

Abstract
A kinetic comparison of the hydrolase and transferase activities of two bacterial phospholipase D (PLD) enzymes with little sequence homology provides insights into mechanistic differences and also the more general role of Ca2+ in modulating PLD reactions. Although the two PLDs exhibit similar substrate specificity (phosphatidylcholine preferred), sensitivity to substrate aggregation or Ca2+, and pH optima are quite distinct. Streptomyces sp. PMF PLD, a member of the PLD superfamily, generates both hydrolase and transferase products in parallel, consistent with a mechanism that proceeds through a covalent phosphatidylhistidyl intermediate where the rate-limiting step is formation of the covalent intermediate. For Streptomyces chromofuscus PLD, the two reactions exhibit different pH profiles, a result consistent with a mechanism likely to involve direct attack of water or an alcohol on the phosphorus. Ca2+, not required for monomer or micelle hydrolysis, can activate both PLDs for hydrolysis of PC unilamellar vesicles. In the case of Streptomyces sp. PMF PLD, Ca2+ relieves product inhibition by interactions with the phosphatidic acid (PA). A similar rate enhancement could occur with other HxKx4D-motif PLDs as well. For S. chromofuscus PLD, Ca2+ is absolutely critical for binding of the enzyme to PC vesicles and for PA activation. That the Ca2+-PA activation involves a discreet site on the protein is suggested by the observation that the identity of the C-terminal residue in S. chromofuscus PLD can modulate the extent of product activation.
Keywords: Phospholipase D, 31P NMR, phosphatidylcholine, phosphatidic acid, Ca2+ activation
Many phospholipase D (PLD) enzymes can catalyze two reactions: (1) hydrolysis of a phospholipid to produce phosphatidic acid (PA) and a free alcohol, and (2) transphosphatidylation of one phospholipid with an alcohol to form a new phosphatidylalcohol. The hydrolysis product PA is an important second messenger in mammalian signal transduction pathways (English 1996). Although products and derivatives of the transphosphatidylation reaction may also have physiological roles in vivo (Exton 1997), the production of a nonnaturally occurring phosphatidylalcohol by PLD has been used as a specific assay for detecting PLD activity in a variety of cells (Heller 1978). This reaction, often carried out in heterogeneous microemulsion systems (e.g., in ether–water), has also been used to synthesize rare phospholipids, unusual nonnaturally occurring lipids, and isotopically labeled phospholipids from naturally abundant PC (Dawson 1967; Eibl and Kovatchev 1981).
Given the usefulness of PLD in generating novel or labeled phospholipids, there have been a number of studies screening different organisms for PLD activities with high transphosphatidylation specific activity. In particular, many Streptomyces PLD enzymes, for example, Streptomyces sp. PMF, have higher transphosphatidylation activity than plant or many other documented sources of the enzyme (Juneja et al. 1988; Hagishita et al. 1999). A notable exception to this is Streptomyces chromofuscus PLD which has significantly lower transphosphatidylation activity compared to its hydrolysis activity (Juneja et al. 1988; Hagishita et al. 1999). Unlike the other Streptomyces PLDs, the S. chromofuscus enzyme does not contain the characteristic HxKx4D (HKD) domains (Yang and Roberts 2002), but instead has tightly bound iron that is necessary for catalytic activity (Zambonelli and Roberts 2003), suggesting that this PLD catalyzes the hydrolysis of phospholipids via a different mechanism. S. chromofuscus can carry out a transphosphatidylation reaction but requires higher concentrations of primary alcohols (Geng et al. 1999). There are few kinetic studies of the transphosphatidylation reaction, particularly in homogeneous solutions. With this in mind, we have examined both hydrolase and transferase activities of both S. chromofuscus and Streptomyces sp. PMF PLD enzymes using NMR spectroscopy (for example, Fig. 1 ▶ for hydrolysis of PI vesicles). Properties of the two different PLDs, including the pH optimum, Ca2+ dependence, substrate specificity, and effect of substrate aggregation state on activity have been determined. A comparison of the two enzymes strongly suggests that they have quite different mechanisms—the Streptomyces sp. PMF uses covalent catalysis while S. chromofuscus PLD utilizes direct attack of water or alcohol on the phosphorus atom to generate PA or new phospholipid products. However, the effect of Ca2+ on the kinetics of both enzymes suggests that caution is needed in implicating Ca2+ as necessary for activity of PLD enzymes.
Figure 1.

500 MHz 1H spectra (2.9–3.8 ppm) of 5 mM PI SUVs (in 20 mM imidazole, pH 7.0, with 1 mM Ca2+, 23°C) as a function of incubation time (in minutes) with 135 μg S. chromofuscus rPLD. Note the appearance of the sharp myo-inositol peaks (identified by carbon number) in the spectrum.
Results
Phosphohydrolase activity of Streptomyces sp. PMF PLD
The substrate specificity and sensitivity to interfaces of Streptomyces sp. PMF PLD, a member of the PLD superfamily (Ponting and Kerr 1996), were examined. Although a variety of phospholipids could be hydrolyzed by Streptomyces sp. PMF PLD, the highest specific activity was for PC with anionic phospholipids relatively poor substrates. This Ca2+-independent PLD (5 mM Ca2+ had no effect on diC4PC hydrolysis) showed good activity toward monomeric diC4PC (Table 1). The dependence of specific activity on chain length at fixed substrate concentration (Table 1) suggested that the longer the chain length, the lower the specific activity of the enzyme. However, if the concentration dependence of this PLD activity were examined, the enzyme activity toward short-chain micelles would have increased substantially (but not in the discreet two-phase model fashion often seen for phospholipases). For diC7PC, both the hydrolase and transferase data (Fig. 2A ▶) could be fit by a single apparent Km of 1.6 ± 0.3 mM, a value rather close to the CMC of that PC (1.5 mM). Pure diC6PC has a CMC of 14 mM (Bian and Roberts 1992). The activity of this PLD as a function of diC6PC concentration was not hyperbolic (Fig. 2B ▶). If data ≤5 mM PC was used to extract an apparent Km, the value was ∼9 mM, a value not too far off the CMC of diC6PC. The dramatic increase in an apparent Km that tracks the substrate CMC strongly suggests that the presence of micelles enhances PLD activity (possibly enhancing product release), and that the enzyme is likely to lower the CMC of the substrate. However, the affinity of the enzyme for the surfaces of micelles is not high because the kinetic effect is gradual (at least with this particular substrate). Thus, the Streptomyces sp. PMF PLD appears to exhibit some interfacial behavior.
Table 1.
Streptomyces sp. PMF PLD activity toward phospholipids in monomer, micelles, vesicles, and mixed micelles at 37°C
| Substrate | Conc. (mM) | Ca2+ (mM) | Physical state | Specific hydrolasea | Activity transferasea |
| diC4PC | 5 | 0 | Monomer | 12.9 | 5.20 |
| diC6PC | 5 | 0 | Monomer | 9.94 | 3.26 |
| diC7PC | 5 | 0 | Micelle | 6.92 | 1.69 |
| POPC | 10 | 0 | SUV | 0.93b | |
| 10 | 5 | SUV | 2.7b | ||
| 10 | 5 | LUV | 0.78b | ||
| POPC/chol | 5/5 | 0 | SUV | 0.33b,c | |
| 5/5 | 5 | SUV | 2.7b,c | ||
| POPC/POPA | 9/1 | 0 | SUV | 0.012b | |
| 9/1 | 5 | SUV | 1.93b | ||
| POPC/TX-100 | 5/20 | 0 | Mixed micelle | 155d | |
| POPC/Zwitte | 5/15 | 0 | Mixed micelle | 0.23 |
a Hydrolase (generation of PA) and transferase (generation of PG) activities of Streptomyces sp. PMF PLD toward 5 mM diCnPC with 5 mM glyerol present in 50 mM MES-NaOH buffer, pH 6.5; 31P NMR was used to monitor PA and PG production.
b The specific activity of the enzyme was measured by 1H NMR spectroscopy, which monitored choline production, and hence, is a combination of hydrolase and transphosphatidylation reaction rates. Duplicate assay samples were used to obtain rates; errors in the rates were <15%.
c The specific activity is based on the rate over the first 10 min; unlike with pure POPC SUVs, the rate decreased over longer times when cholesterol was present in the vesicles.
d With this amount of TX-100 present, the major (>90%) product for Streptomyces sp. PMF PLD is the transphosphatidylation product with the Triton molecule.
e Zwitt = zwittergent 3–14.
Figure 2.
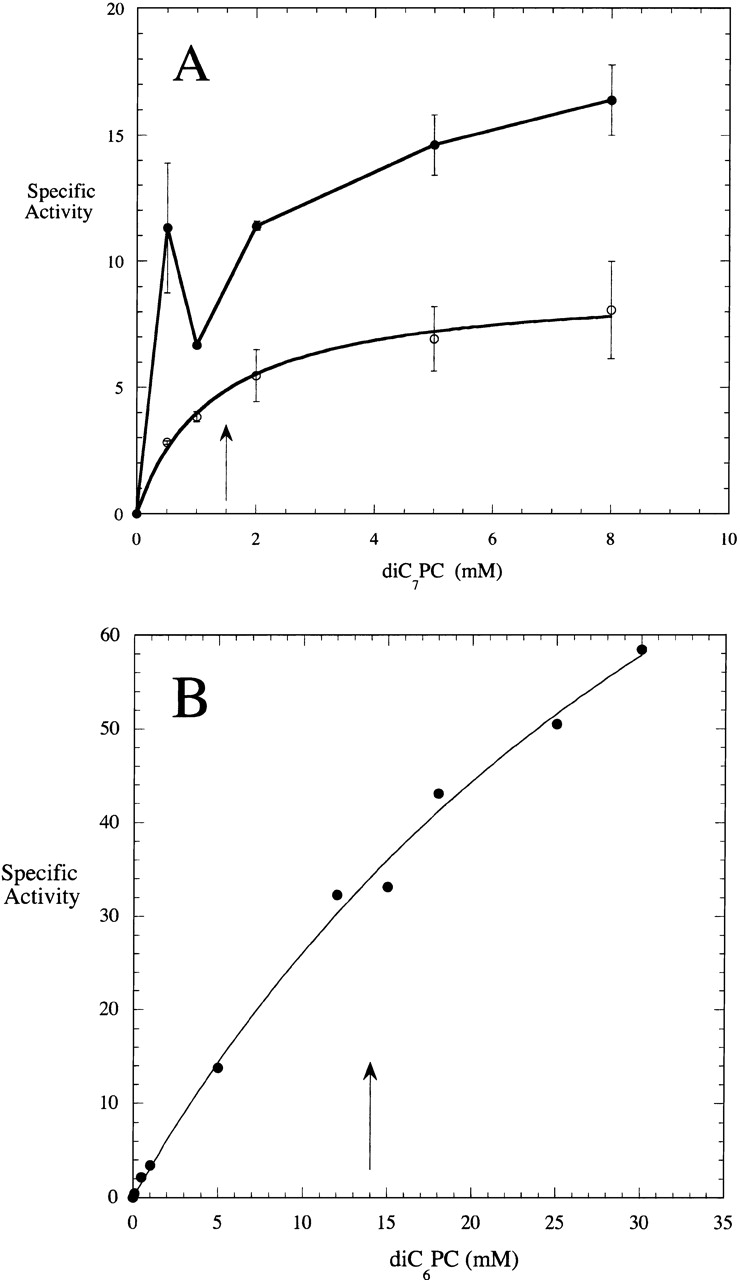
Specific activity (μmole min−1 mg−1) of (A) Streptomyces sp. PMF PLD (open circles) and S. chromofuscus rPLD (filled circles) toward diC7PC in 50 mM Tris HCl, pH 8.0, as a function of substrate concentration. Error bars indicate standard deviations in activity for each PC concentration. (B) Specific activity (μmole min−1 mg−1) of Streptomyces sp. PMF PLD cleavage of diC6PC as a function of substrate concentration. The arrows indicate the CMC of the pure PC.
PC vesicles were also substrates for Streptomyces sp. PMF PLD with much higher activity toward highly curved SUVs than LUVs (Table 1). In the absence of Ca2+, incorporation of 10 mole % PA into the PC vesicles significantly inhibited PMF PLD (leading to an 78-fold decrease in activity). Although Ca2+ was not required for the hydrolysis of monomer and micellar short-chain PC substrates by this PLD, addition of Ca2+ could enhance Streptomyces sp. PMF PLD activity toward PC packed in vesicles. Ca2+ (5 mM) increased the activity of Streptomyces sp. PMF PLD 2.9-fold toward 10 mM POPC SUVs. Ca2+ added to POPC/POPA (9:1) SUVs gave rise to a PLD specific activity that increased 161-fold but was still less than for PC SUVs with Ca2+. With detergent mixed micelle substrates, Streptomyces sp. PMF PLD activity could be either activated or inhibited depending on the type of detergent was used. POPC solubilized in Triton X-100 was a much better substrate than when presented in SUVs (even with Ca2+ added). With PC/Triton X-100 (5 mM/20 mM), the transphosphatidylation reaction led to the formation of a different phosphodiester (the chemical shift of the product peak, shifted slightly downfield of PC, was invariant to pH) that was assumed to be the PA–Triton adduct (Triton X-100 has a hydroxyl group). This new phospholipid represented >90% of the product and was produced at a rate that was significantly activated, 57-fold, above the hydrolysis of PC vesicles with 5 mM Ca2+. The increased specific activity likely represents the high interfacial concentration of Triton. In contrast to Triton X-100, zwittergent 3–14 as a matrix for PC solubilization inhibited hydrolysis of the PC by the enzyme.
Transphosphatidylation activity of Streptomyces sp. PMF PLD
Because diC4PC was a good monomeric substrate for Streptomyces sp. PMF PLD, it was used to examine the transphosphatidylation reaction of this PLD. The enzyme is typically stored in 10% (w/w) glycerol. Assay mixtures with 5 mM diC4PC contained 5 mM glycerol from dilution of enzyme stock into the assay medium. Both diC4PA and diC4PG were formed as products with the rate for diC4PG contributing ∼40% of the phospholipid products. Methanol added to the assay mixture competed effectively with water and the glycerol as a nucleophile to generate diC4PMe from the covalent intermediate (Fig. 3 ▶). The mole fraction of diC4PMe increased with increasing methanol concentration in hyperbolic fashion with an effective Km for methanol of 0.045 ± 0.013 M (Fig. 3A ▶). The sum of hydrolase and transphosphatidylation reactions of Streptomyces sp. PMF was roughly constant as a function of methanol. The ability of water-miscible alcohols to compete effectively with water is a trait similar to that of other HKD-domain PLD enzymes, where low concentrations of 1-butanol or ethanol (e.g., 0.3% to 2% or 0.04 to 0.43 M) have been routinely used to assay PLD activity (Whatmore et al. 1996; Meacci et al. 2002; Xie et al. 2002).
Figure 3.
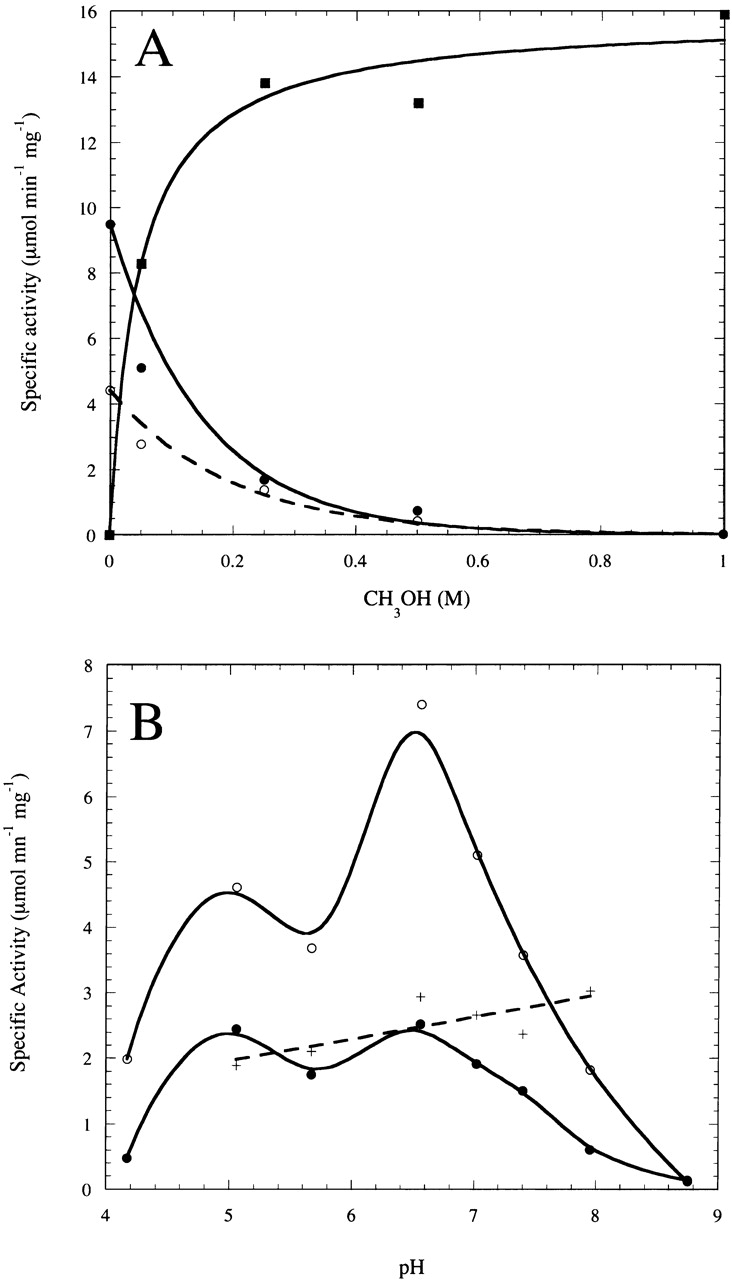
(A) Rates of diC4PA (filled circles), diC4PMe (filled squares), and diC4PG (open circles) production (μmole min−1 mg−1) by Streptomyces sp. PMF PLD (4 μg) as a function of CH3OH concentration at fixed (5 mM) diC4PC (assay mixtures also contained 5 mM glycerol from the enzyme stock). (B) Rates of diC4PA (filled circles), diC4PMe, and diC4PG production (open circles) from diC4PC (5 mM) as function of pH with 5 mM glycerol and 50 mM CH3OH; (+) represents the mole fraction transferase products (diC4PG and diC4PMe) generated by Streptomyces sp. PMF PLD as a function of pH.
The pH dependence of Streptomyces sp. PMF PLD activity toward diC4PC was examined in the presence of 50 mM methanol, a concentration that allows rates of diC4PA, diC4PMe, and diC4PG formation to be measured. Under these conditions, the activity exhibits a maxima ∼ pH 6.5 (Fig. 3B ▶). As might be expected for a mechanism that proceeds through a covalent intermediate, the rates for diC4PA and diC4PMe/diC4PG production behaved in parallel. The ratio of transphosphatidylation product to PA in 50 mM methanol changed very little in this pH range (0.65 at pH 5 to 0.75 at pH 8; Fig. 3B ▶, +). This suggests that the slow step at each pH must be formation of the covalent intermediate with rapid decomposition to either PA or PMe, depending on relative concentrations of the two nucleophiles water and methanol (or glycerol).
Phosphohydrolase activity of S. chromofuscus rPLD
In contrast to PLD from Streptomyces sp. PMF, PLD from S. chromofuscus requires Ca2+ for binding to (Stieglitz et al. 1999) and hydrolysis of zwitterionic PC bilayers (Geng et al. 1998), and is activated by its product PA in the presence of Ca2+ (Geng et al. 1998). Although it carries out a transphosphatidylation reaction, this PLD has been reported to require significantly higher alcohol concentrations than HKD-type PLD enzymes (Juneja et al. 1988). Recent work has shown that S. chromofuscus PLD contains iron as well as zinc or manganese, and that the tightly bound iron ion is critical for catalysis (Zambonelli and Roberts 2003). Addition of EDTA to reaction mixtures interacts with these transition metals as well as any Ca2+, and inactivates the enzyme. These observations suggest that this bacterial PLD carries out PC cleavage with a different mechanism than that of the HKD-type PLD enzymes. With this in mind, we examined the activity of a recombinant S. chromofuscus PLD (rPLD) toward a variety of substrates for comparison with the Streptomyces sp. PMF enzyme.
As with the rPLD cloned from the ATCC S. chromofuscus strain, Ca2+ caused a biphasic decrease in the USDA rPLD intrinsic fluorescence. The decrease was characterized by an S50 value of 0.51 mM (∼5 times higher than the S50 of ATCC rPLD). However, even though this rPLD could bind Ca2+ ions, they were not necessary for catalytic activity under some circumstances. At pH 8, rPLD cloned from the USDA strain of S. chromofuscus was active toward diC4PC in the absence of Ca2+ (the Km for this monomeric substrate was ∼0.3 mM). Addition of Ca2+ had no effect on the hydrolase activity under these conditions. The lack of activation by Ca2+ is in contrast to the enzyme cloned from the ATCC strain of the organism, which was significantly activated (5.6-fold) by 5 mM Ca2+ under the same conditions (Yang and Roberts 2002). With increasing monomeric substrate fatty acyl chain length (Table 2), rPLD activity (in the absence of Ca2+ and at pH 8) increased (from 14 to 54 μmole min−1 mg−1 as chain length increased from four to seven). A monomeric lyso-PC (1-C6-PC) was almost as good a substrate as the diacyl-PC (diC6PC). The activity of the enzyme toward various diC7PC concentrations was examined in the absence of Ca2+ (Fig. 2A ▶) to check for interfacial activation. In the absence of Ca2+, S. chromofuscus rPLD exhibited a small (25%) increase in activity upon micellization of diC7PC (comparison of 0.5 and 5 mM concentrations). Similar behavior was observed for the PLD purified from S. chromofuscus media (Geng et al. 1999). This very small activation was much less than that of other phospholipases (Roberts 1999), and, with the exception of the activity at 1 mM diC7PC (right around the effective CMC in the assay mixture), could be accounted for by a Km ∼0.3–0.5 mM. The decreased specific activity at 1 mM (near the pure diC7PC CMC of 1.5 mM; Bian and Roberts 1992) could indicate that when micelles form, they initially lower the number of particles (monomers plus micelles) available to interact with the enzyme (effectively reducing the particle concentration). Such behavior would be consistent with "hopping" mode kinetics (nonprocessive catalysis in the presence of an interface) for this particular phospholipase (Jain and Gelb 1991).
Table 2.
S. chromofuscus rPLD activity toward phosphatidylcholine substrates
| Substrate | Concentration (mM) | Physical state | Specific activitya (μmole min−1 mg−1) |
| diC4PC | 5 | Monomer | 14b |
| diC6PC | 5 | Monomer | 41b |
| diC7PC | 0.5 | Monomer | 54b |
| 5 | Micelle | 68b | |
| 1-C6PC | 5 | Monomer | 33b |
| POPC | 10 | SUVs | 11.7c |
| 10 | LUVs | 12.5c | |
| POPE | 10 | SUVs | 3.7c |
| 10 | LUVs | 1.5c | |
| DOPMe | 10 | SUVs | 0.44c |
| 10 | LUVs | 0.80c | |
| POPG | 10 | SUVs | 0.013c |
| POPC/DOPMe | 5/5 | SUVs | 11.9 (PC)/0.46 (PMe)c |
| POPC/DOPMe/chol | 5/5/5 | SUVs | 11.5 (PC)/0.93 (PMe)c |
| POPS | 10 | SUVs | 0.0020c |
| PI | 5 | SUVs | 0.058c |
| SM | 5 | SUVs | 0.0021c |
a Errors in specific activities (rates for given assay samples were done in duplicate) are <15%.
b Activities determined by pH-stat at pH 8.0 with 5 mM Ca2+ present.
c Activities determined by 31P NMR at pH 7.0 in 20 mM imidazole and 5 mM Ca2+ (except for PI, which had 1 mM Ca2+).
Although no Ca2+ was needed for hydrolysis of the short-chain PC substrates by this recombinant S. chromofuscus PLD, this divalent cation was absolutely critical for hydrolysis of PC vesicles (Table 2). With 5 mM Ca2+ present, S. chromofuscus PLD specific activities toward PC vesicles were similar to that for diC4PC hydrolysis. There was also little difference in enzyme activity toward PC SUVs and LUVs. This latter observation is unusual in that most phospholipases (including Streptomyces sp. PMF PLD) show higher activity toward PC substrates packed in small, highly curved vesicles than towards LUVs where the interface is relatively flat (Geng et al. 1998; Gadd and Biltonen 2000). Previous studies have shown that S. chromofuscus PLD prefers monomeric diC6PC over similar chain length anionic phospholipids, although most of these are reasonable substrates for the enzyme (Geng et al. 1999; Martin et al. 2000). As shown in Table 2, zwitterionic phospholipid vesicles (e.g., PC and PE) were much better substrates than anionic phospholipid vesicles (PMe, PS, PI, PG) for this recombinant enzyme. The severely reduced activity of the recombinant S. chromofuscus protein toward anionic phospholipid vesicles does not reflect differences in binding of the enzyme to vesicles because in binary vesicles of PC and PMe, the rates of hydrolysis of each substrate were comparable to the rate toward the single component vesicle. Decreasing the flexibility of phospholipids by incorporation of cholesterol (Table 2) did not affect PLD hydrolysis of PC and PMe in the binary vesicle, an observation consistent with a "hopping" mode for this enzyme.
It has been previously reported (Imamura and Horiuti 1979) that sphingomyelin is a substrate for S. chromofuscus PLD. rPLD has very low sphingomyelinase activity (∼5000 times lower than that toward PC SUVs). This ratio was significantly different from the reported ratio of 22% for sphingomyelinase activity compared to PLD activity in experiments that used PLD purified from culture medium (Imamura and Horiuti 1979), and hence, could have been contaminated with sphingomyelinase.
Effect of the C terminus on rPLD phosphohydrolase activity and vesicle binding
Although substrate physical state has little effect on PLD activity, modification of the C-terminal residue can dramatically affect the enzyme specific activity. The specific activities of rPLD and rPLD-E510 toward diC4PC (5 mM) and POPC SUVs (10 mM) are shown in Table 3. Replacement of the C-terminal valine with glutamate yielded protein with much higher specific activity. Previous studies of the PLD purified from S. chromofuscus culture media suggested that the C-terminal region was important for PA activation of the enzyme toward PC vesicles (Stieglitz et al. 1999). Therefore, the recombinant PLDs were screened for PA (10 mole %) activation of PC SUVs (Table 3). rPLD-E510, with higher specific activity toward diC4PC, also exhibited a larger PA activation (threefold) compared to rPLD with a C-terminal valine (which exhibited 1.8-fold PA activation). As with the authentic S. chromofuscus PLD (Geng et al. 1998), other anionic phospholipids either had no effect (PI) or were inhibitors (oleic acid) of PC hydrolysis. Replacement of the C-terminal valine with a lysine residue (rPLD-K510) did not affect the specific activity toward diC4PC but did further reduce the extent of PA activation of the enzyme (at most the rate increased 20% with PA added).
Table 3.
Comparison of the specific activities of C-terminal mutants of S. chromofuscus PLD toward diC4PC (5 mM) and POPC (10 mM) vesicles
| rPLD-V510 | rPLD-E510 | rPLD-K510 | |
| Substrate | Specific activitya | ||
| diC4PCb | 13.5 | 56.2 | 21.0 |
| POPCc | 11.7 | 47.4 | 14.4 |
| POPC/POPAc | 21.1 | 142 | 18.7 |
| Bindingd | % | ||
| POPC + EDTA | 32 | 4 | 19 |
| POPC + Ba2+ | 100 | 100 | 100 |
| OPC/POPA (9:1) + EDTA | 51 | 66 | 10 |
| POPA + EDTA | 94 | 100 | 70 |
a Typical errors in measuring PLD activity <15%.
b Specific activities of PLD enzymes toward this monomeric substrate where measured by pH-stat at pH 8.0, with 5 mM Ca2+.
c Specific activities of PLD toward SUVs were measured by 1H NMR in 20 mM imidazole, 5 mM Ca2+, pH 7.0.
d All binding assays were carried out with small sonicated vesicles containing 2 mM total phospholipids in 10 mM Tris HCl, pH 8.0; the error in integrating the free PLD on SDS-gels was typically 10% of the total PLD intensity.
Previous studies have shown that divalent metal ions and PA can enhance PLD partitioning to the surfaces of POPC vesicles (Stieglitz et al. 1999; Yang and Roberts 2002). Similar experiments were carried out for this recombinant USDA strain PLD, and rPLD-E510, and rPLD-K510. Like ATCC PLD (Yang and Roberts 2002) and authentic S. chromofuscus PLD purified from growth media (Stieglitz et al. 1999), rPLD partitioned weakly to mM concentrations of PC vesicles in the presence of EDTA. Addition of 10 mM Ba2+ dramatically promoted PLD partitioning to the lipid surfaces. With 1–2 mM PC vesicles the added Ba2+ caused all the enzyme to translocate to the vesicle surface (Table 3). In the absence of any divalent metal ion (5 mM EDTA in buffer), rPLD had high binding affinity for PA vesicles. Incorporation of 10% PA with PC led to ∼51% rPLD enzyme partitioning to the PC/PA (9:1) vesicles under these conditions. With rPLD-E510, divalent metal ions were still needed for the enzyme to bind to PC vesicles. However, binding to PC/PA (9:1) SUVs in the absence of Ba2+ was even stronger than for rPLD with a C-terminal valine. Moreover, the rPLD-K510 mutant with the positively charged C-terminal residue had the weakest binding affinity to PC/PA (9:1) and PA SUVs (Table 3). This suggests that a negative charge at the C terminus alters the enzyme conformation in a way that it has stronger interactions with a PA surface. Whatever the interaction of PLD with anionic vesicles, the C terminus cannot be directly involved because a positively charged C terminus leads to weaker interactions with a surface with negative charges.
Transphosphatidylation activity of rPLD
Monomeric diC4PC was used as a substrate in the presence of methanol and Ca2+ to examine the transphosphatidylation specific activity of rPLD under different conditions. This is an excellent substrate to use for this PLD because the product, diC4PA, does not precipitate with ≤20 mM Ca2+ added. Screening for optimal methanol concentration was carried out with 5 mM diC4PC and 5 mM Ca2+ (the latter suggested as necessary for transphosphatidylation activity by this PLD; Geng et al. 1999). Below 1 M (3.2% w/w) methanol, there was no detectable diC4PMe. Increasing the methanol concentration led to accumulation of diC4PMe with optimal production of diC4PMe around 10–12 M methanol under these conditions (Fig. 4 ▶). Higher methanol concentrations caused denaturation of the recombinant PLD and decreased the production of both diC4PA and diC4PMe. Interestingly, addition of methanol to the assay mixtures also activated the phosphohydrolase activity of rPLD (e.g., with 6 M CH3OH, diC4PA production was increased 3.8-fold). DiC4PA production reached a maximum around 10 M methanol (Fig. 4A ▶). This is in dramatic contrast with HKD-motif PLDs (including the Streptomyces sp. PMF PLD examined here) where 1% ethanol or other small alcohols could block the phosphodiesterase activity to yield only transphosphatidylation product (Juneja et al. 1988; Xie et al. 2002).
Figure 4.
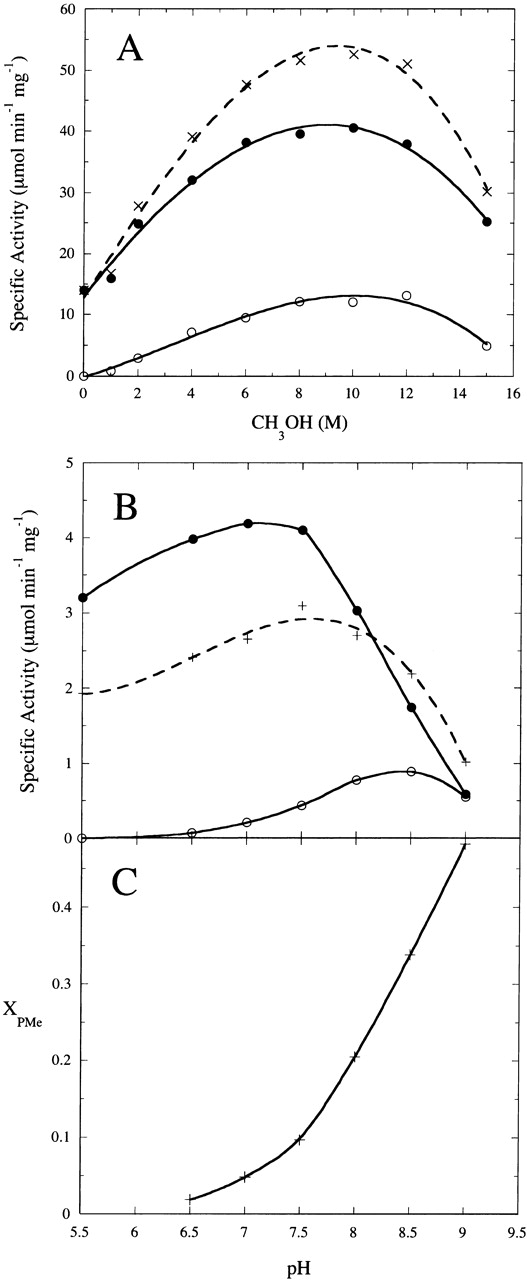
(A) Rate of diC4PA (filled circles) and diC4PMe (open circles) production by S. chromofuscus rPLD from 5 mM diC4PC in 50 mM Tris HCl, 5 mM Ca2+, pH 7.5, as a function of added CH3OH. Enzyme specific activity for generating both products is shown as (X). (B) The pH dependence of the rate of diC4PA production in the absence (+) and the presence (filled circles) of 2 M CH3OH; the rate of diC4PMe (open circles) production in the presence of 2 M CH3OH is also shown. (C) Mole fraction of PMe produced as a function of pH with 5 mM diC4PC, 5 mM Ca2+ and 2 M methanol, pH 8.0.
Because S. chromofuscus PLD from the USDA strain does not need Ca2+ for diC4PC hydrolysis, the requirement for Ca2+ in the transphosphatidylation reaction was examined. At pH 5.5 and 6 M methanol, no transphosphatidylation product, PMe, was produced from diC4PC alone. Addition of 5 mM Ca2+ activated the hydrolysis reaction around fourfold at this pH but there was still little diC4PMe generated. At pH 8.0, addition of 5 mM Ca2+ had little effect on PLD phosphohydrolase activity toward diC4PC but significantly enhanced the transphosphatidylation reaction (Fig. 5 ▶). Increasing the Ca2+ concentration further to 20 mM slightly inhibited the hydrolysis reaction of rPLD at pH 8.0 but had little effect on diC4PMe production. At pH 9.0, Ca2+ was not necessary for either hydrolysis or transphosphatidylation reactions. In fact, addition of Ca2+ inhibited both activities with much higher inhibition of PA production. The highest ratio of diC4PMe to diC4PA generated by S. chromofuscus rPLD was 2.9 at pH 9.0 in the presence of 20 mM Ca2+. By contrast, Streptomyces sp. PMF PLD with 1 M methanol yielded exclusively diC4PMe as a product.
Figure 5.
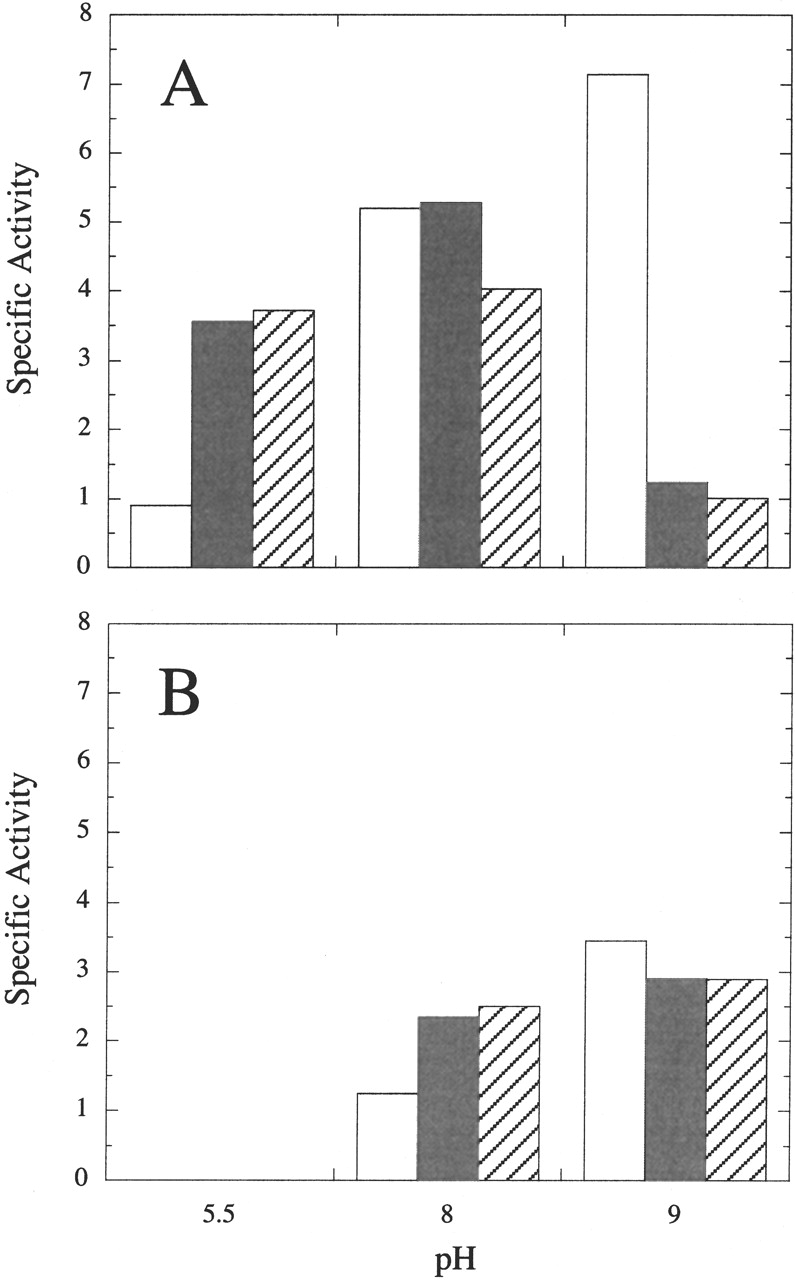
Effect of Ca2+ concentration on the (A) hydrolysis and (B) transphosphatidylation reactions of S. chromofuscus rPLD. Assay conditions included 5 mM diC4PC, 6 M CH3OH, and 9 μg rPLD. The empty rectangles represent assays in the absence of Ca2+; the filled rectangles represent assays with 5 mM Ca2+; diagonally striped rectangles represent assays with 20 mM Ca2+ added.
A more careful investigation of the pH dependence of hydrolase and transferase activities was undertaken for comparison to the pH profile for Streptomyces sp. PMF PLD. Similar to the pH profile for the hydrolysis reaction measured by pH-stat (Geng et al. 1999), which appeared to have broad peak from pH 6 to 8, the optimum pH for rPLD hydrolase activity measured by 31P NMR in the presence 5 mM Ca2+ was around 7.5. In the presence of 2 M CH3OH, which generates some transphosphatidylation product, the optimum pH for the phosphohydrolase activity was slightly more acidic. In contrast, the optimum pH for the transphosphatidylation reaction was 8.5 (Fig. 4B ▶). The mole fraction of transphosphatidylation product as a function of pH increased steadily with increasing pH (Fig. 5C ▶) in contrast to the HKD-motif Streptomyces sp. PMF PLD, which exhibited a relatively constant mole fraction of PMe and PG over a comparable pH range.
Discussion
We have examined two Streptomyces PLDs—one a member of the PLD superfamily (see Stuckey and Dixon 1999, for the first structure of a PLD family member) whose mechanism involves covalent catalysis and for which a crystal structure exists (Leiros et al. 2000), and the other a very disparate protein with virtually no sequence similarity and whose activity, dependent on the presence of tightly bound iron, is also activated by Ca2+ (Zambonelli and Roberts 2003)—for sensitivity to substrate physical state, substrate specificity, pH effects, and whether or not Ca2+ can enhance activities. A comparison of these two PLDs provides insights into mechanistic differences and also the more general role of Ca2+ in modulating PLD reactions.
Both bacterial PLDs exhibited a preference for PC, although each could catalyze the hydrolysis of a range of phosphodiester bonds. A preference for PC when presented in bilayers has been observed in most eukaryotic HKD-type PLDs identified so far, although there are reports that some PLDs prefer phospholipids with other headgroups (e.g., a recently identified intracellular PLD from Streptoverticillium cinnamoneum is reported to prefer PE followed by PS and PI, while it is inert for PC and PG; Ogino et al. 2001). When a preference for PC is observed for substrate presented in a bilayer, it may reflect properties of the phospholipid in the bilayer (e.g., accessibility of headgroup, local dielectric constant) rather than an inherent weaker efficiency for the other phospholipids.
Where the two Streptomyces PLD enzymes begin to differ is in the sensitivity to substrate physical state. Streptomyces sp. PMF PLD, like a more typical phospholipase (Roberts 1999), exhibited moderate interfacial activation (the "Km" for the enzyme toward diC7PC correlates with the CMC of that lipid, and a comparison of specific activities for monomeric and micellar diC6PC clearly showed rate enhancement for micellar substrate) and reduced activity toward PC packed in vesicles compared to PC solubilized in mixed micelles. In contrast, S. chromofuscus PLD was insensitive to aggregation of short-chain PC substrates and equally active towards PC packed in both small unilamellar and large unilamellar vesicles. This insensitivity to PC physical state (lack of classical interfacial activation, no increase in PMe or PC cleavage when binary vesicles used, insensitivity to the inclusion of cholesterol in the vesicles) strongly indicates that S. chromofuscus PLD works in a hopping mode and is easily able to isolate the phosphodiester bond of an individual PC molecule in an interface.
Effects of Ca2+ on PLD activities
Both Ca2+-dependent and Ca2+-independent PLD enzymes have been identified in mammals (Exton 1997, 2000), yeast (Mayr et al. 1996; Hammond et al. 1997; Waksman et al. 1997) and bacteria (Imamura and Horiuti 1979; Juneja et al. 1988; Saito et al. 1990; Hasagawa et al. 1992; Shimbo et al. 1993), and nearly all plant PLDs require micro- to millimolar Ca2+ for stimulation (Wang 2000). When a Ca2+ requirement exists, the question is whether it reflects a direct binding of that cation to the enzyme (causing a change that enhances substrate binding, optimizes catalysis, promotes product release from the enzyme, etc.), or whether it reflects physical effects on the product PA. The PLD product PA has a moderately high affinity for Ca2+; addition of this ion will cause PA to cluster in bilayers, and in some situations lead to vesicle fusion (Swairjo et al. 1994). PA could also be a competitive inhibitor of substrate and inclusion of Ca2+ in assay systems could interact with PA and effect its release from the active site. With S. chromofuscus PLD, we have shown that added Ba2+ and Ca2+ reduce the high affinity of that enzyme for PA surfaces (Stieglitz et al. 1999).
S. chromofuscus PLD purified from growth medium (Geng et al. 1999; Stieglitz et al. 1999; El Kirat et al. 2002) and recombinant PLD from the ATCC strain of S. chromofuscus (Yang and Roberts 2002) have been demonstrated to require Ca2+ for binding to lipid surfaces and enhancing catalysis (both phosphohydrolase and phosphatidylation reactions). Ca2+ is also an activator when other soluble substrates (e.g., p-nitrophenylphosphocholine, bis-p-nitrophenylphosphocholine) are examed (Zambonelli and Roberts 2003). rPLD cloned from a second S. chromofuscus strain also requires Ca2+ for binding to PC vesicles. However, with short chain diC4PC as substrate the dependence of rPLD activity on Ca2+ is not absolute. At acidic pH, Ca2+ enhanced the hydrolysis reaction, while there was no phosphatidylation product diC4PMe even in the presence of 6 M methanol. Under basic conditions (most notably pH 9), Ca2+ inhibited both phosphohydrolase and transferase reactions. A similar pH-dependent Ca2+ stimulation was also observed in plant PLD, although the trend was reversed with protein activated by millimolar range Ca2+ at neutral pH but by 50 μM Ca2+ at pH 4.5–5.0 (Pappan and Wang 1999). This confirms that Ca2+ is not absolutely necessary for S. chromofuscus PLD hydrolase activity, although it can enhance catalysis. Similar to S. chromofuscus, Streptomyces sp. PMF PLD is Ca2+-independent with diC4PC as a substrate at neutral or more basic pH. The only activation observed with monomeric PC is at acidic pH values.
Where Ca2+ has a major role is in the PLD-catalyzed hydrolysis of PC vesicles. S. chromofuscus PLD (recombinant protein from both strains of the organism) does not bind well to PC vesicles without that cation, so the observed PLD specific activity is very Ca2+-dependent. In contrast, Streptomyces sp. PMF PLD could bind and hydrolyze PC SUVs well without Ca2+, although there was a threefold increase in rate with Ca2+ added. Part of the Ca2+ activation observed for S. chromofuscus PLD (and probably most of the activation seen with Streptomyces sp. PMF PLD) is likely due to relief of inhibition of PLD by product PA. Both enzymes were inhibited by PA; for the Ca2+-independent PMF PLD, the rate was ∼78-fold lower with 10 mole % PA in the PC vesicle. Addition of Ca2+ enhanced the rate to a value approaching that observed for Streptomyces sp. PMF PLD action on PC SUVs with Ca2+ present. These observations are consistent with Ca2+ enhancing Streptomyces sp. PMF PLD activity towards vesicles by complexation of PA and preventing it from binding to the enzyme active site. This may be a major factor in explaining the Ca2+-enhancing effects of many other PLDs as well.
For S. chromofuscus PLD, PA inhibition depends on the Ca2+ concentration. With ∼0.2 mM Ca2+, which saturates at least one binding site on the enzyme (Stieglitz et al. 2001) and is more than enough for the maximum activity toward diC4PC (Geng et al. 1998), S. chromofuscus activity toward PC vesicles with 10 mole % PA was inhibited ∼10-fold compared to pure PC vesicles. Addition of 5 mM Ca2+ to the PC/PA (9:1) vesicles led to a rate enhancement over and above that observed for single-component PC vesicles and Ca2+ (a twofold increase with rPLD), consistent with a specific interaction of Ca2+-PA and the enzyme. For S. chromofuscus PLD, the Ca2+-PA activation appears to involve the C terminus of the protein. Interestingly, the charge of the C terminus is key. There was a fourfold difference in specific activity and an enhanced PA activation when the C terminus was changed from neutral and hydrophobic valine to a negatively charged glutamate side chain. In the presence of EDTA, binding of rPLD-E510 to PC/PA bilayers was enhanced by this negatively charged side chain at the C terminus (a result that might seem counterintuitive because the membrane surface already has a negative charge due to the PA). Switching the side chain to positively charged lysine reduces PA activation while having little effect on specific activity.
S. chromofuscus PLD appears to have a binuclear metal ion site responsible for phosphodiester bond cleavage with at least Cys123, Asp151, Tyr154, and His391 residues critical for iron content (Zambonelli and Roberts 2003). Only one residue thus far, His226, has been linked to PA activation (Yang and Roberts 2002). H226A, with roughly 0.10 times the activity of rPLD towards PC substrates, exhibited a greatly enhanced PA activation, while binding to vesicles was not significantly affected. The results with C-terminal mutants also suggest that there really is a distinct site on the enzyme for PA binding that leads to kinetic activation. Whether or not either the C terminus or His226 is a direct part of this site is unclear at present, and likely will require detailed structural information.
Covalent versus noncovalent catalysis
The mechanism of HKD-type of PLD enzymes has been proposed to center around production of a phosphatidyl–histidine intermediate (Leiros et al. 2000). Indeed, for other members of the PLD superfamily, a phosphoryl–enzyme intermediate has been detected (Gottlin et al. 1998). Furthermore, it has been proposed that this first step, forming the phosphoryl–histidyl intermediate, should be the rate-limiting step (Leiros et al. 2000). The second nucleophilic attack by a water molecule or methanol should have little significant effect on catalytic rate. Such a mechanism predicts production of PA and the transphosphatidylation product in parallel as long as it is formation of the intermediate and not its decomposition that is rate limiting. Generating a pH profile for the phosphatidylation reaction is difficult to do in a heterogeneous assay system (e.g., a solvent system of ether–water mixed with alcohol is typically used). DiC4PC is the ideal substrate for activities with mixed water-miscible solvents because it does not form aggregates (Bian and Roberts 1992). The present work with this monomeric substrate shows that the two reactions of Streptomyces sp. PMF PLD have the same pH optima. However, the two reactions catalyzed by S. chromofuscus PLD exhibit different pH optima with a considerably more basic optimum pH for the transphosphatidylation activity such that the ratio of the two products is not constant but increases dramatically with increasing pH. The requirement for Ca2+ for production of PA at acidic pH values is also not consistent with decomposition of a covalent intermediate becoming rate limiting. The shifted pH profiles and differential Ca2+ requirement for these two reactions either indicate that S. chromofuscus PLD catalyzes PC cleavage in a single step (where either water or the alcohol is the nucleophile and a more basic pH is needed to increase the nucleophilicity of the alcohol) or forming the substrate–enzyme intermediate is not rate limiting and the ionization status of the second nucleophile critically affects the reaction rate.
Although most of the identified PLDs have both phosphodiesterase and transferase activities, the transphosphatidylation reaction is regarded as the hallmark of the enzyme–substrate intermediate (Exton 1997, 2000). There are some PLDs identified, including a bacterial PLD (Ogino et al. 2001), a yeast Ca2+-dependent PLD (Mayr et al. 1996; Waksman et al. 1997), and a mammalian mitochondrial PLD (Madesh and Balasubramanian 1997), that have no transferase activity (or at least none under conditions where other PLDs show efficient transferase actvity). For eukaryotic PLDs that catalyze both reactions, 1% ethanol could block the hydrolysis reaction and produce only the phosphatidylation product, phosphatidylethanol. In marked contrast, the hydrolysis activity of the yeast PLD identified from Saccharomyces cerevisiae is stimulated by alcohol (Ella et al. 1995). Similar to this yeast PLD, our recombinant USDA PLD was also activated by alcohol. In the presence of 6 M methanol, the hydrolysis activity of USDA PLD is activated 3.8-fold. In general, addition of methanol will decrease the activity of water molecules and inhibit the hydrolysis reaction. Activation by methanol likely reflects the influence of a more hydrophobic environment at the PLD active site that can enhance substrate binding and catalysis.
Cofactors and activators of a non-HKD-motif PLD
The work presented on the S. chromofuscus PLDs strongly supports a direct attack of water (or alcohol) on the phosphorus and regulation of enzyme activity via the interaction of two functionally distinct phospholipid binding sites on the enzyme: (1) the active site where a binuclear metal center is absolutely essential for catalysis (Zambonelli and Roberts 2003), and (2) an activator site that is specific for PA or other amphiphilic phosphomonoesters (Geng et al. 1998; Stieglitz et al. 1999). Ca2+ binds to this PLD but under some conditions (e.g., high pH) is not needed for catalysis with monomer or micelle substrates. A major effect of Ca2+ binding is to cause a conformational change in the protein that enhances binding of the protein to zwitterionic interfaces. Previous work suggested PLD binding to PC vesicles with Ba2+ as the noncatalytic Ca2+ substitute was insensitive to pH over the range 5–9. PA-driven binding to vesicles, however, was sensitive to pH with a pKa ∼7 (Stieglitz et al. 1999). Deprotonation of a key side chain (possibly a histidine) on the enzyme as the solution pH increases shifts the equilibrium toward a vesicle-bound form. Ca2+ may be critical (but not absolutely necessary) at acidic pH values (for even monomeric substrates) by lowering the pKa2 of active site residues. In the absence of divalent cations (and the presence of EDTA), product PA can drive the PLD to the membrane surface. This binding mode is complicated because PA can occupy the active site as well as another site. The Ca2+ site (or sites) on S. chromofuscus PLD could resemble a C2 domain and the activating interaction could involve a Ca2+-PA complex. Indeed, Ca2+ binding to rPLD does cause a loss of secondary structure as measured by ellipticity at 222 nm (C. Zambonelli, unpubl.) as has been seen for plant PLD C2 domains (Zheng et al. 2000). Looking for sequence homologies to known C2 sequences does not identify a C2 domain in the S. chromofuscus PLD sequence; however, such a β-sandwich structural domain could still occur. The combination of Ca2+ and PA could alter the PLD conformation to (1) destabilize active site binding of PA and (2) allow more productive binding to substrate and/or release of product. That PA can bind to the enzyme at other than the active site and enhance catalysis is confirmed with mutants with altered PA activation. Neither His226 nor Val510 are near residues in the sequence known to be critical for iron binding in the protein. However, mutation of either of these positions can affect PLD specific activity and more importantly the extent of PA activation.
Materials and methods
Chemicals
All phospholipids were obtained from Avanti and used without further purification. The long-chain phospholipids used were all 1-palmitoyl-2-oleoyl phospholipids with the exception of dioleoylphosphatidylmethanol (DOPMe). Triton X-100 was obtained from Sigma while Zwittergent 3–14 was obtained from Calbiochem. The Streptomyces sp. PMF PLD, 95% pure, was obtained from Biomol. All the other chemicals were of the highest purity available. The DNA extraction kit was obtained from Stratagene. Oligonucleotide primers, for amplification of the pld gene from S. chromofuscus DNA, were purchased from Operon Technology. The Advantage GC genomic PCR kit was purchased from Clonetech. Restriction enzymes and plasmids pTYB11 and ER2566 Escherichia coli strains were purchased from New England BioLabs.
PLD overexpression and purification
PLD cloned from the ATCC S. chromofuscus strain 23616 (Yang and Roberts 2002) has a slightly different sequence (87% identity) from the S. chromofuscus PLD published previously (Yoshioka et al. 1991) Differences in some kinetic parameters could be related to the sequence changes, so the S. chromofuscus pld gene was cloned from the originally sequenced strain of the organism (strain NRRL 11098 from the USDA) using primers based on the published sequence for this enzyme (Yang and Roberts 2002). To optimize purification, the pld gene was inserted into pTYB11 to generate an intein-PLD fusion protein. The PLD proteins rPLD, rPLD-E510 and rPLD-K510 (where the C-terminal residue was valine as in the original sequence, changed to negatively charged glutamate, or to positively charged lysine residue using a Quik-Change mutagenesis kit) were purified as described previously (Yang and Roberts 2002). About 4 to 11 mg rPLD protein (and similar amounts of mutants) could be obtained from 1 L cell culture.
Analysis of the secondary structure of rPLD, rPLD-E510, and rPLD-K510 by CD spectroscopy revealed the same secondary structure content and Tm (59°C) as the ATCC recombinant PLD (Yang and Roberts 2002). High concentrations of the protein also exhibited a visible absorption band (λmax ∼500 nm) similar but not identical to that of the S. chromofuscus PLD cloned previously from the ATCC strain of the organism (λmax ∼570 nm; Zambonelli and Roberts 2003). This pink color was also correlated with the PLD activity. Dialyzing rPLD solution extensively against 20 mM Tris-HCl buffer with 2 mM EDTA, pH 8.0, caused loss of the pink color and loss of the PLD activity.
31P NMR assays of PLD activity
PLD specific activities toward short-chain diacylphosphatidylcholine (diCnPC), including diC4PC, diC6PC and diC7PC, and detergent/1-pamitoyl-2-oleoyl-phosphatidylcholine (POPC) mixed micelles were measured by 31P NMR spectroscopy in the absence of Ca2+ as described previously (Geng et al. 1999). Samples typically included 5–10 mM phospholipid in 50 mM buffer (variable depending on pH) in a volume of 0.5 mL, of which 0.1 mL is D2O. The diCnPA product was easily detectable as a pH-dependent resonance (δP = 1.9, 3.2, and 3.8 ppm at pH = 6.4, 7.1, and 8.0, respectively) downfield from the PC resonance (−0.5 ppm). The transphosphatidylation reaction was examined with diC4PC and CH3OH as substrates. The chemical shift for dibutyroylphosphatidylmethanol (diC4PMe) did not vary significantly over the pH range studied (pH 5.5 to 9). Most rates were determined from fixed time point assays (3–20 min) where the Streptomyces sp. PMF PLD reaction was stopped by heating at 98°C in a DNA Thermal Cycler 480 (Perkin-Elmer Cetus) and the S. chromofuscus rPLD reaction was stopped by the addition of 50 mM EDTA. Amounts of enzymes added varied between 1 and 18 μg. All rates are based on assays run at least in duplicate.
1H NMR assays of PLD activity
1H NMR spectra were acquired to monitor the release of the water-soluble base from various phospholipids presented as small unilamellar vesicles (SUVs). For some of the phospholipids, large unilamellar vesicles (LUVs) prepared by multiple passages of the aqueous lipid solutions through polycarbonate membranes (100 nm pore diameter) using a Lipofast extruder from Avestin were also examined as substrates. The PA generated by PLDs in the presence of Ca2+ (needed for S. chromofuscus activity toward phospholipid vesicles; Geng et al. 1998) interacts with the divalent cation, and this interaction leads to broadening of the 31P resonance for the PA. The presence of Ca2+-PA also leads to vesicle fusion, making accurate integrations of substrate and product difficult. However, the water-soluble product choline is not affected by Ca2+, and can be easily detected by sharp resonances in the 1H spectrum. Although this technique has been used to monitor choline release from POPC SUVs (Dorovska-Taran et al. 1996; Geng et al. 1998, 1999), it is also useful for monitoring the water-soluble hydrolase product generated by PLD from other phospholipids as long as product protons can be easily distinguished from the broader substrate vesicle resonances. Phosphatidylethanolamine (PE) hydrolysis yields ethanolamine whose CH2N and CH2OH protons are detected at 3.02 and 3.69 ppm, respectively. Phosphatidylserine (PS) hydrolysis can be measure by the appearance of a sharp resonance (the CH2O protons) at 3.72 ppm, while phosphatidylmethanol (PMe) hydrolysis yields a singlet for methanol at 3.23 ppm. Hydrolysis of phosphatidylglycerol (PG) can be detected from the glycerol resonance at 3.81 ppm (—CH2OH), and PI hydrolysis can be measured by integrating the myo-inositol reonances (e.g., the C[5]H at 3.15 ppm is well separated from PI peaks). With each phospholipid, the amount of soluble alcohol generated was quantified by comparing the integrals for the sharp product resonances to an internal standard of 20 mM Tris HCl (the —CH2OH protons) added with PLD enzyme. As an example, Figure 1 ▶ shows the time-dependent changes in 1H spectra upon incubation of PI SUVs with PLD. Intensities of the C(5)H triplet (3.15 ppm), C(3)H/C(1)H doublet (3.42 ppm), and C(4)H/C(6)H triplet (3.50 ppm) increase with time. The C(2)H resonance at 3.95 ppm is not shown in these spectra. Amounts of enzyme added were similar to what was used for the 31P assays.
rPLD intrinsic fluorescence
Ca2+ binding to rPLD (∼20 μg/mL) was measured by changes in the intrinsic fluorescence of the protein as Ca2+ ions were titrated into the solution (25°C) as described previously (Stieglitz et al. 2001; Yang and Roberts 2002). The protein was excited at 290 nm and emission monitored at 337 nm, the wavelength for maximum emission.
Vesicle binding assays
Aliquots of PLD (48 μg/mL final concentration) were mixed with POPC/POPA vesicles (total phospholipid concentration 2 mM with 0, 10, or 100 mole % POPA) in 10 mM Tris-HCl, 5 mM EDTA, pH 8.0. Ba2+ (15 mM) was added to this buffer when the effect of Ba2+ (a competitive inhibitor of Ca2+ binding to PLD; Geng et al. 1998). This yielded a net Ba2+ concentration of 10 mM. The mixtures were placed in Amicon centricon-100 filters and centrifuged (14,000 g) to separate vesicle-bound from free PLD in the filtrate. The filtrates were lyophilized and the amount of free PLD (PLDf) was quantified with SDS-PAGE by comparing band intensities for samples incubated with vesicles to controls without phospholipids. The percent of PLD bound to vesicles was estimated as (PLDo − PLDf)/PLDo where PLDo is the total amount of PLD.
Acknowledgments
This work was supported by NIH grant GM 26762 (to M.F.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03192503.
References
- Bian, J. and Roberts, M.F. 1992. Thermodynamic comparison of lyso- and diacylphosphatidylcholines. J. Colloid Interface Sci. 153 420–428. [Google Scholar]
- Dawson, R.M.C. 1967. The formation of phosphatidylglycerol and other phospholipids by the transferase activity of phospholipase D. Biochem. J. 102 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorovska-Taran, V., Wick, R., and Walde, P. 1996. A 1H nuclear magnetic resonance method for investigating the phosholipase D-catalyzed hydrolysis of phosphatidylcholine in liposomes. Anal. Biochem. 240 37–47. [DOI] [PubMed] [Google Scholar]
- Eibl, H. and Kovatchev, S. 1981. Preparation of phospholipids and their analogues by phospholipase D. Methods Enzymol. 72 632–639. [DOI] [PubMed] [Google Scholar]
- El Kirat, K., Besson, F., Prigent, A.F., Chauvet, J.P., and Roux, B. 2002. Role of calcium and membrane organization on phospholipase D localization and activity. Competition between a soluble and insoluble substrate. J. Biol. Chem. 277 21231–21236. [DOI] [PubMed] [Google Scholar]
- Ella, K.M., Dolan, J.W., and Meier, K.E. 1995. Characterization of a regulated form of phospholipase D in the yeast Saccharomyces cerevisiae. Biochem. J. 307 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, D. 1996. Phosphatidic acid: A lipid messenger involved in intracellular and extracellular signaling. Cell Signal. 8 341–347. [DOI] [PubMed] [Google Scholar]
- Exton, J.H. 1997. Phospholipase D: Enzymology, mechanisms of regulation, and function. Physiol Rev. 77 303–320. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Phospholipase D. Ann. N.Y. Acad. Sci. 905 61–68. [DOI] [PubMed] [Google Scholar]
- Gadd, M.E. and Biltonen, R.L. 2000. Characterization of the interaction of phospholipase A2 with phosphatidylcholine-phosphatidylglycerol mixed lipids. Biochemistry 39 9623–9631. [DOI] [PubMed] [Google Scholar]
- Geng, D., Chura, J., and Roberts, M.F. 1998. Activation of phospholipase D by phosphatidic acid. Enhanced vesicle binding, phosphatidic acid-Ca2+ interaction, or an allosteric effect? J. Biol. Chem. 273 12195–12202. [DOI] [PubMed] [Google Scholar]
- Geng, D., Baker, D.P., Foley, S.F., Stieglitz, K., and Roberts, M.F. 1999. A 20-kDa domain is required for phosphatidic acid-induced allosteric activation of phospholipase D from Streptomyces chromofuscus. Biochim. Biophys. Acta 1430 234–244. [DOI] [PubMed] [Google Scholar]
- Gottlin, E.B., Rudolph, A.E., Zhao, Y., Matthews, H.R., and Dixon, J.E. 1998. Catalytic mechanism of the phospholipase D superfamily proceeds via a covalent phosphohistidine intermediate. Proc. Natl. Acad. Sci. 95 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagishita, T., Nishikawa, M., and Hatanaka, T. 1999. A spectrophotometric assay for the transphosphatidylation activity of phospholipase D enzyme. Anal Biochem. 276 161–165. [DOI] [PubMed] [Google Scholar]
- Hammond, S.M., Jenco, J.M., Nakashima, S., Cadwallader, K., Gu, Q., Cook, S., Nozawa, Y., Prestwich, G.D., Frohman, M.A., and Morris, A.J. 1997. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-α. J. Biol. Chem. 272 3860–3868. [DOI] [PubMed] [Google Scholar]
- Hasagawa, M., Ohta, Y., and Aisaka, K. 1992. Japanese patent no. H4-88981.
- Heller, M. 1978. Phospholipase D. Adv. Lipid Res. 16 267–326. [DOI] [PubMed] [Google Scholar]
- Imamura, S. and Horiuti, Y. 1979. Purification of Streptomyces chromofuscus phospholipase D by hydrophobic affinity chromatography on palmitoyl cellulose. J. Biochem. 85 79–95. [DOI] [PubMed] [Google Scholar]
- Jain, M.K. and Gelb, M.H. 1991. Phospholipase A2-catalyzed hydrolysis of vesicles: Uses of interfacial catalysis in the scooting mode. Methods Enzymol. 197 112–125. [DOI] [PubMed] [Google Scholar]
- Juneja, L.R., Kazuoka, T., Yamane, T., and Shimizu, S. 1988. Kinetic evaluation of phosphatidylcholine to phosphatidylethanolamine by phospholipase D from different sources. Biochim. Biophys. Acta 960 334–341. [DOI] [PubMed] [Google Scholar]
- Leiros, I., Secundo, F., Zambonelli, C., Servi, S., and Hough, E. 2000. The first crystal structure of a phospholipase D. Struct. Fold. Des. 8 655–667. [DOI] [PubMed] [Google Scholar]
- Madesh, M. and Balasubramanian, K.A. 1997. Metal ion stimulation of phospholipase D-like activity of isolated rat intestinal mitochondria. Lipids 32 471–479. [DOI] [PubMed] [Google Scholar]
- Martin, S.F., LeBlanc, R.L., and Hergenrother, P.J. 2000. Determination of the substrate specificity of the phospholipase D from Streptomyces chromofuscus via an inorganic phosphate quantitation assay. Anal. Biochem. 278 106–110. [DOI] [PubMed] [Google Scholar]
- Mayr, J.A., Kohlwein, S.D., and Paltauf, F. 1996. Identification of a novel, Ca2+-dependent phospholipase D with preference for phosphatidylserine and phosphatidylethanolamine in Saccharomyces cerevisiae. FEBS Lett. 393 236–240. [DOI] [PubMed] [Google Scholar]
- Meacci, E., Becciolini, L., Nuti, F., Donati, C., Cencetti, F., Farnararo, M., and Bruni, P. 2002. A role for calcium in sphingosine 1-phosphate-induced phospholipase D activity in C2C12 myoblasts. FEBS Lett. 521 200–204. [DOI] [PubMed] [Google Scholar]
- Ogino, C., Negi, Y., Daido, H., Kanemasu, M., Kondo, A., Kuroda, S., Tanizawa, K., Shimizu, N., and Fukuda, H. 2001. Identification of novel membrane-bound phospholipase D from Streptoverticillium cinnamoneum, possessing only hydrolytic activity. Biochim. Biophys. Acta 1530 23–31. [DOI] [PubMed] [Google Scholar]
- Pappan, K. and Wang, X. 1999. Molecular and biochemical properties and physiological roles of plant phospholipase D. Biochim. Biophys. Acta 1439 151–166. [DOI] [PubMed] [Google Scholar]
- Ponting, C.P. and Kerr, I.D. 1996. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: Identification of duplicated repeats and potential active site residues. Protein Sci. 5 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, M.F. 1999. Phospholipases: Generation of lipid-derived second messengers. In Signal transduction (ed. S. Ari), pp. 89–146. Birkhauser, Boston.
- Saito, T., Ouchi, H., and Ohta, E. 1990. Japanese patent no. H2-222679.
- Shimbo, K., Iwasaki, Y., Yamane, T., and Ina, K. 1993. Purification and properties of phospholipase D from Streptomyces antibioticus. Agric. Biol. Chem. 57 1946–1948. [Google Scholar]
- Stieglitz, K., Seaton, B., and Roberts, M.F. 1999. The role of interfacial binding in the activation of Streptomyces chromofuscus phospholipase D by phosphatidic acid. J. Biol. Chem. 274 35367–35374. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Binding of proteolytically processed phospholipase D from Streptomyces chromofuscus to phosphatidylcholine membranes facilitates vesicle aggregation and fusion. Biochemistry 40 13954–13963. [DOI] [PubMed] [Google Scholar]
- Stuckey, J.A. and Dixon, J.E. 1999. Crystal structure of a phospholipase D family member. Nat. Struct. Biol. 6 278–284. [DOI] [PubMed] [Google Scholar]
- Swairjo, M.A., Seaton, B.A., and Roberts, M.F. 1994. Effect of vesicle composition and curvature on the dissociation of phosphatidic acid in small unilamellar vesicles—A 31P NMR study Biochim. Biophys. Acta 1191 354–361. [DOI] [PubMed] [Google Scholar]
- Waksman, M., Tang, X., Eli, Y., Gerst, J.E., and Liscovitch, M. 1997. Identification of a novel Ca2+-dependent, phosphatidylethanolamine-hydrolyzing phospholipase D in yeast bearing a disruption in PLD1. J. Biol. Chem. 272 36–39. [DOI] [PubMed] [Google Scholar]
- Wang, X. 2000. Multiple forms of phospholipase D in plants: The gene family, catalytic and regulatory properties, and cellular functions. Prog. Lipid Res. 39 109–149. [DOI] [PubMed] [Google Scholar]
- Whatmore, J., Morgan, C.P., Cunningham, E., Collison, K.S., Willison, K.R., and Cockcroft, S. 1996. ADP-ribosylation factor 1-regulated phospholipase D activity is localized at the plasma membrane and intracellular organelles in HL60 cells. Biochem J. 320 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z., Ho, W.T., and Exton, J.H. 2002. Functional implications of post-translational modifications of phospholipases D1 and D2. Biochim. Biophys. Acta 1580 9–21. [DOI] [PubMed] [Google Scholar]
- Yang, H. and Roberts, M.F. 2002. Cloning, overexpression, and characterization of a bacterial Ca2+-dependent phospholipase D. Protein Sci. 11 2958–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka, K., Mizoguchi, M., Takahara, M., Imamura, S., Beppu, T., and Horinuchi, S. 1991. DNA having the genetic information of phospholipase D and its use. Euopean patent 0435725B1.
- Zambonelli, C. and Roberts, M.F. 2003. An iron-dependent bacterial phospholipase D reminiscent of purple acid phosphatases. J. Biol. Chem. 278 13706–13711. [DOI] [PubMed] [Google Scholar]
- Zheng, L., Krishnamoorthi, R., Zolkiewski, M., and Wang, X. 2000. Distinct Ca2+ binding properties of novel C2 domains of plant phospholipase D α and β. J. Biol. Chem. 275 19700–19706. [DOI] [PubMed] [Google Scholar]