

Abstract
Mycothiol is the predominant low-molecular weight thiol produced by actinomycetes, including Mycobacterium tuberculosis. The last reaction in the biosynthetic pathway for mycothiol is catalyzed by mycothiol synthase (MshD), which acetylates the cysteinyl amine of cysteine–glucosamine–inositol (Cys–GlcN–Ins). The crystal structure of MshD was determined in the presence of coenzyme A and acetyl–CoA . MshD consists of two tandem-repeated domains, each exhibiting the Gcn5-related N-acetyltransferase (GNAT) fold. These two domains superimpose with a root-mean-square deviation of 1.7 Å over 88 residues, and each was found to bind one molecule of coenzyme, although the binding sites are quite different. The C-terminal domain has a similar active site to many GNAT members in which the acetyl group of the coenzyme is presented to an open active site slot. However, acetyl–CoA bound to the N-terminal domain is buried, and is apparently not positioned to promote acetyl transfer. A modeled substrate complex indicates that Cys–GlcN–Ins would only fill a portion of a negatively charged channel located between the two domains. This is the first structure determined for an enzyme involved in the biosynthesis of mycothiol.
Keywords: Mycothiol biosynthesis, GNAT fold, acetyltransferase
Mycothiol is the major low-molecular weight thiol in actinomycetes, including Mycobacterium tuberculosis, where it serves a function similar to glutathione found in Gram-negative bacteria and eukaryotes. These functions include maintenance of the cellular redox state and the detoxification of powerful electrophiles (Fahey 2001). Actinomycetes deficient in mycothiol biosynthesis are more susceptible to oxidants and to antimicrobial agents (Rawat et al. 2002), a feature that has focused attention on enzymes of the mycothiol biosynthetic pathway as potential drug targets. Most of the biosynthetic steps leading to mycothiol have been elucidated (Newton and Fahey 2002). Recently, the gene that codes for mycothiol synthase (MshD) was identified (Koledin et al. 2002). MshD, an enzyme unique to actinomycetes, catalyzes the acetylation of the cysteinyl amine of 1-d-myo-inosityl-2-l-cysteinylamido-2-deoxy-α-d-glucopyranoside (Cys–GlcN–Ins), the last step in mycothiol biosynthesis (Fig. 1A ▶). Chromosomal deletion of MshD in Mycobacterium smegmatis resulted in a 290-fold increase in the cellular concentration of Cys–GlcN–Ins and an 83-fold drop in mycothiol over wild type (Koledin et al. 2002). In addition, the cloning and expression of MshD from M. tuberculosis in Escherichia coli resulted in a crude extract with significant mycothiol synthase activity (500-fold over background; Koledin et al. 2002). Sequence analysis suggests that MshD belongs to the ubiquitous Gcn5-related N-acetyltransferase family (GNAT). Interestingly, the sequence of MshD is twice the length of the typical GNAT protein, and exhibits two distinct regions with homology to GNATs, indicating that MshD may be composed of two GNAT domains (Koledin et al. 2002). Here we report the structure of mycothiol synthase, Rv0819, from M. tuberculosis in complex with CoA and acetyl–CoA.
Figure 1.

Structural features of MshD. (A) Reaction catalyzed by MshD, the final step in the biosynthesis of mycothiol. (B) Stereo Cα diagram of MshD with ribbon diagram of the MshD binary acetyl–CoA complex. In the Cα diagram every 10th residue is marked by a black sphere, while every 20th residue is numbered. In the ribbon diagram, the conserved structural elements of the GNAT fold are colored according to Vetting et al. (2002). Dashed lines indicate structural elements with no electron density. Acetyl–CoA is modeled in a stick representation. Figures 1B and 2 ▶ ▶ were prepared with the programs PYMOL (DeLano 2002) and MOLSCRIPT (Kraulis 1991).
Results and Discussion
The crystal packing of MshD determined here suggests that the enzyme functions as a monomer in solution, in agreement with dynamic light scattering and gel filtration results. The MshD polypeptide (315 amino acids) is a two-domain protein in which the N-terminal domain (residues 1–140) and the C-terminal domain (141–315) share a similar α + β fold with a generic topology of β1α1α2β2β3β4α3β5α4β6′ (Fig. 1B ▶). These domains are related to the Gcn5 N-acetyltransferase (GNAT) family of acetyltransferases whose members include enzymes with such diverse functions as transcriptional regulation, protein trafficking, circadian rhythms, bacterial quorum sensing, and antibiotic resistance (Neuwald and Landsman 1997; Dyda et al. 2000; Watson et al. 2002). The central antiparallel β-sheet of each domain (β1–β4) is flanked by helices α1/α2 on one face and helices α3/α4 on the other. The two tandem-repeated domains superimpose with an RMSD of 1.7 Å over 88 structurally similar residues. The largest differences between the two domains are the lack of β-strand 1 in the N-terminal domain, the position of helix 2, and a long loop inserted between α3′ and β5′. Interestingly, the MshD fold exhibits domain strand exchange, in which the last strand, β6/β6′ forms antiparallel β-sheet interactions with β5/β5′ of the opposite domain. The two domains joined in this fashion form a continuous U-shaped β-sheet. Most GNAT proteins are dimers in solution, and like MshD, utilize C-terminal β-strands to form a continuous β-sheet to link their two subunits (Burk et al. 2003). Strand exchange similar to MshD has been observed in glucosamine-6-phosphate N-acetyltransferase (GNA1; Peneff et al. 2001), histone N-acetyltransferase HPA2 (Angus-Hill et al. 1999), and protein N-myristoyltransferase (Nmt; Bhatnagar et al. 1998; Weston et al. 1998). However, only MshD and Nmt are monomers bearing two copies of the GNAT domain. A search using VAST (Gibrat et al. 1996) and the individual domains of MshD indicates that tabtoxin resistance protein (He et al. 2003) is the most similar, exhibiting an RMSD of 1.6 Å with both the N and C-terminal domains of MshD (over 90 and 123 common residues, respectively). Despite similarities in the dimer interfaces among GNATs, they tend to not superimpose over the whole oligomer due to differences in the exact orientation of the two monomers. The details of the interactions between monomers or GNAT domains is important because the active site(s) of most GNAT dimers are located within this interface. In addition, differences in the orientation of α1, and α2 and the length and position of the loop connecting β3 and β4 have a strong influence on the shape and character of this interface.
Several GNAT family members have been determined to date in complex with acetyl–CoA or CoA. The orientation of the cofactor in all cases is very similar, and in general, the GNAT fold can be viewed as a phosphopantetheine binding domain (Dyda et al. 2000; Watson et al. 2002). The hallmarks of the GNAT fold are binding sites for the pyrophosphate moiety and pantetheine arm of CoA. The pyrophosphate binding site is located in a loop joining β4 and α3 and has a signature motif of {Q/R}-x-x-G-x-{G/A} (Neuwald and Landsman 1997). This signature sequence positions several consecutive backbone amides in an orientation to coordinate the pyrophosphate oxygens directly or through a conserved water molecule. The pantetheine arm binding site is located between β-strands 4 and 5, which splay apart to allow the pantetheine arm to make pseudo β-sheet interactions with the exposed backbone atoms of β4. Both the N- and C-terminal GNAT domains of MshD contain the signature pyrophosphate binding motif (RRRGIG and QR RGLG, respectively) and the required splaying of strands β4 and β5.
MshD, crystallized in the presence of acetyl–CoA, exhibited clear electron density for the cofactor in both the N-terminal and C-terminal GNAT domains. In general, the two cofactors are bound in similar conformations, utilizing the loop joining β4 and α3 to coordinate the pyrophosphate moiety and the exposed hydrogen bonds of β4 to coordinate the pantetheine arm. In each domain the adenosine moieties are constrained within a solvent exposed crevice located between α4 and the pyrophosphate binding loop. The largest difference in the conformation of the two bound acetyl–CoA molecules is the positioning of the acetyl function. In the C-terminal domain the pantetheine arm runs completely parallel to β4′ with the acetyl carbonyl hydrogen bonded to β4′ through the backbone amide of Leu 238 (Fig. 2A ▶). In contrast, the pantetheine arm of the acetyl–CoA in the N-terminal domain follows along β4 initially, but then turns 90° and adopts an orientation perpendicular to β4. This difference appears to arise from the shorter distance in the N-terminal domain (∼5 Å) between the pyrophosphate binding loop and the splay between strands β4 and β5, which may prevent the pantetheine arm from adopting a fully extended conformation.
Figure 2.
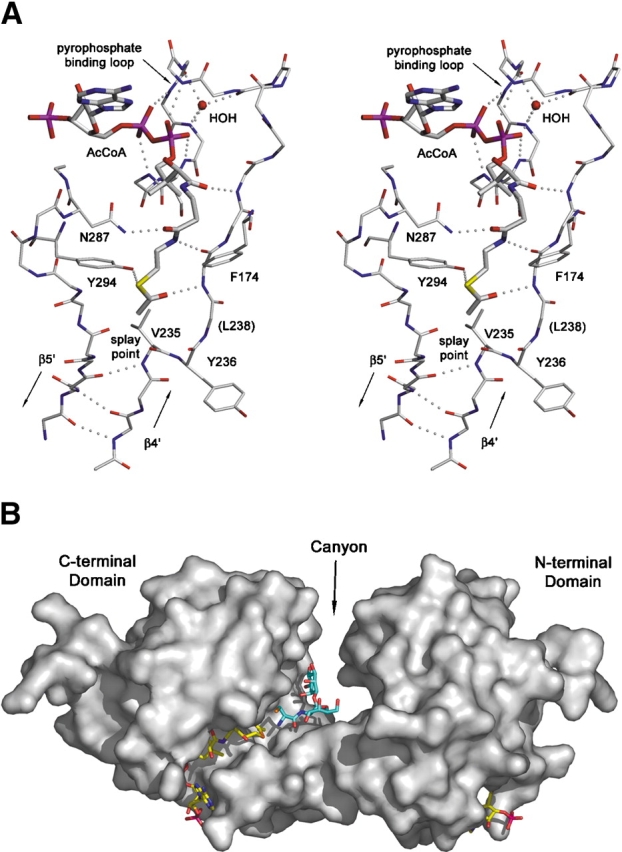
Interaction of MshD with substrates. (A) Stereodiagram of the acetyl–CoA (thick sticks) binding site in the C-terminal domain. (B) Solvent-accessible surface representation of MshD with experimentally determined acetyl–CoA (yellow carbons) and modeled Cys–GlcN–Ins (blue carbons). For visual clarity, four loops (residues 70–75, 176–179, 266–277, 226–230) that exhibit high mobility and overlay the sides of the canyon were removed prior to calculation of the surface.
The acetyl group of the N-terminal acetyl–CoA is completely buried within a hydrophobic pocket created by residues donated from β3,β4,β5,α3, α4 and by atoms of the pantetheine arm itself. There is no direct path by which an acceptor substrate could bind and interact with the acetyl group of the acetyl–CoA bound to the N-terminal GNAT domain. In contrast, the acetyl group of acetyl–CoA bound to the C-terminal domain is presented to a solvent exposed canyon between the two domains (Fig. 2B ▶). The canyon is approximately 25 Å in length, and tends to be wider at its bottom (12–19 Å wide) than at its top (6–12 Å wide). The canyon has an overall electronegative character and is highly polar with approximately 40 waters solvating its surface. The canyon is bound on either side by loops connecting α3′ to β5′, β3′ to β4′, β3 to β4, and α1′ to α2′. Portions of these loops exhibited no electron density or above average B-factors. Flexibility within these loops may be coupled to catalysis, and has been observed in many other GNAT enzymes (Dyda et al. 2000).
Another feature conserved among many GNAT active sites is the β-bulge in strand β4′ located directly adjacent to the splay in strands β4′ and β5′. In general, the β-bulge positions two consecutive carbonyl or two amide backbone atoms towards the active site. The hydrogen bonding potential of these atoms have been implicated in the stabilization of the tetrahedral intermediate and/or coordination of the acceptor substrate (Dyda et al. 2000; Vetting et al. 2002). In the C-terminal MshD GNAT domain the β-bulge presents the carbonyls of Val 235 and Tyr 236 towards the canyon while the β-bulge is absent in the N-terminal domain.
The predominant mechanism of acetyl transfer by members of the GNAT family is through direct nucleophilic attack of the acetyl–CoA cofactor by the acceptor amine (Dyda et al. 2000). Histone acetyltransferase ESA1 is the only GNAT enzyme in which acetylation proceeds through an acetyl–cysteine intermediate (Yan et al. 2002). The lack of any cysteines in the sequence of MshD suggests that the enzyme catalyzes acetyl transfer by a direct nucleophilic attack. The numerous polar groups and water molecules that surround the active site in the C-terminal domain could act as a proton wire to shuttle protons away during the reaction mechanism as proposed for many GNAT family members (Dyda et al. 2000; Vetting et al. 2002). The side chain hydroxyl of Tyr 294, a conserved residue among many GNAT family members, is positioned close (3.6 Å) to the sulfhydryl of CoA in the C-terminal domain, and could act as an active site acid to protonate the sulfhydryl of CoA after breakdown of the tetrahedral intermediate. Interestingly, the corresponding residue in the N-terminal domain is an alanine (Ala 122), and the space afforded by this difference is filled by the acetyl group of acetyl–CoA.
Crystals of MshD grown in the presence of CoA also exhibited clear electron density for the cofactor in both the N-terminal and C-terminal GNAT domains. There are no large changes between the acetyl–CoA and CoA structures, with the two structures exhibiting an RMSD of 0.14 Å over all Cα residues. In the C-terminal domain, the pantetheine arm of CoA is modeled in two conformations, indicating that the loss of a hydrogen bond between the acetyl group and β4′ leads to more flexibility in the pantetheine arm after acetyl transfer. Surprisingly, acetyl–CoA was the form of the coenzyme bound in the N-terminal domain. Acetyl–CoA must have copurified with MshD and remained bound to the N-terminal domain despite competition by excess CoA during crystallization.
The large differences in the two cofactor binding sites suggest they have dissimilar functions. Because the acetyl group of acetyl–CoA in the N-terminal domain is completely inaccessible to an acceptor substrate, it seems unlikely that the N-terminal domain exhibits any acetyltransferase activity. It may be possible that structural rearrangements upon catalysis within the C-terminal domain could make the N-terminal acetyl–CoA accessible (i.e., a reciprocating mechanism); however, the N-terminal domain also lacks many of the catalytic features shared by most GNAT enzymes. As such, the function of the N-terminal bound acetyl–CoA is unclear, but it may act as an effector molecule or function to stabilize the domain. Interestingly, in Nmt, the only other monomeric protein with two GNAT folds, only one molecule of myristol–CoA is bound, and the active GNAT fold is found in the N-terminal domain (Bhatnagar et al. 1998). One of the exceptional features of GNAT family members that utilize different natural substrates is the near absence of conserved residues. The ability to form unique active sites at dimer interfaces would also augment this active site plasticity and could permit GNAT family members to acetylate a much wider variety of substrates. The similarity of the subunit interface of typical GNAT proteins and the interface between the two GNAT domains of MshD suggests that the progenitor of MshD arose from gene duplication and fusion of a homodimeric GNAT, followed by structural rewiring through mutation and selection.
It is difficult to model a Cys–GlcN–Ins MshD complex with any confidence due to substrate rotational freedom, the openness of the lower canyon and the high degree of active site dissimilarity between GNAT family members. However, a rudimentary docking study does indicate that the substrate would be totally engulfed by the canyon such that both rings of substrate would be critical to binding affinity, and that significant room would remain within its confines even after binding. This suggests that upon binding there may be a contraction of the two domains around the substrate, reducing the cavity volume. Mobile loops which surround the edges of the canyon may also take different conformations upon substrate binding that may further reduce cavity volume. These movements may provide a degree of substrate selectivity to the active site. Further analysis of residues critical to activity will require determination of the structure of MshD in complex with its acceptor substrate. These studies are currently being pursued.
Materials and methods
The Rv0819 gene encoding MshD (residues 1–315) was amplified from M. tuberculosis genomic DNA using PCR and cloned into the pET28a(+) plasmid (Novagen). This vector construction adds six histidine residues and a thrombin cleavage sequence to the N terminus to facilitate protein purification. E. coli stain BL21(DE3)pLysS transformed with this plasmid was cultured at 37°C in Luria-Bertani medium. Cells were induced with 1 mM IPTG at an o.d. of 0.8–1.0, and grown for an additional 14–16 h at 18°C.
The frozen bacterial cell pellet was thawed and disrupted by sonication, and MshD was purified to homogeneity by Ni+2-NTA agarose chromatography. Purified protein was treated with catalytic amounts of thrombin (Sigma) to remove the N-terminal hexahistidine tag. The cleaved tag and thrombin were separated from MshD by anion exchange chromatography (Q-Sepharose, Pharmacia). Purified protein was dialyzed against 20 mM triethanolamine (TEA) pH 8.0, concentrated to 15 mg/mL using a YM10 ultrafiltration membrane (Amicon) and stored at −80°C. The protein concentration was estimated by measuring the absorbance at 280 nm, employing the calculated extinction coefficient of 37,410 M−1cm−1. Dynamic light-scattering experiments were performed on a DynaPro-MS/X instrument (Protein Solutions). The data were measured at room temperature with 15 mg/mL MshD in 20 mM TEA, pH 8.0.
Crystallization of MshD was by the vapor diffusion under oil method using 96-well round-bottom assay plates. Various crystallization conditions were screened by mixing 3 μL 15 mg/mL MSHD, 5 mM CoA with 3 μL of screening solution under 100 μL of silicon oil (Fisher). Initial crystals were obtained with 15% (w/v) polyethylene glycol 4000, 100 mM sodium citrate pH 5.6, 20% (v/v) isopropanol. These crystals were crushed, serially diluted, and used to microseed (under oil) drops that contained 7.5 mg/mL protein, 2.5 mM CoA, or acetyl–CoA, 5%–10% (w/v) polyethylene glycol 4000, and 100 mM ADA, pH 6.0. Well-diffracting crystals grew to approximate dimensions 0.2 × 0.4 × 0.4 mm within 4 d. Crystals were stabilized in 28% (w/v) sucrose, 50 mM ADA, pH 6.0, 10% polyethylene glycol 4000, and 200 mM NaCl for 5 min before vitrification by immersion in liquid nitrogen.
All X-ray diffraction data were collected at 100 K using an R-Axis IV++ image plate detector using CuKα radiation from a Rigaku RU-H3R X-ray generator and processed using DENZO/SCALEPACK (Table 1; Otwinowski 1993). The crystals belong to space group P212121 with unit cell parameters a = 59.9 Å, b = 61.7 Å, c = 84.5 Å. There is one molecule per asymmetric unit, with a specific volume of 2.3 Å3 Da−1 and an estimated, solvent content of 46% (Matthews 1968).
Table 1.
Crystallographic data and refinement statistics
| CoA | Acetyl-CoA | TMLAb | |
| Resolution (Å)a | 1.60 (1.60–1.66) | 1.7 (1.70–1.76) | 1.8 (1.80–1.86) |
| Completeness (%)a | 97.8 (93.6) | 97.5 (87.3) | 99.2 (98.1) |
| Redundancy | 4.8 | 3.9 | 3.7 |
| I/σ(I) | 21.3 | 26.8 | 20.3 |
| Rsyma | 0.037 (0.120) | 0.028 (0.125) | 0.042 (0.113) |
| Figure of meritc | — | — | 0.49 (0.44) |
| Phasing powerc | — | — | 1.14 (1.12) |
| Model and refinement statistics | |||
| Rcrysta | 19.7 (25.2) | 19.6 (24.1) | |
| Rfree (5% of data)a | 23.7 (29.8) | 23.5 (29.6) | |
| No. atoms total | 2686 | 2669 | |
| No. residues | 290 | 290 | |
| No. waters | 398 | 378 | |
| Average B-factor | |||
| Protein | 18.4 | 20.4 | |
| CoA/AcCoA | 14.9 | 16.7 | |
| Waters | 30.3 | 31.3 | |
| RMSD | |||
| Bond lengths | 0.021 | 0.020 | |
| Bond angles | 2.04 | 1.95 | |
| Ramachandran plot | |||
| Most favored | 94.9 | 94.5 | |
| Additional allowed | 5.1 | 5.5 | |
| Generously allowed | 0.0 | 0.0 | |
| Disallowed | 0.0 | 0.0 |
a Values in parentheses for highest resolution shell.
b Values calculated without merging Friedel mates.
c As calculated by the program SOLVE (Terwilliger 2002).
The structure of MshD was solved by single isomorphous replacement with anomalous scattering (SIRAS). Heavy atom derivatives of MshD were screened utilizing the quick soak method (Sun et al. 2002). A quality derivative was obtained from a crystal soaked in cryoprotectant buffer supplemented with 100 mM trimethyllead acetate for 4 min and back-soaked in cryoprotectant buffer minus the derivative for 10 sec. Four Pb sites were located by the program SOLVE (Terwilliger 2002) and used to calculate the initial SIRAS phases. The initial figure of merit was 0.49 for the resolution 15–1.8 Å (0.44 in the high-resolution shell). Density modification and phase extension within the program RESOLVE (Terwilliger 2002) yielded a high-quality experimental electron density map that was suitable for automated model building with MAID (Levitt 2001). Electron density map inspection and fitting were carried out in O (Jones et al. 1991) and structural models were refined with CNS (Brünger et al. 1998). No density was located for two N-terminal residues (plus three remaining from the tag), five C-terminal residues, and residues in three loops (71–73, 209–214, 266–274), and were not included in the model. The final model of the "CoA" complex contains one CoA and one acetyl–CoA molecule, while the acetyl–CoA complex contains two acetyl–CoA molecules. All residues are in the allowed region of the Ramachandran plot. Refined atomic coordinates have been deposited in the Protein Data Bank (PDB ID 1OZP, 1P0H).
Acknowledgments
This work was supported by grants from the NIH, no. GM33449 (to J.S.B) and T32GM07288 (to M.Y.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03153703.
References
- Angus-Hill, M.L., Dutnall, R.N., Tafrov, S.T., Sternglanz, R., and Ramakrishnan, V. 1999. Crystal structure of the histone acetyltransferase Hpa2: A tetrameric member of the Gcn5-related N-acetyltransferase superfamily. J. Mol. Biol. 294 1311–1325. [DOI] [PubMed] [Google Scholar]
- Bhatnagar, R.S., Futterer, K., Farazi, T.A., Korolev, S., Murray, C.L., Jackson-Machelski, E., Gokel, G.W., Gordon, J.I., and Waksman, G. 1998. Structure of N-myristoyltransferase with bound myristoylCoA and peptide substrate analogs. Nat. Struct. Biol. 5 1091–1097. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Burk, D.L., Ghuman, N., Wybenga-Groot, L.E., and Berghuis, A.M. 2003. X-ray structure of the AAC(6′)-Ii antibiotic resistance enzyme at 1.8 Å resolution; Examination of oligomeric arrangements in GNAT superfamily members. Protein Sci. 12 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. The PyMOL molecular graphics system. http://www.pymol.org.
- Dyda, F., Klein, D.C., and Hickman, A.B. 2000. GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 29 81–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey, R.C. 2001. Novel thiols of prokaryotes. Annu. Rev. Microbiol. 55 333–356. [DOI] [PubMed] [Google Scholar]
- Gibrat, J.F., Madej, T., and Bryant, S.H. 1996. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 6 377–385. [DOI] [PubMed] [Google Scholar]
- He, H., Ding, Y., Bartlam, M., Sun, F., Le, Y., Qin, X., Tang, H., Zhang, R., Joachimiak, A., Liu, J., et al. 2003. Crystal structure of tabtoxin resistance protein complexed with acetyl coenzyme A reveals the mechanism for β-lactam acetylation. J. Mol. Biol. 325 1019–1030. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for finding protein models in electron density maps and location of error in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Koledin, T., Newton, G.L., and Fahey, R.C. 2002. Identification of the mycothiol synthase gene (mshD) encoding the acetyltransferase producing mycothiol in actinomycetes. Arch. Microbiol. 178 331–337. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Levitt, D.G. 2001. A new software routine that automates the fitting of protein X-ray crystallographic electron-density maps. Acta Crystallogr. D 57 1013–1019. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F. and Landsman, D. 1997. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22 154–155. [DOI] [PubMed] [Google Scholar]
- Newton, G.L. and Fahey, R.C. 2002. Mycothiol biochemistry. Arch. Microbiol. 178 388–394. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. 1993. Data collection and processing. In Proceedings of the CCP4 Study Weekend (ed. L. Sawer et al.), pp. 55–62. Daresbury Laboratory, Warrington.
- Peneff, C., Mengin-Lecreulx, D., and Bourne, Y. 2001. The crystal structures of Apo and complexed Saccharomyces cerevisiae GNA1 shed light on the catalytic mechanism of an amino-sugar N-acetyltransferase. J. Biol. Chem. 276 16328–16334. [DOI] [PubMed] [Google Scholar]
- Rawat, M., Newton, G.L., Ko, M., Martinez, G.J., Fahey, R.C., and Av-Gay, Y. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 46 3348–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, P.D., Radaev, S., and Kattah, M. 2002. Generating isomorphous heavy-atom derivatives by a quick-soak method. Part I: Test cases. Acta Crystallogr. D Biol. Crystallogr. 58 1092–1098. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2002. Automated structure solution, density modification and model building. Acta Crystallogr. D Biol. Crystallogr. 58 1937–1940. [DOI] [PubMed] [Google Scholar]
- Vetting, M.W., Hegde, S.S., Javid-Majd, F., Blanchard, J.S., and Roderick, S.L. 2002. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 9 653–658. [DOI] [PubMed] [Google Scholar]
- Watson, W.T., Minogue, T.D., Val, D.L., von Bodman, S.B., and Churchill, M.E. 2002. Structural basis and specificity of acyl-homoserine lactone signal production in bacterial quorum sensing. Mol. Cell 9 685–694. [DOI] [PubMed] [Google Scholar]
- Weston, S.A., Camble, R., Colls, J., Rosenbrock, G., Taylor, I., Egerton, M., Tucker, A.D., Tunnicliffe, A., Mistry, A., Mancia, F., et al. 1998. Crystal structure of the anti-fungal target N-myristoyl transferase. Nat. Struct. Biol. 5 213–221. [DOI] [PubMed] [Google Scholar]
- Yan, Y., Harper, S., Speicher, D.W., and Marmorstein, R. 2002. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat. Struct. Biol. 9 862–869. [DOI] [PubMed] [Google Scholar]