

Abstract
The [H26N, H33N] mutant of horse heart cytochrome c was expressed in E. coli during growth on isotopically enriched minimal media. Complete resonance assignments of both the diamagnetic reduced (spin zero) and paramagnetic oxidized (spin ½) states of the protein were obtained using standard triple resonance and total correlation spectroscopy using the previously determined 1H chemical shifts of the wild-type protein as a guide. The correspondence of chemical shifts between the wild type and the mutant protein is excellent, indicating that they have nearly identical structures. The expanded library of chemical shifts for both redox states in both proteins allowed the refinement of the electron spin g-tensor of the oxidized states. The g-tensors of the oxidized states of the wild-type and [H26N, H33N] mutant proteins are closely similar, indicating that the subtle details of the ligand fields are nearly identical. The refined g-tensors were then used to probe for redox-dependent structure change in the two proteins.
Keywords: NMR resonance assignments, labeling hemeproteins, g-tensor, hyperfine shifts, paramagnetic shifts
Horse cytochrome c (Mr ∼12.36 kD) functions as a soluble mediator of electron transfer between redox proteins in an electron transfer cascade and contains a covalently attached heme prosthetic group. Cytochrome c has emerged as a paradigm for a range of biophysical studies, particularly for issues in protein folding and stability (Englander 2001). NMR spectroscopy has played a significant role in these works and has been aided by an extensive library of proton resonance assignments of the wild-type protein in both redox states (Feng et al. 1989; Wand et al. 1989). Unfortunately, the utility of cytochrome c as a model system has been hindered by an inability to prepare biophysical quantities of isotopically enriched protein (Pollock et al. 1998), which is necessary for use in modern NMR-based investigations. Expression of eukaryotic cytochrome c has been particularly problematic. Fortunately, this barrier has now largely been overcome (Dolgikh et al. 1998; Pollock et al. 1998; Patel et al. 2001; Jeng et al. 2002; Martin et al. 2002; Rumbley et al. 2002).
Here we examined the [H26N, H33N] double mutant of horse cytochrome c, which has superior expression properties but is otherwise largely indistinguishable from the wild-type protein (Rumbley et al. 2002 and below). The [H26N, H33N] cytochrome c and mutants thereof are being used as model systems for a variety of studies of the folding, stability, and dynamics of c-type cytochromes. Standard heteronuclear triple resonance methods were employed to obtain essentially complete resonance assignments of the reduced (diamagnetic, spin 0) and oxidized (paramagnetic, spin ½) states of the holoprotein. Comparison to natural abundance 13C- and 15N-HSQC spectra led to extensive cross-assignments of the natural wild-type horse heart cytochrome c in its two redox states. These data then allowed the electronic g-tensors of the oxidized state of both the wild-type and [H26N, H33N] mutant to be determined and compared. Knowledge of the g-tensor also allowed for redox-dependent changes in both proteins to be detected.
Results and Discussion
NMR assignments of ferro- and ferricytochrome c
Sequence-specific assignments of the backbone 1H, 13Cα, 13Cβ, 13CO, and 15N resonances were obtained using the HNCACB (Wittekind and Mueller 1993), CBCA(CO)NH (Wittekind and Mueller 1993), and CT-HNCO (Grzesiek and Bax 1992; Wittekind and Mueller 1993) three-dimensional NMR spectra. Starting from the published proton assignments of the wild-type protein (Feng et al. 1989; Wand et al. 1989), backbone 1HN, 13Cα, 13Cβ, 13CO, and 15N resonance assignments were obtained for all residues except Gly1 and Gly56 in the reduced state, and Gly1 and Thr28 in the oxidized state. Side-chain methine, methylene, and methyl resonances were assigned using the three-dimensional C(CO)NH (Montelione et al. 1992), HC(CO)NH (Bax et al. 1990), and HCCH3-TOCSY (Uhrin et al. 2000) experiments. Stereospecific assignments of the methyl groups of leucine and valine were obtained using the limited glucose labeling approach of Neri et al. (1989). The resonance assignments for the [H26N, H33N]-horse cytochrome c have been deposited with BioMagResBank under accession 5827 (reduced form) and 5828 (oxidized form).
Wild-type horse cytochrome c is commercially available. Natural abundance 15N-HSQC and 13C-HSQC spectra were obtained at 750 MHz (1H) and compared to the resonance assignments obtained for the recombinant mutant protein. Because of the close correspondence of the spectra of the two proteins, essentially complete cross-assignment of the natural horse cytochrome c in both redox states could be achieved (BMRB accession 5829 and 5830). The high degree of correspondence of the solution structures of the two proteins is illustrated by the high degree of correlation of chemical shifts of corresponding resonances as shown in Figure 1 ▶, panels a–d for the reduced state and in Figure 1 ▶, panels e–h for the oxidized protein. Significant deviations are found to be highly localized to the sites of the two mutations.
Figure 1.
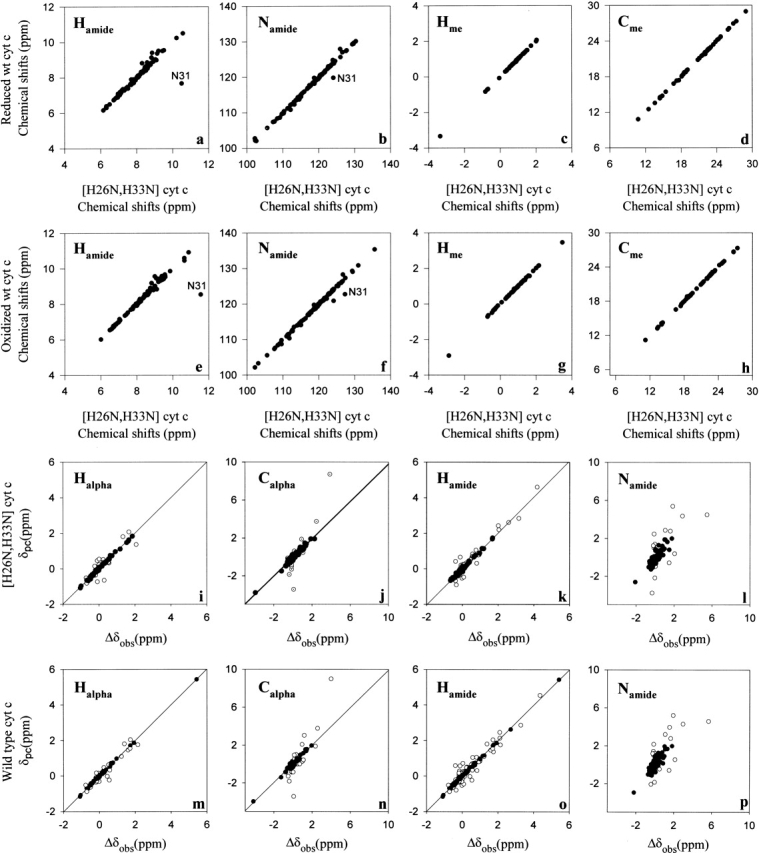
Comparison of the solution structures of natural wild-type horse cytochrome c and recombinant [H26N, H33N] horse cytochrome c. Correlation of the chemical shifts of reduced natural wild-type horse cytochrome c with those of reduced recombinant [H26N, H33N] horse cytochrome c (panel a, amide hydrogens; b, amide nitrogens; c, methyl protons; d, methyl carbons). Correlation of the chemical shifts of oxidized natural wild-type horse cytochrome c with those of oxidized recombinant [H26N, H33N] horse cytochrome c (panel e, amide hydrogens; f, amide nitrogens; g, methyl protons; h, methyl carbons). Refinement of the electronic g-tensor parameters for recombinant [H26N, H33N] horse cytochrome c (panels i–l) and natural wild-type horse cytochrome c (panels m–p). Solid circles correspond to those sites used to determine the parameters of the g-tensor by minimizing the difference between the observed and calculated pseudocontact shifts essentially as described by Feng et al. (1990). A similar plot is shown for recombinant [H26N, H33N] horse cytochrome c in panels m–p.
Electronic g-tensor parameters
The paramagnetic iron center of ferricytochrome c contributes directly to the chemical shift of neighboring nuclei via hyperfine interactions termed the Fermi contact (δc) and the through-space pseudocontact (δpc) shifts (Bertini et al. 2002). The difference of the chemical shift of a nucleus in the two oxidation states may also reflect diamagnetic shift effects due to structural change (δstr). For a given nucleus, the observed redox-dependent change in chemical shift, Δδobs can then be expressed as:
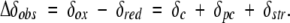 |
The pseudocontact shifts are determined by the electronic g-tensor (Kurland and McGarvey 1970; Horrocks Jr. and Greenberg 1973).
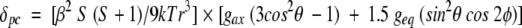 |
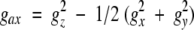 |
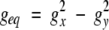 |
where β is the Bohr magneton, S is the electron spin quantum number (½ for ferricytochrome c), and T is the absolute temperature. The position of each proton is defined by its polar coordinates (r,θ,φ) in the reference system of the electron spin g tensor. The principal g-tensor component gax, geq, and three Euler angles, α, β, and γ for horse cytochrome c are obtained using a least-squares fitting method (Feng et al. 1990).
In the past, reference to the crystal structure and knowledge of the chemical shifts of parent nuclei in the paramagnetic oxidized and diamagnetic reduced states have been used to determine the g-tensor of the oxidized heme (e.g. Feng et al. 1990; Boyd et al. 1999). In that case, a consensus is sought for those sites for which δc + δstr is negligible and can therefore be used to determine the parameters defining the g-tensor (Feng et al. 1990). We have repeated this procedure using the crystal structure of oxidized horse cytochrome c (Berghuis and Brayer 1992; PDB code 1HRC) and the expanded chemical shift library of the wild-type protein (Fig. 1 ▶, panels i–l) and the [H26N,H33N] mutant (Fig. 1 ▶, panels m–p). Excellent convergence of the fit was observed in both cases. The obtained g-tensor parameters for both proteins are essentially identical (Table 1). As pointed out by Boyd et al. (1999), the amide nitrogen sites show considerable variance (Fig. 1 ▶, panels l and p), which can be attributed to the exquisite sensitivity of this nucleus to small changes in the electrostatic environment (Ubbink et al. 2002).
Table 1.
Detemined g-tensor parameters for wild-type and [H26N, H33N]-recombinant cytochrome c
| Number of sites | gx | gy | gz | α | β | γ | χ2red |
| [H26N,H33N] 300 | 2.22 | 2.64 | 2.31 | 106 | 13 | 251 | 0.002 |
| [Wild type] 296 | 2.23 | 2.61 | 2.32 | 106 | 13 | 251 | 0.002 |
| [Wild type] 64a | 2.25 | 2.59 | 2.32 | 106 | 13 | 251 | 0.004 |
a Reference set of Feng et al. (1990).
Redox-dependent structure change
Having determined the parameters defining the electronic g-tensors of both the wild-type and mutant proteins, one can begin to assess the degree of true structure change upon a change in redox state (Feng et al. 1990). The optimized g-tensor parameters were used together with spatial coordinates for the oxidized form to calculate the pseudocontact shift contribution (δpc) to all nuclei. The distributions of an apparent redox-dependent chemical shift discrepancy (Δδobs − δpc) could then be evaluated, and for the most part, the residual chemical shift discrepancies are negligible. Exceptions include the loops containing residues 27–33, 37–43, and 50–60, and regions localized near the axial heme ligands Met80 and His18. The latter class is anticipated to have a large Fermi contact contribution (δc). The remaining regions are consistent with structural changes and correspond closely to those previously found for the wild-type protein (Feng et al. 1990).
In summary, these data strongly indicate that the recombinant [H26N, H33N] mutant of horse cytochrome c is structurally highly similar to the wild-type protein in both redox states. The close correspondence of the g-tensors of the two proteins reinforces the notion that the structural details of heme ligation are nearly identical. Accordingly, the [H26N,H33N] mutant of horse cytochrome c should be considered a highly suitable biophysical model of wild-type cytochrome c.
Materials and methods
The expression vector for [H26N,H33N] horse cytochrome c has been described elsewhere (Rumbley et al. 2002). Cultures were inoculated from glycerol stocks and grown overnight in LB broth (30°C). Cells were pelleted by centrifugation and resuspended in fresh 20 mL minimal media and used to inoculate 1L of minimal media (Morar et al. 1999) containing 1 g of 15NH4Cl and 2 g of 13C-glucose. Cells were grown for 60 h at 30°C and harvested by centrifugation. The cell pellet was suspended in 25 mM potassium phosphate, pH 7, containing PMSF, benzamidine, and DNase, and broken with an Aminco French press. Ammonium sulfate was added to the whole-cell lysate to 25% saturation, and the precipitate was removed by centrifugation. The supernatant was dialyzed against 25 mM potassium phosphate, pH 7. Cytochrome c was oxidized with potassium ferricyanide and isolated on a CM-Sepharose column using a NaCl gradient. The pure cytochrome c was concentrated by pressure filtration. NMR samples (650 μL) of uniformly 15N-/13C-labeled cytochrome c (1 mM) were prepared in 50 mM potassium phosphate buffer, pH* 5.8 (95% H2O/5% D2O). The yield was generally 6–8 mg per liter of minimal media.
NMR spectra were recorded at 293 K on a Varian INOVA 750 MHz and 500 MHz NMR spectrometers equipped with four RF channels and pulsed field gradients. Triple resonance and total correlation experiments were carried out, and the data were processed essentially as described elsewhere (Liu et al. 2001). 15N and 13C chemical shifts were referenced indirectly using the consensus Xi ratios of gyromagnetic ratios, 0.101329118 and 0.251449528 for 15N/1H and 13C/1H, respectively (Wishart et al. 1995).
Electronic supplemental material
15N- and 13C-HSQC spectra annotated with resonance assignments are given as electronic supplemental material.
Acknowledgments
This work was supported by NIH grant GM35940.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03211303.
References
- Bax, A., Clore, G.M., and Gronenborn, A.M. 1990. Proton–proton correlation via isotropic mixing of carbon-13 magnetization, a new three-dimensional approach for assigning proton and carbon-13 spectra of carbon-13-enriched proteins. J. Magn. Reson. 88 425–431. [Google Scholar]
- Berghuis, A.W. and Brayer, G.D. 1992. Oxidation state-dependent conformational changes in cytochrome c. J. Mol. Biol. 223 959–976. [DOI] [PubMed] [Google Scholar]
- Bertini, I., Luchinat, C., and Parigi, G. 2002. Paramagnetic constraints: An aid for quick solution structure determination of paramagnetic metalloproteins. Concept. Magn. Res. 14 259–286. [Google Scholar]
- Boyd, J., Dobson, C.M., Morar, A.S., Williams, R.J.P., and Pielak, G.J. 1999. 1H and 15N hyperfine shifts of cytochrome c. J. Am. Chem. Soc. 121 9247–9248. [Google Scholar]
- Dolgikh, D.A., Latypov, R.F., Abdullaev, Z.Kh., Kolon, V., Roder, H., and Kirpichnikov, M.P. 1998. Expression of mutant genes for horse cytochrome c in Escherichia coli. Russ. J. of Bioorganic. Chem. 24 672–675. [PubMed] [Google Scholar]
- Englander, S.W. 2001. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29 213–238. [DOI] [PubMed] [Google Scholar]
- Feng, Y., Roder, H., Englander, S.W., Wand, A.J., and Di Stefano, D.L. 1989. Proton resonance assignments of horse ferricytochrome c. Biochemistry 28 195–203. [DOI] [PubMed] [Google Scholar]
- Feng, Y., Roder, H., and Englander, S.W. 1990. Redox-dependent structure change and hyperfine nuclear magnetic resonance shifts in cytochrome c. Biochemistry 29 3494–3504. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1992. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114 6291–6293. [Google Scholar]
- Horrocks Jr., W.D. and Greenberg, S. 1973. Evaluation of dipolar nuclear magnetic resonance shifts in low-spin hemin systems. Ferricytochrome c and metmyoglobin cyanide. Biochim. Biophys. Acta 322 38–44. [DOI] [PubMed] [Google Scholar]
- Jeng, W.-Y., Chen, C.-Y., Chang, H.-C., and Chuang, W.-J. 2002. Expression and characterization of recombinant human cytochrome c in E. coli. J. Bioenerg. Biomemb. 34 423–431. [DOI] [PubMed] [Google Scholar]
- Kurland, R.J. and McGarvey, B.R.J. 1970. Isotropic NMR shifts in transition metal complexes: Calculation of the Fermi contact and pseudocontact terms. J. Magn. Reson. 2 286–301. [Google Scholar]
- Liu, W., Flynn, P.F., Fuentes, E.J., Kranz, J.K., McCormick, M., and Wand, A.J. 2001. Main chain and side chain dynamics of oxidized flavodoxin from Cyanobacterium anabaena. Biochemistry 40 14744–14753. [DOI] [PubMed] [Google Scholar]
- Martin, A.G. and Fearnhead, H.O. 2002. Apocytochrome c blocks caspase-9 activation and Bax-induced apoptosis. J. Biol. Chem. 277 50834–50841. [DOI] [PubMed] [Google Scholar]
- Montelione, G.T., Lyons, B.A., Emerson, S.D., and Tashiro, M. 1992. An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J. Am. Chem. Soc. 114 10974–10975. [Google Scholar]
- Morar, A.S., Kakouras, D., Young, G.B., Boyd, J., and Pielak, G.J. 1999. Expression of 15N-labeled eukaryotic cytochrome c in Escherichia coli. J. Biol. Inorg. Chem. 4 220–222. [DOI] [PubMed] [Google Scholar]
- Neri, D., Szyperski, T., Otting, G., Senn, H., and Wüthrich, K. 1989. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional carbon-13 labeling. Biochemistry 28 7510–7516. [DOI] [PubMed] [Google Scholar]
- Patel, C.N., Lind, M.C., and Pielak, G.J. 2001. Characterization of horse cytochrome c expressed in Escherichia coli. Protein Expr. Purif. 22 220– 224. [DOI] [PubMed] [Google Scholar]
- Pollock, W.B.R., Rosell, F.I., Twitchett, M.B., Dumont, M.E., and Mauk, A.G. 1998. Bacterial expression of a mitochondrial cytochrome c. Trimethylation of Lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry 37 6124–6131. [DOI] [PubMed] [Google Scholar]
- Rumbley, J.N., Hoang, L., and Englander, S.W. 2002. Recombinant equine cytochrome c in Escherichia coli: High-level expression, characterization, and folding and assembly mutants. Biochemistry 41 13894–13901. [DOI] [PubMed] [Google Scholar]
- Ubbink, M., Worrall, J.A.R., Canters, G.W., Groenen, E.J.J., and Huber, M. 2002. Paramagnetic resonance of biological metal centers. Annu. Rev. Biophys. Biomol. Struct. 31 393–422. [DOI] [PubMed] [Google Scholar]
- Uhrin, D., Uhrinova, S., Leadbeater, C., Nairn, J., Price, N.C., and Barlow, P.N. 2000. 3D HCCH3-TOCSY for resonance assignment of methyl-containing side chains in 13C-labeled proteins. J. Magn. Reson. 142 288–293. [DOI] [PubMed] [Google Scholar]
- Wand, A.J., Di Stefano, D.L., Feng, Y., Roder, H., and Englander, S.W. 1989. Proton resonance assignments of horse ferrocytochrome c. Biochemistry 28 186–194. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6 135–140. [DOI] [PubMed] [Google Scholar]
- Wittekind, M. and Mueller, L. 1993. HNCACB, A high sensitivity 3D NMR experiment to correlate amide proton and nitrogen resonances with the α-carbon and β-carbon resonances in proteins. J. Magn. Reson. 101B 201–205. [Google Scholar]