

Abstract
Receptor signaling in the growth hormone (GH)–growth hormone receptor (GHR) system is controlled through a sequential two-step hormone-induced dimerization of two copies of the extracellular domain (ECD) of the receptor. The regulatory step of this process is the binding of the second ECD (ECD2) to the stable preassociated 1 : 1 GH/ECD1 complex on the cell surface. To determine the energetics that governs this step, the binding kinetics of 38 single- and double-alanine mutants in the hGH Site2 contact with ECD2 were measured by using trimolecular surface plasmon resonance (TM-SPR). We find that the Site2 interface of hGH does not have a distinct binding hot-spot region, and the most important residues are not spatially clustered, but rather are distributed over the whole binding surface. In addition, it was determined through analysis of a set of pairwise double alanine mutations that there is a significant degree of negative cooperativity among Site2 residues. Residues that show little effect or even improved binding on substitution with alanine, when paired with D116A-hGH, display significant negative cooperativity. Because most of these pairwise mutated residues are spatially separated by ≥10 Å, this indicates that the Site2 binding interface of the hGH–hGHR ternary complex displays both structural and energetic malleability.
Keywords: Human growth hormone, extracellular domain of human growth hormone, regulatory step, binding energetics, alanine scanning, surface plasmon resonance
The common theme in cytokine biology is that the representative activities are triggered through a hormone-induced receptor aggregation process (Ihle et al. 1994; Carpenter et al. 1998; Nicola and Hilton 1999). Although these receptor aggregation mechanisms are conceptually simple, the molecular strategies that are used are complex and hardly predictable (Kossiakoff and de Vos 1998; Syed et al. 1998). The receptor aggregation process can involve formation of complexes with a set of identical receptors, or the production of complexes containing two or more different receptors; the complexes can be quite short-lived or extremely stable (Gertler et al. 1996; Elkins et al. 2000). A similarity among cytokine-induced signaling complexes is that they are usually formed in a programmed sequential order. This is because the binding affinities for the receptors are usually different and require a specific stepwise order of adding the constituent components to form stable intermediates that subsequently form the binding templates for additional receptor interactions (Kossiakoff and de Vos 1998; Wilson and Jolliffe 1999; Liparoto et al. 2002).
Because of the fundamental importance in understanding how the dynamic hormone-receptor association and dissociation process is programmed for optimum biological function, there has been considerable effort to determine the energetic basis for formation of these complexes (Wells and de Vos 1996; DeLano 2002; Bernat et al. 2003). These studies have produced important insights into the thermodynamic criteria involved in assembling these complexes and have additionally made significant contributions toward the understanding of the structure/function relationships governing protein–protein interactions in general (Wells and de Vos 1996; Kossiakoff and de Vos 1998).
Within the cytokine superfamily, the growth hormone (GH)/ prolactin (PRL) family of hormones and receptors are arguably among the most extensively studied systems focused on structure/function issues and molecular recognition (Cunningham and Wells 1989, Cunningham and Wells 1991, Cunningham and Wells 1993; Kossiakoff et al. 1994; Somers et al. 1994; Clackson and Wells 1995; Atwell et al. 1997; Chen et al. 1997; Clackson et al. 1998). For this family, the active signaling form is a product of receptor homodimerization: a complex containing one hormone and two copies of the extracellular domain (ECD) of the cognate receptor. A notable feature of these hormones is that their asymmetric tertiary structures play an integral role in how they regulate receptor activation. The GHs and PRLs are in the structural class of long-chain four α-helix bundle proteins that contain no tertiary symmetry that might support equivalent binding environments for homodimerization of the receptor ECDs. As a consequence, the two ECDs bind at two distinctly different topographical surfaces designated Site1 and Site2. Site1 is a high-affinity site for receptor binding (Kd ∼1 nM; Cunningham and Wells 1993) and is located along a large surface cleft formed between helix 1 and helix 4 of the hormone (Fig. 1 ▶). In contrast, Site2 is a low-affinity site, which is located along a relative flat surface on the opposite side of the molecule.
Figure 1.
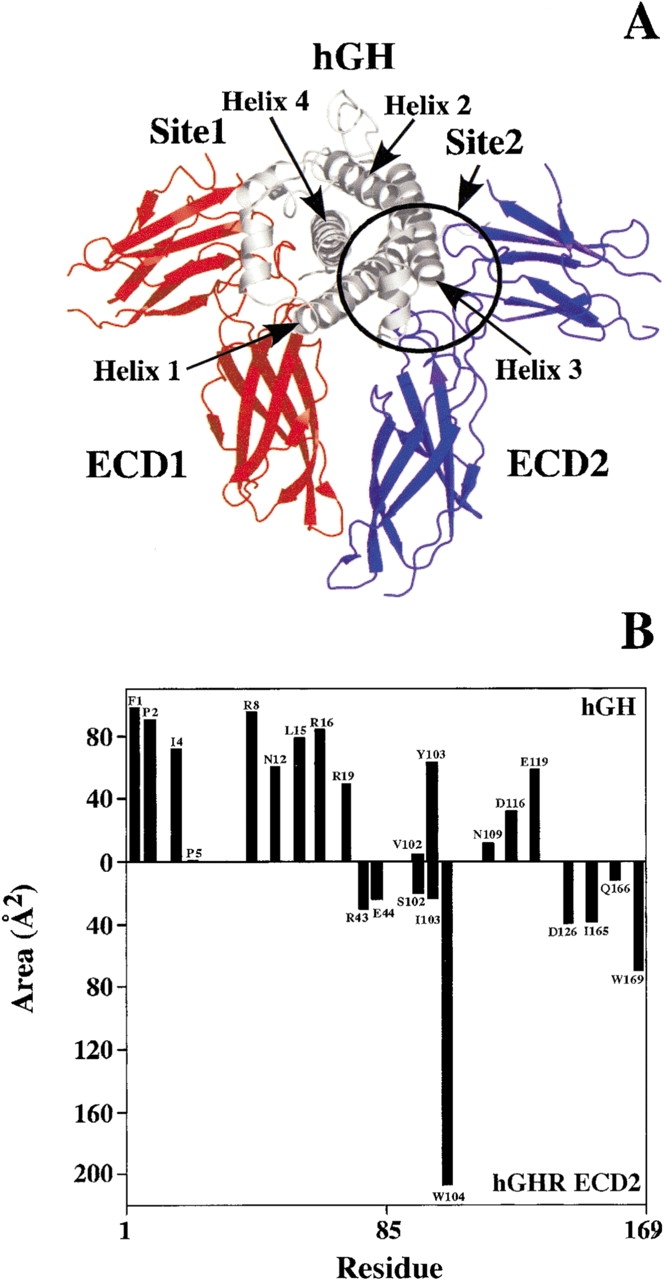
(A) Ribbon diagram of the ternary complex of human growth hormone (hGH) bound to two copies of the extracellular domain (ECD) of the human growth hormone receptor (hGHR; de Vos et al. 1992). The hGHR ECD1 and ECD2 are colored red and blue, respectively. The Site2 binding interface between hGH and the hGHR ECD2 is circled. (B) Accessible surface area burial of Site2 interface residues of hGH (top bars) and hGHR ECD2 (bottom bars). All structural figures were generated by using the program Pymol (Warren DeLano, www.pymol.org).
The different binding affinities for receptor ECDs at Site1 (ECD1) and Site2 (ECD2) are mechanistically important in the receptor activation process. The high-affinity site, Site1, is always occupied first by the receptor ECD (ECD1; Fuh et al. 1992). This forms a stable 1 : 1 hormone-receptor intermediate (hGH-ECD1). The formation of this stable intermediate is essential step because productive binding of ECD2 at Site2 of the hormone also requires additional contacts to a patch of the C-terminal domain of ECD1 (Fig. 1 ▶).
The second step involving the binding of ECD2 to the hGH–ECD1 intermediate acts as the programmed regulatory step for triggering biological action, and it entails a set of highly tuned interactions among binding interfaces in two spatially distinct binding sites. The energetic relationship between the hGH Site2–ECD2 interactions and the ECD1–ECD2 contacts are known to be functionally important. A recent study by Bernat et al. (2003) showed that the ECD1–ECD2 contacts contain a significant part of the binding force that drives the receptor homodimerization process; however, their results also made clear that the functional Site2 hormone-receptor contact plays a consequential role in the process.
The nature of this functional hGH Site2 binding epitope has not been fully characterized. By using a fluorescence quenching approach, a partial hGH Ala-scan of Site2 has been performed (Cunningham et al. 1991). However, the EC50 data generated from this study did not provide any kinetic data describing the mechanism of association and dissociation of the receptor ECD as a function of the binding properties of hGH residues in Site2. In addition, to understand the energetics driving this homodimerization step, it is important to determine whether this step is fully under control of binding additivity effects, or whether, as was the case for the ECD2 Site2 interface, there exists some degree of binding cooperativity (Bernat et al. 2003).
We report here an Ala-scanning study of hGH Site2 using trimolecular surface plasmon resonance (TM-SPR), which describes the characteristic properties of the binding energy surface between the hormone and receptor at Site2. The binding kinetics of 15 single hGH Site2 alanine mutations, as well as a series of pairwise double alanine mutations were determined. In addition, the energetics of the complementary ECD2 Site2 binding interface were explored by determining the binding kinetics of eight ECD2 alanine mutations.
We find that that the energetics of this site are functionally different than that determined for the high-affinity Site1 in hGH (Cunningham and Wells 1993) in three mechanistically important ways. First, whereas Site1 has been shown to have a binding hot-spot where the residues that are the major contributors to binding are focused on a relatively small area in the binding interface (Cunningham and Wells 1993), in Site2 there is no equivalent hot-spot. Rather the binding energy is distributed throughout the interface, with no single residue being crucial for ECD2 binding. Second, in Site1 the binding hot-spot residues of the hormone and receptor form a spatially distinct core in the interface between them. That is, hot-spot residues of the hormone directly contact the hot-spot residues of the receptor. In contrast, in Site2, no spatial complementary is present; the residues of the hormone that contribute most to binding do not overlay those of the receptor. Third, in Site2, there appears to be in operation a significant degree of cooperativity between certain sets of residues. This may be due to the inherent structural plasticity that has been observed for the Site2–ECD2 interface (Schiffer et al. 2002). This contrasts to the extremely additive nature of Site1, in which the individual binding contributions are independent of the other residues in the interface (Cunningham and Wells 1993; Clackson and Wells 1995; Weiss et al. 2000).
Results and Discussion
Site2 Ala-scan of hGH
The binding kinetics for 15 hGH Site2 Ala-scan mutants were determined by using the TM-SPR procedure (Table 1; Bernat et al. 2003). The TM-SPR method is capable of accurately decoupling the kinetics of the sequential binding steps in forming the 1 : 2 hormone-receptor ternary complex as shown in Figure 2 ▶. The Ala-scanned hGH residues involved in the interface with the receptor ECD2 are located in the N terminus (residues 1–6) and in helices 1 and 3 (circled in Fig. 1A ▶).
Table 1.
Site2 alanine scan of hGH to the hGHR ECD2
| kon (M−1 s−1, ×105) | koff (s−1, ×10−4) | Kda (nM) | Kd(Ala)/Kd(wt) | |
| wt-hGH | 2.0 | 7.6 | 3.8 | 1 |
| F1A-hGH | 0.77 | 25.6 | 33 | 9 |
| P2A-hGH | 1.2 | 22.4 | 19 | 5 |
| I4A-hGH | 3.3 | 295 | 89 | 23 |
| P5A-hGH | 1.1 | 19.8 | 18 | 5 |
| L6A-hGH | 2.3 | 13.8 | 6.0 | 2 |
| R8A-hGH | 1.4 | 32.8 | 23 | 6 |
| N12A-hGH | 2.8 | 7.4 | 2.6 | 0.7 |
| L15A-hGH | 1.2 | 22.1 | 18 | 5 |
| R16A-hGH | 2.0 | 96.1 | 48 | 13 |
| R19A-hGH | 1.6 | 20.3 | 13 | 3 |
| V102A-hGH | 1.8 | 19.6 | 11 | 3 |
| Y103A-hGH | 2.2 | 18.6 | 8.5 | 2 |
| N109A-hGH | 1.7 | 120 | 71 | 19 |
| D116A-hGH | 2.0 | 6.2 | 3.1 | 0.8 |
| E119A-hGH | 2.6 | 25.5 | 9.8 | 3 |
| L6A,N12A-hGH | 3.0 | 8.1 | 2.7 | 0.71 (1)b |
| L6A,D116A-hGH | 1.9 | 240 | 126 | 33 (2) |
| N12A,D116A-hGH | 2.2 | 290 | 132 | 35 (0.6) |
| R16A,D116A-hGH | 2.6 | 170 | 65 | 17 (11) |
| R19A,D116A-hGH | 2.5 | 270 | 108 | 28 (2) |
| Y103A,D116A-hGH | 3.6 | 110 | 31 | 8 (1) |
| N109A,D116A-hGH | 2.2 | 45.4 | 21 | 6 (13) |
| E119A,D116A-hGH | 2.0 | 253 | 127 | 33 (4) |
| L6A,N12A,D116A-hGH | 1.9 | 510 | 268 | 71 (1) |
| D116N-hGH | 2.6 | 33.4 | 13 | 3.68 |
| D116V-hGH | 2.5 | 23.0 | 9.2 | 2.68 |
| L6A,D116V-hGH | 1.5 | 220 | 147 | 39 (4) |
| N12A,D116V-hGH | 3.1 | 220 | 71 | 19 (1) |
| R16A,D116V-hGH | 2.0 | 67.3 | 34 | 9 (26) |
| R19A,D116V-hGH | 2.0 | 237 | 119 | 31 (6) |
| Y103A,D116V-hGH | 1.9 | 143 | 75 | 20 (4) |
| N109A,D116V-hGH | 1.7 | 65 | 38 | 10 (38) |
| E119A,D116V-hGH | 1.1 | 160 | 145 | 38 (6) |
Experiments were done in 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, 0.005% TWEEN-20 at 25°C.
a Equilibrium dissociation constant: Kd = koff/kon.
b Values in parentheses are the calculated binding additivities based on the single point mutations: [Kd(Ala1)/Kd(wt)] × [Kd(Ala2)/Kd(wt)].
Figure 2.
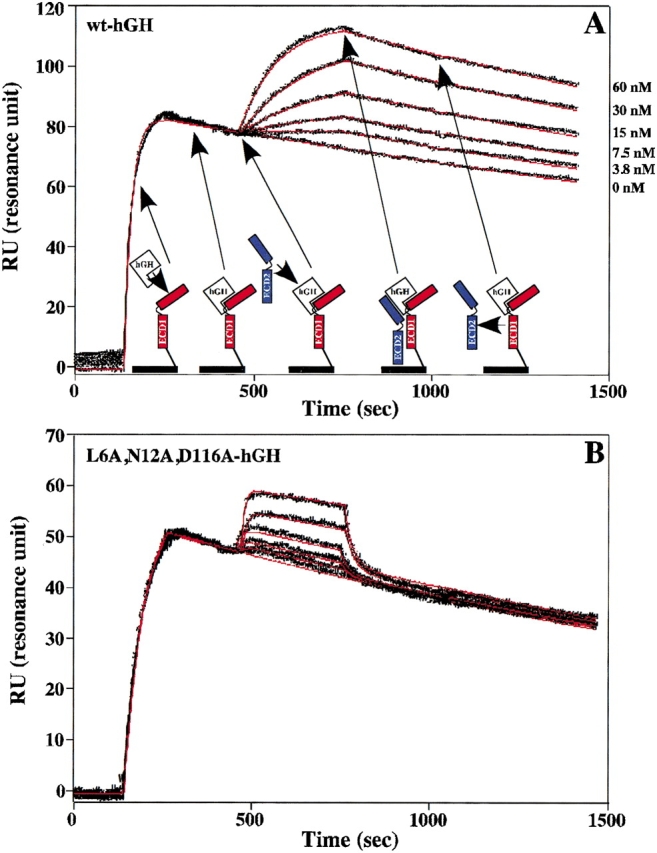
Trimolecular surface plasmon resonance (TM-SPR) to measure the binding kinetics of the hGHR ECD2 to the 1 : 1 intermediate complex of hGH–hGHR ECD1. (A) Sensorgrams showing the Site2 binding kinetics of wt-hGH to wt-hGHR ECD2. Also shown is a cartoon representation of TM-SPR method. The hGHR ECD1 (red) is coupled to the sensor chip through a disulfide bond at the C-terminal domain. This coupling allows for minimal lateral movement and proper orientation of ECD1. A saturating concentration of free hGH is injected over the sensor chip to form the 1 : 1 intermediate hGH–hGHR ECD1 complex. A second injection of free hGHR ECD2 (blue) is followed to determine the binding kinetics of the 1 : 2 ternary complex. Twofold dilution series with concentrations of 0, 3.75, 7.5, 15, 30, and 60 nM for the second hGHR ECD2 concentrations were typically used. This results in the case of the highest concentration of about one half of the potential sites bind a soluble ECD. The six sensorgrams were pooled and globally fit (red lines) to the decaying surface model (Joss et al. 1998) by using the program Clamp99 (Myszka and Morton 1998) to determine the on (kon) and off (koff) binding rate constants. (B) The fitted Site2 binding sensorgrams for the L6A,N12A,D116A-hGH triple mutant to the hGHR ECD2 (concentrations of 15.63, 31.25, 62.5, 125, and 250 nM were used).
The wild type (wt) ECD2 binds to the 1 : 1 wt-hGH–ECD1 complex with an Kd of 3.8 nM, which is similar to the binding affinity at the high-affinity Site1 (∼1.2 nM; Cunningham and Wells 1993). Flowing saturating amounts of the ECD over the sensor chip shows that no binding occurs between the soluble and chip immobilized receptor ECDs in the absence of the hormone (data not shown). We note the distinction that Site1 involves a direct interaction between the hormone and receptor, whereas the binding of ECD2 is accomplished by the energetic coupling between two spatial distinct sites. This is an example of binding additivity, where the relatively weak energies derived from two spatially distinct sites (hGH Site2 and the ECD1–ECD2 contact) add to produce a strong interaction.
The Ala-scan data indicate that no single hGH Site2 residue is absolutely critical for the binding of ECD2 to the hGH–ECD1 intermediate (Table 1). Overall, most of the alanine substitutions produced twofold to 10-fold effects on binding affinity (Kd[Ala]/Kd[wt]). The largest decreases in binding affinity were found for the I4A, R16A, and N109A variants with 23-, 13-, and 19-fold decreases, respectively. N12A and D116A mutants showed a slight increase in binding affinity to ECD2 (Table 1). Generally, the kon rate constants show little fluctuation (<2.5-fold) over the 15-residue Ala-scan; the differences in binding result mainly from changes in the koff rate constants. A similar trend of rate dependence due to off-rate kinetics has been seen in other cytokine systems: Site1 binding mutants of hGH (Cunningham and Wells 1993) and ECD1 (Clackson et al. 1998), interleukins 2 (Liparoto and Ciardelli 1999) and 4 (Shen et al. 1996; Wang et al. 1997), erythropoietin (Hensley et al. 2000), and granulocyte colony-stimulating factor (G-CSF) (Nice et al. 1993).
The binding energetics based on the Ala-scan data are color-coded and mapped on the hGH Site2 molecular surface in Figure 3A ▶. The color-coding of this energy surface shows that there is no concentrated single hot-spot organization of binding energy. Rather the energy is distributed throughout the surface. The most important contributors to binding are I4, R16, and N109, which are separated by >10 Å. I4 buries a large amount of surface area (∼70 Å2), mainly through its van der Waals contact with P106E2, and it is probable that the 15-fold decrease in binding is caused by a disruption of this packing interaction (Figs. 1B, 4 ▶ ▶; residues on the ECD2 are designated with a subscript E2 and a superscript E1 for ECD1 residues). However, the 20-fold decrease seen for the N109A-hGH mutant is probably due to some indirect role at the Site2 interface, because N109 buries only ∼15 Å2 of surface area and does not form any H-bonds with the ECD2 (Fig. 1B ▶).
Figure 3.
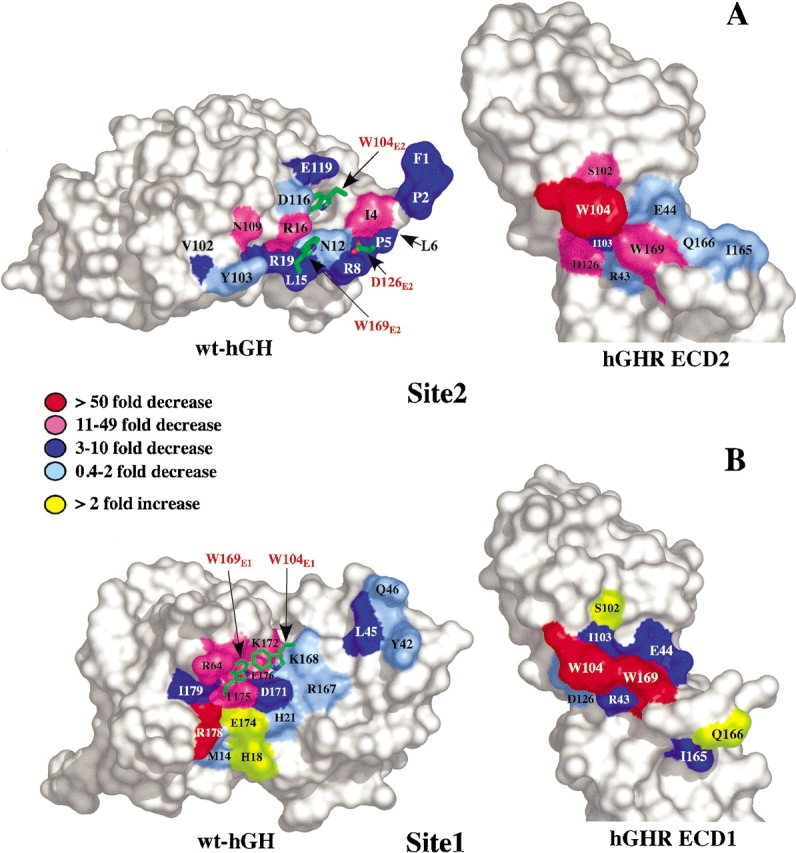
Site2 and Site1 alanine scanning mutagenesis of hGH and the hGHR ECDs. (A) Peeled away surface diagram of the Site2 Ala-scan of wt-hGH and the hGHR ECD2 shown as a function of fold decrease or increase (Kd[Ala]/Kd[wt-hGH]) determined by using TM-SPR. Also depicted on the hGH surface are the Site2 hGHR ECD2 residues of W104E2, D126E2, and W169E2 (red letters). L6, depicted by the arrow, is located behind P5 in this orientation. (B) Surface representation of the Site1 Ala-scan of wt-hGH (Cunningham and Wells 1989, 1993) and hGHR ECD1 (Clackson and Wells 1995; Clackson et al. 1998) plotted on the same fold decrease/increase scale. W104E1 and W169E1 (red letters) are shown on the wt-hGH Site1 interface.
Figure 4.

Cross-eye stereo diagram showing the Site2 interface between hGH and hGHR ECD2. The wt-hGH residues are in green with black labels, and the hGHRE2 residues are in yellow with red labels. Intermolecular hydrogen bonds are depicted as broken yellow sticks for N12Oδ1 (wt-hGH) to R43Nη2E2, N12Nδ2 to D126Oδ2E2, and R16Nη1 to E44Oɛ2E2. Oxygen and nitrogen atoms are colored red and blue, respectively.
The side-chain of R16 participates in an intermolecular H-bond with the side-chain of E44E2 (Fig. 4 ▶). The aliphatic portion of arginine side-chain forms one side of a hydrophobic cleft making van der Waals contact with the indole ring of W169E2 (Fig. 5A ▶, see below). Note, however, the complementary mutation to the ECD2 residue, E44E2A, has no measurable effect on binding (Table 2). Thus, this indicates that the 13-fold effect of the R16A-hGH mutant is produced through the loss of the van der Waals contact to W169E2 (Fig. 1B ▶), rather than the loss of the H-bond to E44E2. The other side of the hydrophobic cleft for the indole ring of W169E2 is created by the side-chain of N12 forming 2 intermolecular H-bonds to the side-chains of R43E2 and D126E2 (Figs. 4, 5A ▶ ▶). It is noteworthy that N12A-hGH shows an unexpected result of a small increase in binding affinity (Table 1).
Figure 5.
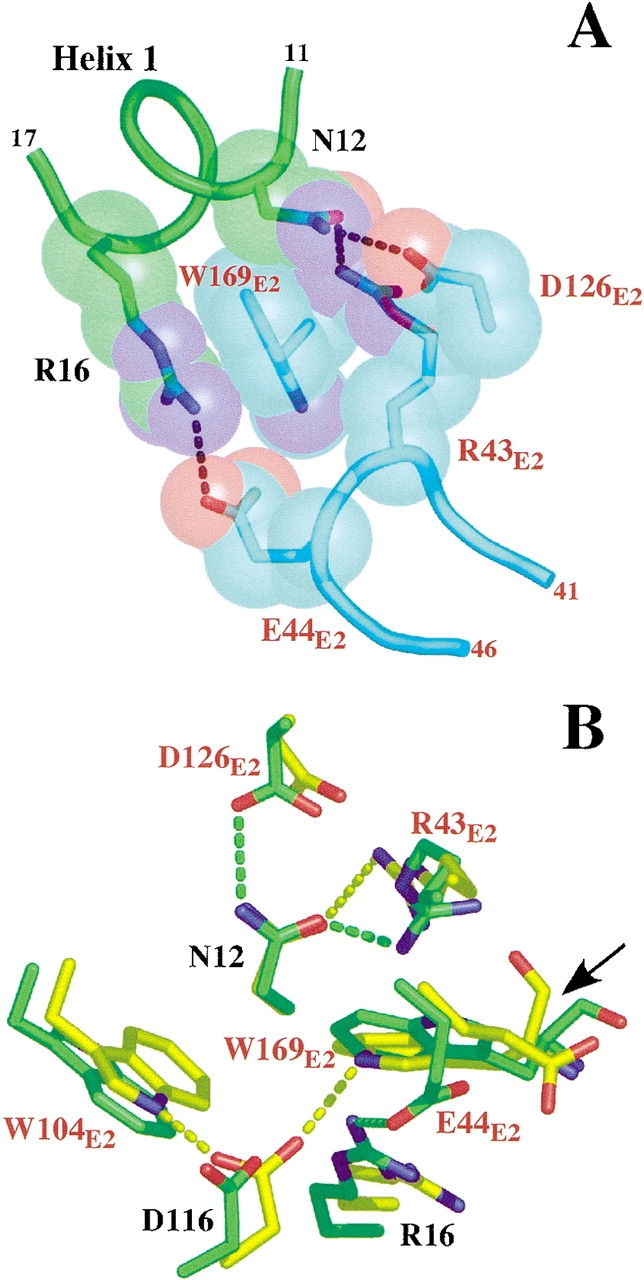
(A) Van der Waals packing interactions formed by the three intermolecular H-bonds around W169E2 at the Site2 interface of the wt-hGH–hGHR ECD ternary complex. The hGHR residues are labeled in red. (B) Superimposed diagram showing the interactions of D116 in the wt-hGH–hGHR ECD complex (green) and of the Site1 phage displayed optimized hGHv–hGHR ECD complex (yellow; Schiffer et al. 2002). The arrow depicts the peptide bond flip of W169E2 between the two crystal structures.
Table 2.
Site2 single and pairwise alanine scan of hGHR ECD2 to hGH variants
| kon (M−1 s−1, × 105) | koff (s−1, ×10−4) | Kdb (nM) | Kd(Ala)/Kd(wt) | |
| wt-hGH | ||||
| wt-hGHR ECD2 | 2.0 | 7.6 | 3.8 | 1 |
| R43A-ECD2b | 2.3 | 20.4 | 8.9 | 2 |
| E44A-ECD2b | 3.4 | 16.1 | 4.7 | 1 |
| S102A-ECD2 | 0.91 | 45.8 | 50 | 13 |
| I103A-ECD2 | 2.0 | 37.0 | 19 | 5 |
| W104A-ECD2b | nd | nd | >1000 | >263 |
| D126A-ECD2b | 2.0 | 280 | 140 | 37 |
| I165A-ECD2 | 1.7 | 12.9 | 7.6 | 2 |
| Q166A-ECD2b | 3.1 | 11.7 | 3.8 | 1 |
| W169A-ECD2b | 1.9 | 76.0 | 40 | 11 |
| W104F-ECD2 | 0.65 | 20 | 308 | 81 |
| W169F-ECD2 | 1.0 | 17.7 | 18 | 4.7 |
| N12A-hGHd | 2.8 | 7.4 | 2.6 | 0.7 |
| R43A-ECD2 | 2.3 | 6.5 | 2.8 | 0.74 (1)c |
| D126A-ECD2 | 6.8 | 256 | 38 | 10 (7) |
| R16A-hGHd | 2.0 | 96.1 | 48 | 13 |
| E44A-ECD2 | 13.6 | 301 | 22 | 6 (13) |
| D116A-hGHd | 2.0 | 6.2 | 3.1 | 0.8 |
| W104F-ECD2 | nd | nd | >1000 | >263 (66) |
| W169F-ECD2 | 1.6 | 18.3 | 11 | 3 (4) |
| D116N-hGHd | 2.6 | 33.4 | 13 | 3 |
| W104F-ECD2 | nd | nd | >1000 | >263 (243) |
| W169F-ECD2 | 1.2 | 68 | 57 | 15 (14) |
| D116V-hGHd | 2.5 | 23.0 | 9.2 | 2 |
| W104F-ECD2 | nd | nd | >1000 | >263 (162) |
| W169F-ECD2 | 1.2 | 110 | 92 | 24 (9) |
nd indicates not determined.
Experiments were done in 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, 0.005% TWEEN-20 at 25°C.
a Equilibrium dissociation constant: Kd = koff/kon.
b Values taken from Bernat et al. (2003).
c Values in parentheses are the calculated binding additivities based on the single point mutations: [Kd(Ala1)/Kd(wt)] × [Kd(Ala2)/Kd(wt)].
d Pairwise alanine mutations on both hGH and ECD2.
Site2 Ala-scan of the hGHR ECD2
A subset of the ECD2 residues contributing to binding of hGH at Site2 has been Ala-scanned by Bernat et al. (2003). This set has been expanded here to include residues S102E2, I103E2, and I165E2, which have been implicated by structural studies to be directly involved in binding to Site2 of hGH (Sundstrom et al. 1996; Schiffer et al. 2002). The effects to binding of homolog substitutions (Trp–Phe) of the two important ECD2 tryptophans, W104E2 and W169E2, were also evaluated. The ECD2 Site2 Ala-scan kinetics are given in Table 2 and the distribution of the binding energy among these residues in the ECD2 binding interface is shown in Figure 3A ▶.
Among the Ala-scanned ECD2 residues, only the W104E2A mutant produces a variant having no detectable binding (Bernat et al. 2003). S102E2A, which makes van der Waals interactions on top of W104E2 (Fig. 3A ▶), shows a 13-fold decrease in binding (Table 2). It has been observed that the S102E2 hydroxyl side-chain can adopt several possible conformations, one of which forms an H-bond with E119 (Sundstrom et al. 1996). S102E2 along with D126E2, W104E2, and W169E2 (a 11-fold decrease, dark blue on this scale) form a spatially focused hot-spot region on the ECD2 surface (Fig. 3A ▶). I103E2 packs underneath W104E2 and shows a fivefold decrease in binding affinity (Table 2). I165E2, which buries ∼40 Å2 of surface area (Figs. 1B, 3A ▶ ▶), shows only a minimal twofold decrease in binding affinity (Table 2).
A major structural component in the Site2 hormone-receptor interface is the H-bonding and packing environment that forms the binding cleft for the W169E2 side-chain (Fig. 5A ▶). This cleft is structured by three H-bonds that position the aliphatic portions of the H-bonding side-chains into direct contact with the hydrophobic part of W169 E2, resulting in the burial of ∼300 Å2 of surface area among the groups involved (Figs. 1B, 5A ▶ ▶). However, surprisingly, considering the amount of surface burial involved, the W169E2A mutation results only in a 11-fold decrease in binding affinity. The role of the H-bonding groups was also assessed by both single and pairwise mutations (Table 2), with the general finding that this region of the binding interface contributes disproportionately little to the overall binding considering its surface area and extensive structure.
Comparison of the hGH Site1 and Site2 binding energy epitopes
The energetics of Site1 hGH–ECD1 binding have been extensively characterized. Figure 3B ▶ shows the hGH Site1 Ala-scan determined by Cunningham and Wells (1993) colored in the same manner as the Site2 hGH Ala-scan determined here (Fig. 3A ▶). The most distinguishing feature of the hGH Site1 binding epitope is the spatial organization of the binding energy into a so-called binding hot-spot. This hot-spot contains residues (R64, K172, T175, F176, and R178) that are clustered together, and although encompassing only ∼25% of the total epitope, the cluster provides >80% of the total binding energy.
Figure 3 ▶ emphasizes two distinguishing features between hGH Site1 and Site2 binding epitopes. First, for Site2 there is no equivalent hot-spot; rather the energy is generally distributed throughout the surface. This general pattern was observed by Cunningham et al. (1991) using EC50 data; however, there were some distinct differences in the absolute and relative magnitudes from the effects determined in this study. Second, the magnitudes of the largest contributors to Site2 binding are considerably less than those in hGH Site1. The absence of a set of residues that make a significant contribution to binding is an indication of why the hGH Site2 binding interface alone cannot support binding of ECD2.
A set of distinguishing traits is found for the ECD residues forming the interactions to the hormone in Site1 and Site2. A pertinent feature of these interactions is that the same residues on the ECDs are used in both hGH Site1 and 2 binding (de Vos et al. 1992). Structurally, the C-terminal FG loop (residues in the 160s loop) of the receptors undergoes ∼8 Å backbone displacement going from its configuration in the Site1-to-Site2 interface (see Q166 in Fig. 3 ▶). In addition, W104 is found in a different rotamer state in the two interfaces (Fig. 3 ▶). The Site1 ECD1 Ala-scan shows the key binding determinants are W104E1 and W169E1 (Fig. 3B ▶; Clackson and Wells 1995). Alanine mutations at either of W104E1 or W169E1 result in complete loss of hGH Site1 binding and function (Clackson and Wells 1995; Clackson et al. 1998). Moreover, mutation of W104E1 and W169E1 to phenylalanine still results in no detectable Site1 binding (data not shown).
Site1 and Site2 also differ in how the energetically important residues are organized at the contact interface. In Site1, the hot-spots of the respective hormone and receptor surfaces are spatially matched; that is, the hot-spot residues, W104E1 and W169E1, interact with their counterpart hot-spot residues of R64, K172, T175, and F176, which are located in helix 4 of the hGH (Fig. 3B ▶). In contrast, the relationships between the most important residues for Site2 binding do not have a similar spatial complementarity. It is surprising, for instance, that although W104E2 is essential for binding at Site2, removing hormone residues that contact it has a rather small or modest influence on binding. Similarly, the W169AE2 has a modest effect on binding, but removing the contacting residue in the hormone (R16A) has a significantly larger effect. This indicates that the spatial complementarity of the hot-spot residues might be an important attribute for providing very tight binding interactions, which is the case for Site1. Conversely, such an arrangement is not necessary, or perhaps desirable, for weaker protein–protein interactions such as the hGH Site2 interface, the function of which is to fine-tune the transient protein association processes, which regulate the activity of the system. In this regard, it was determined that it is the receptor–receptor contact that contributes most energy to formation of the final ternary complex, and although the Site2 hGH-receptor interaction is required for receptor signaling, the energy derived from it is somewhat less (Bernat et al. 2003).
Binding additivity of pairwise interactions
Although the residues constituting the hGH Site1 interface display general additivity with regard to their individual contributions to the binding energy (Cunningham and Wells 1993; Lowman and Wells 1993), Bernat et al. (2003) found that several receptor residues of ECD2 act together through some cooperative mechanism. To explore whether there is a similar interplay of additivity and cooperativity (nonadditive) effects contributing to the energy surface in the hGH Site2 interface, a series of pairwise hGH double mutants were constructed and their binding kinetics measured. Previous structural work identified the Site2 hGH D116 carboxyl side-chain as having two distinct conformational states, which can apparently trigger a switch from an indirect role in the wt-hGH complex to forming two intermolecular H-bonds to the indole nitrogens of W104E2 and W169E2 (Fig. 5B ▶; Schiffer et al. 2002). D116 is located in the middle of helix 3, which contains several residues implicated in Site2 binding. Thus, based on its central location in the binding epitope and its observed involvement in the conformational flexibility of Site2, most of our work focused on pairing D116A with other Site2 mutations.
Seven double mutations were constructed by pairing D116A with other alanine hGH Site2 mutations. These residues included groups that were both near and distant from D116, and by themselves displayed a range of effects on binding as single mutants. Of the pairwise D116A mutants evaluated, only those with R16A and N109A resulted in hGH variants displaying binding additivity (Table 1). In contrast, the L6A, N12A, E119A, R19A, and Y103A double mutants with D116A show strong nonadditive behavior (Table 1). There is no apparent pattern with respect to proximity; R16 and E119 are close to D116, but have different additivity characteristics when paired with D116A. Interestingly, these data show that groups separated by at least 10 to 15 Å (as is the case for positions L6, N12, R19, Y103, and N109) and on different secondary structural elements appear to be coupled, at least in the context of binding energy additivity. We note that none of the stereochemically diverse residue types substituted at position 116 (D, A, N, V) display any measurable cooperativity with complementary mutations at W169E2, an ECD2 residue in close proximity to 116 (Fig. 4 ▶; Tables 1, 2). W169E2A or W169E2F have similar binding constants whether the hGH 116 residue is either an Asp, Ala, Asn, or Val (Tables 1, 2). Thus, the nonadditive effects observed for residue 116 of hGH appear to result from intramolecular factors and are not similarly induced through pairwise intermolecular hormone-receptor mutations.
The strong nonadditivity effects for most of these pairwise mutants are surprising, given the fact that the D116A single mutant actually showed slightly increased binding. It is noteworthy that the pattern of nonadditivity of the D116A pairwise mutations is essentially paralleled when the mutations are paired with D116V (Table 1). Thus, the nonadditivity properties are not due to some special feature of having an alanine at residue 116, but rather its positioning in the interface (Fig. 4 ▶). Interestingly, the two largest nonadditive effects are seen in combination with the L6A and N12A mutants, which also showed a slightly increased binding as single mutants. The pairwise N12A,D116A-hGH double mutant, which should result in a tighter binding variant (0.5-fold decrease predicted based on additivity), actually produces a 35-fold decrease in binding affinity—a 70-fold discrepancy in additivity (Table 1). Similarly, the L6A, D116A-hGH variant shows a 33-fold decrease in binding affinity (predicted twofold decrease, Table 1). It is noteworthy that in contrast, the L6A,N12A-hGH double mutant obeys general additivity (Table 1). Thus, the nonadditive effect seen when the residues are combined with D116A is the product of the mutation at 116, and not that either the L6A or N12A has some inherent character that triggers nonadditivity. In addition, when all three mutations are combined: L6A,N12A,D116A-hGH, binding is decreased by ∼70-fold (Fig. 2B ▶; Table 1), appreciably less than what would be expected for full additivity of the two double mutants. These results indicate that the factor(s) responsible for the D116A effect plays a similar role in both the double mutants with L6A and N12A and, thus, is not double-counted in the context of the triple mutant.
Implications of functional nonadditivity binding effects
In the cases of both the hGH and hPRL receptors binding at Site1 of hGH, there was a distinct spatial localization of the residues contributing most to binding, and the individual residues in this group contributed in an additive fashion to the binding (Cunningham and Wells 1991, 1993; Clackson et al. 1998). Pairwise correlations between hGH residues in Site1 were explored using the shot-gun Ala-scanning approach, which further demonstrated that this binding surface is highly additive (Weiss et al. 2000). In contrast, the pattern of nonadditivity observed for the pairwise mutations involving D116A indicates that the energy surface responsible for Site2 binding has some functionally different properties than those observed for hGH Site1 binding. It is relevant, perhaps, that Site2 does not have a binding hot-spot or a particular single residue that makes an essential interaction. It might be argued that subtle mutations in Site2 would not produce major changes in the energetics or kinetics of binding and therefore would not disrupt the persistence times of the signaling complex. However, although single mutations in the Site2 interface of hGH generally lead to small effects, our data show that certain combinations of double mutants can act cooperatively to produce effects greater than what would be expected through additivity. Thus, it is noteworthy that position 116, which we show here is an apparent focal point for the propagation of nonadditive effects, is highly conserved across species. It appears that a mutation on almost any part of the Site2 binding surface maybe be hypersensitive to a mutation at position 116. Whether position 116 is unique in this regard is unknown, and identification of other potential foci of binding cooperativity needs to be more fully explored.
The anomalous behavior of hGH N12A is also noteworthy. Based on biophysical criteria, an Asn side-chain at position 12 should play a critical binding role. It makes two H-bonds to the receptor, which structure a side of a pocket that effectively buries the hydrophobic portion of the W169E2 side-chain (Figs. 4, 5A ▶ ▶). Nevertheless, the N12A mutation appears to slightly increase binding affinity, and the pairwise mutations with it and its H-bonding partners, R43A and D126A, show strict additivity (Table 2). Thus, although interactions with residues in direct contact exhibit additivity, it is quite surprising that there are clear cooperativity effects with D116A, which is 10 Å away.
Although the functional origin of the nonadditive effects in Site2 is unclear, its existence may be a general property of topography of the Site2 interface itself. A recent structure of a high-affinity Site1 variant of hGH showed that the tertiary contacts between the hormone and ECD2 are totally reorganized, even though there were no mutations of any groups in the interface itself (Fig. 5B ▶; Schiffer et al. 2002). Several segments of polypeptide chain containing the Site2 binding determinants on both the hormone and ECD2 undergo conformational changes up to 8 Å. Incredibly, of the eight H-bonds that are made in the interface of this variant with ECD2, only one is found in the wild-type interaction. These observations give compelling evidence for the existence of an inherent plasticity among the groups within the Site2 interface, and thus, it is perhaps not too surprising that there exist anomalous binding energetic relationships that can be triggered by one or several changes.
Materials and methods
Sample preparation
hGH was expressed and purified as described previously (Chang et al. 1987). A truncated form (residues 29–238) of the ECD of the hGHR was expressed and purified as described (Fuh et al. 1990; Clackson et al. 1998), except the receptor was eluted with 4.5 M MgCl2 off a hGH affinity column. All mutations in hGH and the hGHR ECD were made by using Kunkel mutagenesis (Kunkel et al. 1987) and confirmed by DNA sequencing. All protein purity and molecular weights were analyzed by analytical HPLC and mass spectrometry, respectively.
Surface plasmon resonance
Surface plasmon resonance experiments were performed by using a Biacore 2000 instrument at 25°C. The hGHR ECD1 was coupled by using a disulfide bond to a Pioneer C1 sensor chip through an engineered cysteine (S237C-hGHR) residue at the C-terminal end (Fig. 2A ▶). Sensor chip preparations and experiments were carried out in HBS-EP (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, 0.005% TWEEN-20 at pH 7.4) buffer. The C1 sensor chip was prepared with a 15 μL injection of 100 mM glycine and 0.3% Triton-X100 (pH 12; 10 μL/min), followed by activation of each flow cell with 25 μL (at 5 μL/min) of NHS/EDC (N-hydroxysuccinimide/N-ethy-N′-[3-dimethyl-amino-propyl]-carbodiimide, 75 mg/mL : 11.5 mg/mL) and a 40 μL injection of PDEA (2-[2-pyridinyldithio]ethaneamine hydrochloride, 18 mg/mL), and finally a 100 μL injection of reduced S237C-hGHR (50 to 100 μg/mL, typically 30 μL of hGHR plus 100 μL of 10 mM sodium acetate at pH 4.5). Flow cells 2–4 were coupled with S237C-hGHR, whereas flow cell 1 was activated but no S237C-hGHR was injected (served as the reference cell). The flow cells were blocked with 30 μL of 50 mM reduced glutathione in 20 mM sodium acetate, 1 M NaCl (pH 4.5). The coupling strategy produced a low-density surface with a typical functional response of ∼60 to 90 resonance units.
Trimolecular SPR experiments (flow rate of 50 μL/min) were begun with a saturating concentration injection of 100 μL of 250 nM of a hGH variant and then a 250 μL injection of free hGHR, followed by a 700-s dissociation period (Fig. 2 ▶). The surface was regenerated for subsequent injections with a 5 μL injection of 4.5 M MgCl2. This regeneration procedure allowed for the measurement of ∼200 to 300 protein injections on a single chip. The sensorgrams were analyzed by using the global decaying surface model (Joss et al. 1998) in the program Clamp (Myszka and Morton 1998) to determine on (kon) and off (koff) rate constants for the hGHR ECD2. Each protein variant was measured in triplicate, and the average kon and koff rate constants are reported. The rate constants show differences <5%. There was minimal chip-to-chip variability as assayed by measuring the kinetics of the wt-hGH–ECD ternary complex formation.
Acknowledgments
We thank Drs. B. Bernat and G. Pal for SPR assistance and many discussions. This work was supported by the NIH grant DK-61602 (to A.A.K.). S.T.R.W. is a Burroughs Wellcome Fund fellow of the Life Sciences Research Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03133903.
References
- Atwell, S., Ultsch, M., de Vos, A.M., and Wells, J.A. 1997. Structural plasticity in a remodeled protein–protein interface. Science 278 1125–1128. [DOI] [PubMed] [Google Scholar]
- Bernat, B., Pal, G., Sun, M., and Kossiakoff, A.A. 2003. Determination of the energetics governing the regulatory step in growth hormone–induced receptor homodimerization. Proc. Natl. Acad. Sci. 100 952–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter, L.R., Yancopoulos, G.D., and Stahl, N. 1998. General mechanisms of cytokine receptor signaling. Adv. Protein Chem. 52 109–140. [DOI] [PubMed] [Google Scholar]
- Chang, C.N., Rey, M., Bochner, B., Heyneker, H., and Gray, G. 1987. High-level secretion of human growth hormone by Escherichia coli. Gene 55 189–196. [DOI] [PubMed] [Google Scholar]
- Chen, T.-J., Kuo, C.B., Tsai, K.F., Liu, J.-W., Chen, D.-H., and Walker, A.M. 1997. Development of recombinant human prolactin receptor antagonists by molecular mimicry of the phosphorylated hormone. Endocrinology 139 609–616. [DOI] [PubMed] [Google Scholar]
- Clackson, T. and Wells, J.A. 1995. A hot spot of binding energy in a hormone-receptor interface. Science 267 383–386. [DOI] [PubMed] [Google Scholar]
- Clackson, T., Ultsch, M.H., Wells, J.A., and de Vos, A.M. 1998. Structural and functional analysis of the 1 : 1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J. Mol. Biol. 277 1111–1128. [DOI] [PubMed] [Google Scholar]
- Cunningham, B.C. and Wells, J.A. 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244 1081–1085. [DOI] [PubMed] [Google Scholar]
- 1991. Rational design of receptor-specific variants of human growth hormone. Proc. Natl. Acad. Sci. 88 3407–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1993. Comparison of a structural and a functional epitope. J. Mol. Biol. 234 554–563. [DOI] [PubMed] [Google Scholar]
- Cunningham, B.C., Ultsch, M., de Vos, A.M., Mulkerrin, M.G., Clauser, K.R., and Wells, J.A. 1991. Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science 254 821–825. [DOI] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 12 14–20. [DOI] [PubMed] [Google Scholar]
- de Vos, A.M., Ultsch, M., and Kossiakoff, A.A. 1992. Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 255 306–312. [DOI] [PubMed] [Google Scholar]
- Elkins, P.A., Christinger, H.W., Sandowski, Y., Sakal, E., Gertler, A., de Vos, A.M., and Kossiakoff, A.A. 2000. Ternary complex between placental lactogen and the extracellular domain of the prolactin receptor. Nat. Struct. Biol. 7 808–815. [DOI] [PubMed] [Google Scholar]
- Fuh, G., Mulkerrin, M.G., Bass, S., McFarland, N., Brochier, M., Bourell, J.H., Light, D.R., and Wells, J.A. 1990. The human growth hormone receptor. J. Biol. Chem. 265 3111–3115. [PubMed] [Google Scholar]
- Fuh, G., Colosi, P., Wood, W.I., and Wells, J.A. 1992. Rational design of potent antagonists to the human growth hormone receptor. Science 268 5376–5381. [DOI] [PubMed] [Google Scholar]
- Gertler, A., Gosclaude, J., Strasburger, C.J., Nir, S., and Dijane, J. 1996. Real-time kinetic measurements of the interaction between lactogenic hormones and prolactin-receptor extracellular domains from several species support the model of hormone-induced transient receptor dimerization. J. Biol. Chem. 271 24482–24491. [DOI] [PubMed] [Google Scholar]
- Hensley, P., Doyle, M.L., Myszka, D.G., Woody, R.W., Brigham-Burke, M.R., Erickson-Miller, C.L., Griffin, C.A., Jones, C.S., McNulty, D.E., O’Brien, S.P., et al. 2000. Evaluating energetics of erythropoietin ligand binding to homodimerized receptor extracellular domains. Methods Enzymol. 323 177–207. [DOI] [PubMed] [Google Scholar]
- Ihle, J.N., Witthuhn, B.A., Quelle, F.W., Yamamoto, K., Thierfelder, W.E., Kreider, B., and Silvennoinen, O. 1994. Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem. Sci. 19 222–227. [DOI] [PubMed] [Google Scholar]
- Joss, L., Morton, T.A., Doyle, M.L., and Myszka, D.G. 1998. Interpreting kinetic rate constants from optical biosensor data recorded on a decaying surface. Anal. Biochem. 261 203–210. [DOI] [PubMed] [Google Scholar]
- Kossiakoff, A.A. and de Vos, A.M. 1998. Structural basis for cytokine hormone-receptor recognition and receptor activation. Adv. Protein Chem. 52 67–108. [DOI] [PubMed] [Google Scholar]
- Kossiakoff, A.A., Somers, W., Ultsch, M., Andow, K., Muller, Y.A., and de Vos, A.M. 1994. Comparison of the intermediate complexes of human growth hormone bound to the human growth hormone and prolactin receptors. Protein Sci. 3 1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel, T.A., Roberts, J.D., and Zakour, R.A. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154 367–382. [DOI] [PubMed] [Google Scholar]
- Liparoto, S.F. and Ciardelli, T.L. 1999. Biosensor analysis of the interleukin-2 receptor complex. J. Mol. Recognit. 12 316–321. [DOI] [PubMed] [Google Scholar]
- Liparoto, S.F., Myszka, D.G., Wu, Z., Goldstein, B., Laue, T.M., and Ciardelli, T.L. 2002. Analysis of the role of the interleukin-2 receptor γ chain in ligand binding. Biochemistry 41 2543–2551. [DOI] [PubMed] [Google Scholar]
- Lowman, H.B. and Wells, J.A. 1993. Affinity maturation of human growth hormone by monovalent phage display. J. Mol. Biol. 234 564–578. [DOI] [PubMed] [Google Scholar]
- Myszka, D.G. and Morton, T.A. 1998. Clamp: A biosensor kinetic data analysis program. Trends Biochem. Sci. 23 149–150. [DOI] [PubMed] [Google Scholar]
- Nice, E., Layton, J., Fabri, L., Hellman, U., Engstrom, A., Persson, B., and Burgess, A.W. 1993. Mapping of the antibody- and receptor-binding domains of granulocyte colony-stimulating factor using an optical biosensor: Comparison with enzyme-linked immunosorbent assay competition studies. J. Chromatogr. 646 159–168. [DOI] [PubMed] [Google Scholar]
- Nicola, N.A. and Hilton, D.J. 1999. General classes and functions of four-helix bundle cytokines. In Cytokines (ed. J.A. Wells), pp. 1–65. Academic Press, San Diego, CA. [DOI] [PubMed]
- Schiffer, C., Ultsch, M., Walsh, S., Somers, W., de Vos, A.M., and Kossiakoff, A. 2002. Structure of a phage display-derived variant of human growth hormone complexed to two copies of the extracellular domain of its receptor: Evidence for strong structural coupling between receptor binding sites. J. Mol. Biol. 316 277–289. [DOI] [PubMed] [Google Scholar]
- Shen, B.J., Hage, T., and Sebald, W. 1996. Global and local determinants for the kinetics of interleukin-4/interleukin-4 receptor α chain interaction: A biosensor study employing recombinant interleukin-4-binding protein. Eur. J. Biochem. 240 252–261. [DOI] [PubMed] [Google Scholar]
- Somers, W., Ultsch, M., de Vos, A.M., and Kossiakoff, A.A. 1994. The X-ray structure of a growth hormone-prolactin receptor complex. Nature 372 478–481. [DOI] [PubMed] [Google Scholar]
- Sundstrom, M., Lundqvist, T., Rodin, J., Giebel, L.B., Milligan, D., and Norstedt, G. 1996. Crystal structure of an antagonist mutant of human growth hormone, G120R, in complex with its receptor at 2.9 Å resolution. J. Biol. Chem. 271 32197–32203. [DOI] [PubMed] [Google Scholar]
- Syed, R.S., Reid, S.W., Li, C., Cheetham, J.C., Aoki, K.H., Liu, B., Zhan, H., Osslund, T.D., Chirino, A.J., Zhang, J., et al. 1998. Efficiency of signaling through cytokine receptors depends critically on receptor orientation. Nature 395 511–516. [DOI] [PubMed] [Google Scholar]
- Wang, Y., Shen, B.J., and Sebald, W. 1997. A mixed-charge pair in human interleukin 4 dominates high-affinity interaction with the receptor α chain. Proc. Natl. Acad. Sci. 94 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, G.A., Watanabe, C.K., Zhong, A., Goddard, A., and Sidhu, S.S. 2000. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. 97 8950–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, J.A. and de Vos, A.M. 1996. Hematopoietic receptor complexes. Ann. Rev. Biochem. 65 609–634. [DOI] [PubMed] [Google Scholar]
- Wilson, I.A. and Jolliffe, L.K. 1999. The structure, organization, activation and plasticity of the erythropoietin receptor. Curr. Opin. Struct. Biol. 9 696–704. [DOI] [PubMed] [Google Scholar]