M.A., Schlessinger, J., and Sigler, P.B. 1995. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83 1037–1046. [DOI] [PubMed] [Google Scholar]
- Ferguson, K.M., Kavran, J.M., Sankaran, V.G., Fournier, E., Isakoff, S.J., Skolnik, E.Y., and Lemmon, M.A. 2000. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell 6 373–384. [DOI] [PubMed] [Google Scholar]
- Fushman, D., Najmabadi-Haske, T., Cahill, S., Zheng, J., LeVine III, H., and Cowburn, D. 1998. The solution structure and dynamics of the pleckstrin homology domain of G protein–coupled receptor kinase 2 (βadrenergic receptor kinase 1): A binding partner of Gβγ subunits. J. Biol. Chem. 273 2835–2843. [DOI] [PubMed] [Google Scholar]
- Gallagher, K. and Sharp, K. 1998. Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys. J. 75 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, P., Gupta, R., Shah, S., Morris, A.J., Rudge, S.A., Scarlata, S., Petrova, V., McLaughlin, S., and Rebecchi, M.J. 1995. The pleckstrin homology domain of phospholipase C-δ1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry 34 16228– 16234. [DOI] [PubMed] [Google Scholar]
- Gilson, M.K., Sharp, K.A., and Honig, B.H. 1987. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J. Comp. Chem. 9 327–335. [Google Scholar]
- Henry, R.A., Boyce, S.Y., Kurz, T., and Wolf, R.A. 1995. Stimulation and binding of myocardial phospholipase C by phosphatidic acid. Am. J. Physiol. 269 C349–C358. [DOI] [PubMed] [Google Scholar]
- Hirose, K., Kadowaki, S., Tanabe, M., Takeshima, H., and Iino, M. 1999. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science 284 1527–1530. [DOI] [PubMed] [Google Scholar]
- Honda, A., Nogami, M., Yokozeki, T., Yamazaki, M., Nakamura, H., Watanabe, H., Kawamoto, K., Nakayama, K., Morris, A.J., Frohman, M.A., et al. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99 521–532. [DOI] [PubMed] [Google Scholar]
- Honig, B. and Nicholls, A. 1995. Classical electrostatics in biology and chemistry. Science 268 1144–1149. [DOI] [PubMed] [Google Scholar]
- Hurley, J.H. and Meyer, T. 2001. Subcellular targeting by membrane lipids. Curr. Opin. Cell. Biol. 13 146–152. [DOI] [PubMed] [Google Scholar]
- Kanematsu, T., Takeya, H., Watanabe, Y., Ozaki, S., Yoshida, M., Koga, T., Iwanaga, S., and Hirata, M. 1992. Putative inositol 1,4,5-trisphosphate binding proteins in rat brain cytosol. J. Biol. Chem. 267 6518–6525. [PubMed] [Google Scholar]
- Kanematsu, T., Misumi, Y., Watanabe, Y., Ozaki, S., Koga, T., Iwanaga, S., Ikehara, Y., and Hirata, M. 1996. A new inositol 1,4,5-trisphosphate binding protein similar to phospholipase C-δ1. Biochem. J. 313 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus, K. and Hu, B. 2001. Evaluation of protein multiple alignments by SAM-T99 using the BAliBASE multiple alignment test set. Bioinformatics 17 713–720. [DOI] [PubMed] [Google Scholar]
- Kawabata, T., Fukuchi, S., Homma, K., Ota, M., Araki, J., Ito, T., Ichiyoshi, N., and Nishikawa, K. 2002. GTOP: A database of protein structures predicted from genome sequences. Nucleic Acids Res. 30 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, G.G., Reks, S.E., Ondrako, J.M., and Smrcka, A.V. 2001. Phospholipase CE: A novel Ras effector. EMBO J. 20 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J.E. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- Kim, C.G., Park, D., and Rhee, S.G. 1996. The role of carboxyl-terminal basic amino acids in Gqα-dependent activation, particulate association, and nuclear localization of phospholipase C-β1. J. Biol. Chem. 271 21187–21192. [DOI] [PubMed] [Google Scholar]
- Kim, J., Mosior, M., Chung, L.A., Wu, H., and McLaughlin, S. 1991. Binding of peptides with basic residues to membranes containing acidic phospholipids. Biophys. J. 60 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni, S., Das, S., Funk, C.D., Murray, D., and Cho, W. 2002. Molecular basis of the specific subcellular localization of the C2-like domain of 5-lipoxygenase. J. Biol. Chem. 277 13167–13174. [DOI] [PubMed] [Google Scholar]
- Laux, T., Fukami, K., Thelen, M., Golub, T., Frey, D., and Caroni, P. 2000. GAP43, MARCKS, and CAP23 Modulate PI(4,5)P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell. Biol. 149 1455–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, M.A. and Ferguson, K.M. 2000. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 350 1–18. [PMC free article] [PubMed] [Google Scholar]
- Lemmon, M.A., Ferguson, K.M., O’Brien, R., Sigler, P.B., and Schlessinger, J. 1995. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. 92 10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T., Tsukada, S., Satterthwaite, A., Havlik, M.H., Park, H., Takatsu, K., and Witte, O.N. 1995. Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity 2 451–460. [DOI] [PubMed] [Google Scholar]
- Lietzke, S.E., Bose, S., Cronin, T., Klarlund, J., Chawla, A., Czech, M.P., and Lambright, D.G. 2000. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol. Cell 6 385–394. [DOI] [PubMed] [Google Scholar]
- Lodowski, D.T., Picher, J.A., Capel, W.D., Lefkowitz, R.J., and Tessmer, J.J.G. 2003. Keeping G proteins at bay: A complex between G protein-coupled receptor kinase 2 and Gβγ. Science 300 1256–1262. [DOI] [PubMed] [Google Scholar]
- Luthy, R., Bowie, J.U., and Eisenberg, D. 1992. Assessment of protein models with three-dimensional profiles. Nature 356 83–85. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Panchenko, A.R., Ariel, N., and Bryant, S.H. 2002. Comparison of sequence and structure alignments for protein domains. Proteins 48 439–446. [DOI] [PubMed] [Google Scholar]
- Martelli, A.M., Gilmour, R.S., Bertagnolo, V., Neri, L.M., Manzoli, L., and Cocco, L. 1992. Nuclear localization and signaling activity of phospholipase C β in Swiss 3T3 cells. Nature 358 242–245. [DOI] [PubMed] [Google Scholar]
- Matsuda, M., Paterson, H.F., Rodriguez, R., Fensome, A.C., Ellis, M.V., Swann, K., and Katan, M. 2001. Real time fluorescence imaging of PLCγ translocation and its interaction with the epidermal growth factor receptor. J. Cell. Biol. 153 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin, L.J., Bryson, K., and Jones, D.T. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Murray, D. and Honig, B. 2002. Electrostatic control of the membrane targeting of C2 domains. Mol. Cell 9 145–154. [DOI] [PubMed] [Google Scholar]
- Murray, D., Ben-Tal, N., Honig, B., and McLaughlin, S. 1997. Electrostatic interaction of myristoylated proteins with membranes: Simple physics, complicated biology. Structure 5 985–989. [DOI] [PubMed] [Google Scholar]
- Murray, D., Hermida-Matsumoto, L., Buser, C.A., Tsang, J., Sigal, C.T., Ben-Tal, N., Honig, B., Resh, M.D., and McLaughlin, S. 1998. Electrostatics and the membrane association of Src: Theory and experiment. Biochemistry 37 2145–2159. [DOI] [PubMed] [Google Scholar]
- Murray, D., McLaughlin, S., and Honig, B. 2001. The role of electrostatic interactions in the regulation of the membrane association of G protein βγ heterodimers. J. Biol. Chem. 276 45153–45159. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302 205–217. [DOI] [PubMed] [Google Scholar]
- Pawelczyk, T. and Lowenstein, J.M. 1993. Binding of phospholipase C δ1 to phospholipid vesicles. Biochem. J. 291 693–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelczyk, T. and Matecki, A. 1999. Phospholipase C-δ3 binds with high specificity to phosphatidylinositol 4,5-bisphosphate and phosphatidic acid in bilayer membranes. Eur. J. Biochem. 262 291–298. [DOI] [PubMed] [Google Scholar]
- Peitzsch, R.M., Eisenberg, M., Sharp, K.A., and McLaughlin, S. 1995. Calculations of the electrostatic potential adjacent to model phospholipid bilayers. Biophys. J. 68 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip, F., Guo, Y., and Scarlata, S. 2002. Multiple roles of pleckstrin homology domains in phospholipase Cβ function. FEBS Lett. 531 28–32. [DOI] [PubMed] [Google Scholar]
- Provitera, P., Bouamr, F., Murray, D., Carter, C., and Scarlata, S. 2000. Binding of equine infectious anemia virus matrix protein to membrane bilayers involves multiple interactions. J. Mol. Biol. 296 887–898. [DOI] [PubMed] [Google Scholar]
- Razzini, G., Brancaccio, A., Lemmon, M.A., Guarnieri, S., and Falasca, M. 2000. The role of the pleckstrin homology domain in membrane targeting and activation of phospholipase Cβ1. J. Biol. Chem. 275 14873–14881. [DOI] [PubMed] [Google Scholar]
- Rebecchi, M., Peterson, A., and McLaughlin, S. 1992. Phosphoinositide-specific phospholipase C-δ1 binds with high affinity to phospholipid vesicles containing phosphatidylinositol 4,5-bisphosphate. Biochemistry 31 12742–12747. [DOI] [PubMed] [Google Scholar]
- Rebecchi, M.J. and Pentyala, S.N. 2000. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80 1291–1335. [DOI] [PubMed] [Google Scholar]
- Rebecchi, M.J. and Scarlata, S. 1998. Pleckstrin homology domains: A common fold with diverse functions. Annu. Rev. Biophys. Biomol. Struct. 27 503–528. [DOI] [PubMed] [Google Scholar]
- Rhee, S.G. 2001. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70 281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, S.G. and Bae, Y.S. 1997. Regulation of phosphoinositide-specific phospholipase C isozymes. J. Biol. Chem. 272 15045–15048. [DOI] [PubMed] [Google Scholar]
- Romoser, V., Ball, R., and Smrcka, V. 1996. Phospholipase C β2 association with phospholipid interfaces assessed by fluorescence resonance energy transfer: G protein βγ subunit-mediated translocation is not required for enzyme activation. J. Biol. Chem. 271 25071–25078. [DOI] [PubMed] [Google Scholar]
- Rong, S.B., Hu, Y., Enyedy, I., Powis, G., Meuillet, E.J., Wu, X., Wang, R., Wang, S., and Kozikowski, A.P. 2001. Molecular modeling studies of the akt ph domain and its interaction with phosphoinositides. J. Med. Chem. 44 898–908. [DOI] [PubMed] [Google Scholar]
- Rost, B. 1996. PHD: Predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 266 525–539. [DOI] [PubMed] [Google Scholar]
- Runnels, L.W., Jenco, J., Morris, A., and Scarlata, S. 1996. Membrane binding of phospholipases C-β1 and C-β2 is independent of phosphatidylinositol 4,5-bisphosphate and the α and βγ subunits of G proteins. Biochemistry 35 16824–16832. [DOI] [PubMed] [Google Scholar]
- Sali, A. and Blundell, T.J. 1993. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Schultz, J., Milpetz, F., Bork, P., and Ponting, C.P. 1998. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. 95 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seykora, J.T., Myat, M.M., Allen, L.A., Ravetch, J.V., and Aderem, A. 1996. Molecular determinants of the myristoyl-electrostatic switch of MARCKS. J. Biol. Chem. 271 18797–18802. [DOI] [PubMed] [Google Scholar]
- Shi, J., Blundell, T.L., and Mizuguchi, K. 2001. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310 243–257. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Singer, A.U., Waldo, G.L., Harden, T.K., and Sondek, J. 2002. A unique fold of phospholipase C-β mediates dimerization and interaction with G α q. Nat. Struct. Biol. 9 32–36. [DOI] [PubMed] [Google Scholar]
- Sippl, M.J. 1993. Recognition of errors in three-dimensional structures of proteins. Proteins 17 355–361. [DOI] [PubMed] [Google Scholar]
- Smith, R.F. and Smith, T.F. 1992. Pattern-induced multi-sequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for use in comparative protein modelling. Protein Eng. 5 35–41. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Smrcka, A.V. and Sternweis, P.C. 1993. Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C β by G protein α and βγ subunits. J. Biol. Chem. 268 9667–9674. [PubMed] [Google Scholar]
- Stahelin, R.V., Long, F., Diraviyam, K., Bruzik, K.S., Murray, D., and Cho, W. 2002. Phosphatidylinositol 3-phosphate induces the membrane penetration of the FYVE domains of Vps27p and Hrs. J. Biol. Chem. 277 26379–26388. [DOI] [PubMed] [Google Scholar]
- Stahelin, R.V., Burian, A., Bruzik, K.S., Murray, D., and Cho, W. 2003. Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J. Biol. Chem. 278 14469–14479. [DOI] [PubMed] [Google Scholar]
- Sugimoto, K., Mori, Y., Makino, K., Ohkubo, K., and Morii, T. 2003. Functional reassembly of a split PH domain. J. Am. Chem. Soc. 125 5000–5004. [DOI] [PubMed] [Google Scholar]
- Swierczynski, S.L. and Blackshear, P.J. 1996. Myristoylation-dependent and electrostatic interactions exert independent effects on the membrane association of the myristoylated alanine-rich protein kinase C substrate protein in intact cells. J. Biol. Chem. 271 23424–23430. [DOI] [PubMed] [Google Scholar]
- Takeuchi, H., Oike, M., Paterson, H.F., Allen, V., Kanematsu, T., Ito, Y., Erneux, C., Katan, M., and Hirata, M. 2000. Inhibition of Ca(2+) signalling by p130, a phospholipase-C–related catalytically inactive protein: Critical role of the p130 pleckstrin homology domain. Biochem. J. 349 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall, E.G., Spector, I., Pentyala, S.N., Bitter, I., and Rebecchi, M.J. 2000. Dynamics of phosphatidylinositol 4,5-bisphosphate in actin-rich structures. Curr. Biol. 10 743–746. [DOI] [PubMed] [Google Scholar]
- Thodeti, C.K., Adolfsson, J., Juhas, M., and Sjolander, A. 2000. Leukotriene D(4) triggers an association between Gβγ subunits and phospholipase C-γ1 in intestinal epithelial cells. J. Biol. Chem. 275 9849–9853. [DOI] [PubMed] [Google Scholar]
- Thomas, C.C., Dowler, S., Deak, M., Alessi, D.R., and van Aalten, D.M. 2001. Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain–containing protein 1 (TAPP1): Molecular basis of lipid specificity. Biochem. J. 358 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P., Lin, X., Lee, S.B., Tuymetova, G., Bondeva, T., Spat, A., Rhee, S.G., Hajnoczky, G. and Balla, T. 2002. Inositol lipid binding and membrane localization of isolated pleckstrin homology (PH) domains: Studies on the PH domains of phospholipase C δ1 and p130. J. Biol. Chem. 277 27412–27422. [DOI] [PubMed] [Google Scholar]
- Venclovas, C., Zemla, A., Fidelis, K., and Moult, J. 2001. Comparison of performance in successive CASP experiments. Proteins 5 163–170. [DOI] [PubMed] [Google Scholar]
- Wang, J., Gambhir, A., Hangyas-Mihalyne, G., Murray, D., Golebiewska, U., and McLaughlin, S. 2002. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277 34401–34412. [DOI] [PubMed] [Google Scholar]
- Wang, T., Pentyala, S., Rebecchi, M.J., and Scarlata, S. 1999. Differential association of the pleckstrin homology domains of phospholipases C-β1, C-β2, and C-δ1 with lipid bilayers and the βγ subunits of heterotrimeric G proteins. Biochemistry 38 1517–1524. [DOI] [PubMed] [Google Scholar]
- Wang, T., Dowal, L., El Maghrabi, M.R., Rebecchi, M., and Scarlata, S. 2000. The pleckstrin homology domain of phospholipase C-β2 links the binding of gβγ to activation of the catalytic core. J. Biol. Chem. 275 7466–7469. [DOI] [PubMed] [Google Scholar]
- Wheeler, D.L., Church, D.M., Federhen, S., Lash, A.E., Madden, T.L., Pontius, J.U., Schuler, G.D., Schriml, L.M., Sequeira, E., Tatusova, T.A. et al. 2003. Database resources of the National Center for Biotechnology. Nucl. Acids Res. 31 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley, W.C. and White, S.H. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3 842–848. [DOI] [PubMed] [Google Scholar]
- Wing, M.R., Houston, D., Kelley, G.G., Der, C.J., Siderovski, D.P., and Harden, T.K. 2001. Activation of phospholipase C-ɛ by heterotrimeric G protein βγ-subunits. J. Biol. Chem. 276 48257–48261. [DOI] [PubMed] [Google Scholar]
- Wu, C.H., Huang, H., Arminski, L., Castro-Alvear, J., Chen, Y., Hu, Z.Z., Ledley, R.S., Lewis, K.C., Mewes, H.W., Orcutt, B.C., et al. 2002. The Protein Information Resource: An integrated public resource of functional annotation of proteins. Nucleic Acids Res. 30 35–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, C., Bao, Z., Yue, C., Sanborn, B.M., and Liu, M. 2001. Phosphorylation and regulation of G-protein activated phospholipase C-β3 by cGMP-dependent protein kinases. J. Biol. Chem. 276 19770–19777. [DOI] [PubMed] [Google Scholar]
- Xiang, Z. and Honig, B. 2001. Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 311 421–430. [DOI] [PubMed] [Google Scholar]
- Xiang, Z., Soto, C.S., and Honig, B. 2002. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. 99 7432–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagisawa, H., Sakuma, K., Paterson, H.F., Cheung, R., Allen, V., Hirata, H., Watanabe, Y., Hirata, M., Williams, R.L., and Katan, M. 1998. Replacements of single basic amino acids in the pleckstrin homology domain of phospholipase C-δ1 alter the ligand binding, phospholipase activity, and interaction with the plasma membrane. J. Biol. Chem. 273 417–424. [DOI] [PubMed] [Google Scholar]
- Yang, A.S. and Honig, B. 1999. Sequence to structure alignment in comparative modeling using PrISM. Proteins 37 66–72. [DOI] [PubMed] [Google Scholar]
- Yoshida, M., Kanematsu, T., Watanabe, Y., Koga, T., Ozaki, S., Iwanaga, S., and Hirata, M. 1994. D-myo-inositol 1,4,5-trisphosphate-binding proteins in rat brain membranes. J. Biochem. (Tokyo) 115 973–980. [DOI] [PubMed] [Google Scholar]