

Summary
Sterol 14α-demethylases (CYP51) serve as primary targets for antifungal drugs and specific inhibition of CYP51s in protozoan parasites Trypanosoma brucei (TB) and Trypanosoma cruzi (TC) might provide an effective treatment strategy for human trypanosomiases. Primary inhibitor selection is based initially on the cytochrome P450 spectral response to ligand binding. Ligands which demonstrate strongest binding parameters were examined as inhibitors of reconstituted TB and TC CYP51 activity in vitro. Direct correlation between potency of the compounds as CYP51 inhibitors and their antiparasitic effect in TB and TC cells implies essential requirements for endogenous sterol production in both trypanosomes and suggests a novel lead structure with a defined region most promising for further modifications. The approach developed here can be used for further large-scale search for new CYP51 inhibitors.
Introduction
Trypanosomatidae cause endemic infections and countless human deaths over the world (http://www.emedicine.com). Sleeping sickness, or African trypanosomiasis (causative agent Trypanosoma brucei (TB)), and Chagas disease, or American trypanosomiasis (Trypanosoma cruzi (TC)), are most abundant. The parasites have complex life-cycles using insects, tsetse fly (TB) and triatomine bugs (TC), as vectors, humans and a variety of mammals as hosts. In humans TB remains extracellular, the chronic stage of the disease beginning when the pathogen crosses the blood-brain barrier and invades the central nervous system. TC affects the heart and gastrointestinal tract and at the chronic stage is found mainly as an intracellular amastigote. Currently ∼60 million people in Sub-Saharan Africa are at risk of sleeping sickness, 0.3-0.5 million new cases occurring each year. Sixteen to eighteen million people in Central and South America are infected with TC with an annual incidence of 0.2 million new cases. In the US Chagas disease predominantly exists as a result of immigration, blood transfusion or organ transplantation, however autochthonous cases of the infection have also been reported in several states [1-4]. There are no vaccines for these diseases and only a very limited set of drugs; 4 for sleeping sickness (suramin (since 1916), pentamidine (1941), melarsoprol (1949) eflornithine (1990)) and 2 for Chagas disease (nifurtimox (since 1972) and benznidazole (1978)) (Supplemental Data, Figure S1). These drugs are inadequate because of high toxicity, side effects, difficulties with administration, resistance and low or no efficacy at the prevalent chronic stages, which are commonly fatal. New, more efficient medications for antitrypanosomal therapy are urgently needed. [1, 5-8].
One of the approaches for rational design of antitrypanosomal drugs is to specifically block an essential enzyme or metabolic pathway in the parasite. Being required in most eukaryotic kingdoms, sterol biosynthesis is one such possible target. The pathway leads to production of cholesterol in mammals, ergosterol in fungi and a variety of 24-alkylated and olephynated sterols in plants and protists [9, 10]. Cholesterol, ergosterol and sitosterol (plants) are essential structural components of plasma membranes. These structural sterols stabilize membranes, determine their fluidity and permeability, and modulate activity of membrane-bound enzymes and ion channels. In addition, sterols serve as precursors for bioactive molecules, which function at nanomolar hormonal levels as regulators of cell cycle and development [10, 11]. While mammals can accumulate cholesterol from the diet, blocking of ergosterol production in fungi is lethal; it affects cytokinesis, stops cell growth, and eventually leads to a collapse of the cellular membrane [9, 11]. Inhibitors of sterol biosynthesis are currently the most widely used clinical and agricultural antifungal agents [12]. Positive results of use of inhibitors of fungal sterol biosynthetic enzymes for potential treatment of protozoan infections have been obtained for TC [13-22] and Leishmania species [23-25]. As for TB, it has been reported that contrary to procyclic (insect) forms, bloodstream (mammalian) stages of the parasite life-cycle do not synthesize endogenous sterols but use host cholesterol to build their membranes [26, 27]. However, recent experiments have demonstrated that inhibitors of fungal sterol 24-methyltransferase are effective in killing bloodstream forms of TB [28, 29].
Sequencing of TB and TC genomes [7] revealed presence of all sterol biosynthetic enzymes in the parasites including sterol 14α-demethylase (CYP51), a cytochrome P450 which functions at the initial stages of the specific postsqualene portion of the pathway, catalyzing a three-step reaction of oxidative removal of the 14α-methyl group from the newly cyclized sterol precursors [30]. CYP51 is a primary target for azole derivatives in antifungal therapy. Inhibition of the CYP51 reaction in fungi leads to accumulation of 14α-methylated sterols which are unable to replace ergosterol in the membrane because of steric hindrance [11].
CYP51s from TB and TC have only ∼25% amino acid identity to their fungal orthologs and are 83% identical to each other. We have shown that while TCCYP51 expresses preference towards the C4-dimethylated 24-methylenedihydrolanosterol, the natural substrate of CYP51 from filamentous fungi, TBCYP51 is strictly specific toward the C4-monomethylated plant-like substrates (obtusifoliol and norlanosterol) and that based on amino acid sequence all other sequenced protozoan CYP51 will resemble the TBCYP51 in activity [31-33].
In this study correlation between specific inhibition of trypanosomal CYP51 and antiparasitic effect on trypanosomal cells has been investigated. Using antifungal drugs ketoconazole and fluconazole as controls, a selection of fifteen novel imidazole derivatives from Novartis, found to cause strong spectral responses in highly purified TB and TC CYP51 (Kd[P450-azole] < Kd[P450-substrate]), were further examined as inhibitors of reconstituted TB and TC CYP51 activity in vitro. In accordance with binding parameters, the compounds produced a profound effect on the initial rates of catalysis of both trypanosomal CYP51s. In most cases no excess of the inhibitor over the enzyme was required to cause a 2-fold decrease in the rate of substrate conversion. Time-course experiments, however, revealed significant divergence in their potencies, often contradicting the Kd values and suggesting that determination of an inhibitory potency as the influence on maximal CYP51 turnover can be misleading. More detailed analysis of azole structure/inhibitory effect relationship has shown that while the presence of a bulky aromatic structure in either α- or β-position relative to the imidazole ring is required for efficient binding, the azoles having an α-phenyl group can be easily replaced by substrate in the reconstituted enzyme reaction. However, switching the phenyl group to the β-position converts them into much more potent CYP51 inhibitors, sometimes having virtually irreversible action. Ketoconazole and selected experimental compounds were further tested in TB and TC cells and found to be effective growth inhibitors, cellular drug sensitivities correlating with the long-term inhibitory effect observed on CYP51 activity in vitro.
Results and Discussion
CYP51 spectral response to ligand binding as a basis for the initial screening
By mechanism of action, azoles belong to the group of reversible competitive inhibitors of cytochromes P450 [34]. The unsubstituted basic nitrogen forms the sixth (axial) coordination bond with the heme iron blocking binding and activation of molecular oxygen and preventing accommodation of the substrate in the active center. The inhibitory effect strongly depends on the size and configuration of the N-substituted part of the azole molecule, which forms multiple additional interactions with the protein moiety of the cytochrome P450. Thus, the inhibitory effect of small molecules such as imidazole or phenylimidazoles is very weak, while much bulkier ketoconazole serves as a potent antifungal drug but has rather low selectivity; in addition to CYP51 it was also found to strongly inhibit other human P450s [35].
Being highly hydrophobic, membrane bound enzymes metabolizing substrates of extremely low solubility in aqueous solutions, fungal CYP51s are quite resistant to purification (so far purification of only three fungal CYP51 orthologs was reported [30]) and also often to reconstitution of activity in vitro. Because of these problems, inhibitory potencies of azole drugs are mainly compared indirectly as their effects on fungal strain growth [e.g. 11, 36-39]. Though not normalized per equal amount of P450, the data imply that potencies of the same azole on CYP51s from different fungal species can vary significantly, differences up to three orders of magnitude have been reported [11, 34]. The latter means that highly selective strong CYP51 inhibition can be potentially reached as a result of maximal correspondence between the structure of an azole derivative and the topology of the target P450 substrate binding cavity. The needs for selective CYP51 inhibitors are growing sharply because such compounds would increase the efficiency and shorten the time necessary for treatment, which in turn can prevent the development of drug resistance, which is one of the most severe problems in clinical antifungal therapy (especially in treatment of immunocompromised patients) and in agriculture [40].
As a cysteine-coordinated hemoprotein, sterol 14α-demethylase responds spectrally to any perturbations in the area surrounding the heme iron (Figure 1). These spectral responses, type 1 for the interaction with the substrate and type 2 for interaction with an azole inhibitor, can be used to estimate the apparent dissociation constants (Kd) of the enzyme/ligand complexes. Thirty imidazole derivatives from a Novartis program on inhibitors of vitamin D hydroxylases [41, 42] were tested for spectral responses in highly purified TB and TC CYP51. Eleven compounds (Figure 2A) demonstrated high binding affinities (the Kds being close or lower than the Kds calculated from type 1 spectral responses of TB and TC CYP51 to their substrates) and were chosen for further testing as potential inhibitors. Antifungal drugs ketoconazole and fluconazole served as controls. Supporting the notion that the affinities of azole/CYP51 interactions can be species specific, the estimated apparent Kd of these compounds to the protozoan CYP51s are often more than one order of magnitude lower than the values obtained for the CYP51 from Mycobacterium tuberculosis (26% amino acid identity) and in many cases significantly lower than those calculated for the human ortholog (27% identity to TB/TC CYP51) (Supplemental Data, Table S1). Comparing the Kds calculated for the two trypanosomal CYP51s, only compounds 1, 11 and especially ketoconazole show higher affinity toward the TC ortholog, the rest of the azoles produce rather similar Kds in both enzymes, the best values (compounds 4 and 10) being ∼20-fold lower than the Kds for the interaction of TB and TC CYP51 with their substrates.
Figure 1. CYP51 absorbance upon alterations in the heme and its environment.
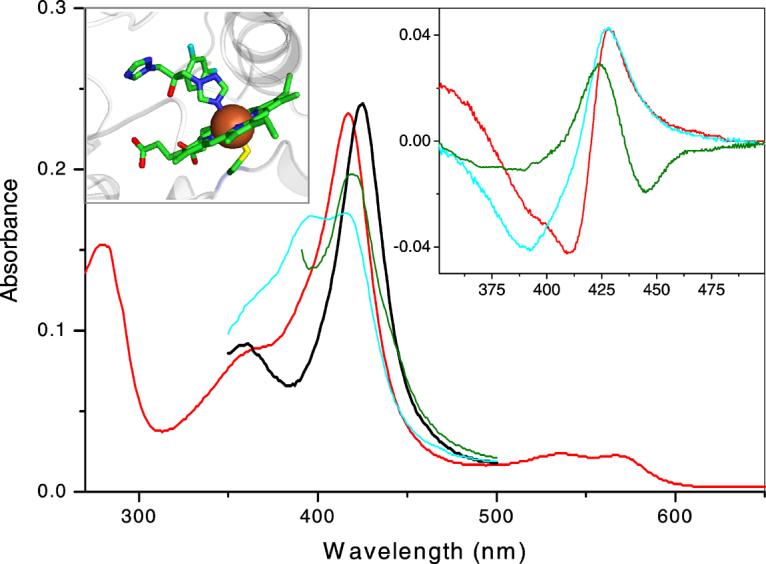
Main panel: Absolute spectra of 2 μM TBCYP51: oxidized ligand-free (red), reduced ligand-free (green), oxidized 47% high-spin obtusifoliol-bound (cyan), azole 4 -bound (black). Blue shift in the Soret band maximum (from 417 to 394 nm) is caused by expulsion of a water molecule from the sixth coordination sphere of the heme iron (e.g. by substrate, which itself does not form a coordination bond with the iron). This spectral response (type 1) reflects transition of the iron from the hexa-coordinated low-spin state to the penta-coordinated high-spin state. Direct coordination of a basic atom (e.g. aliphatic or aromatic nitrogen) to the heme iron causes a red shift in the Soret band (to 426-429 nm) also known as type 2 response in the difference spectra. Right inset: type 2 difference spectra observed upon azole binding to oxidized, reduced and substrate-bound TBCYP51 (color code as above). Left inset: coordination of fluconazole to the heme iron in the crystal structure of the CYP51 from Mycobacterium tuberculosis [1ea1]. The iron is shown as brown sphere, the heme-coordinated Cys residue (fifth axial ligand) is located beneath the iron, the sulfur atom is colored in yellow, protein moiety is shown in grey.
Figure 2. Azole derivatives assayed as potential inhibitors of TB and TCCYP51.

A. Compounds selected based on the binding affinities estimated from the P450 spectral responses. B. Azoles added after structure/activity relation analysis. The compounds used in cellular studies are marked with grey background. *Apparent dissociation constants, μM. Inhibitory effects of azoles with Kds higher than the Kds for the enzyme/substrate complexes (1.2 and 0.8 μM for the interaction of TBCYP51 with obtusifoliol and TCCYP51 with 24-methylenedihydrolanosterol, respectively [31, 32]) were much weaker; compound 5 is included as an example. **molar ratio inhibitor/P450 which produces a two-fold decrease in the activity. *** I/E2 calculated as the influence on the initial rate (5') of catalysis (I/E2 calculated as the influence on the percentage of substrate conversion after 1 hour (60') reaction). The results of four experiments are presented as mean; standard deviation does not exceed 10%.
Identification of the most potent CYP51 inhibitors
Estimation of the inhibitory potency in the reconstituted enzyme reaction in vitro
In accordance with their high binding affinity the compounds produced strong inhibitory effect on the initial rates of catalysis. The molar ratios inhibitor/enzyme required to slow down the reaction two-fold (I/E2) were lower than one in all cases except for compound 5 (for both trypanosomal CYP51s), ketoconazole and fluconazole for the TB ortholog (Figure 2).
The reaction conditions used to reconstitute the CYP51 activity are optimized to give maximal turnover and reproducibility [30]. Low solubility of the sterols limits maximal substrate concentration to 50 μM while decrease in the enzyme concentration affects stability and protein-protein interactions with the electron donor, cytochrome P450 reductase. Because of these restrictions time-course experiments were undertaken to increase sensitivity of the assay in order to distinguish the most potent inhibitors. The results revealed significant divergence in the potencies of the compounds assayed. However, I/E2 determined as the molar ratio inhibitor/enzyme required for reaching a two-fold decrease in the substrate conversion per 1 hour often demonstrated lack of correspondence with the relative apparent Kd values (Figure 2, numbers in brackets). Though certain increase in the I/E2 ratios estimated for long-term incubations was expected as a result of substrate depletion, no such changes were registered for compounds 2 and 4. Stronger inhibitory effect than could be predicted from the Kd was demonstrated by 1, especially on TBCYP51. On the contrary, compounds 6-10, regardless of the variations in their Kds, all show a similar rapid time-dependent increase in the I/E2 .
Clearly the best inhibitors are distinguished over time upon comparison of the effects of the azoles at equimolar ratio to the enzymes (Figure 3). Under this condition, no substrate conversion can be detected even after 1 hour reaction when compounds 2 or 4 are added. Incubation intervals up to 4 hours were tested with the same result, while more than 90% of the enzyme in the reaction remains in the active P450 form. Thus, within the time tested the effect of compounds 2 and 4 on both trypanosomal CYP51s remains irreversible.
Figure 3. CYP51 activity at 1:1 molar ratio inhibitor/enzyme.

The substrates are added at 50-fold molar excess. The results are presented as mean; standard deviation did not exceed 10%. Inhibitor numbers and letters refer to compounds in Figure 2.
B. The affinity of the azole-CYP51 interaction can change under the reaction conditions
To understand the reasons for the observed discrepancies between the apparent Kds and inhibitory effects on the CYP51 activity, we tested stability of compounds 4 (long-term inhibitor) and 10 (short-term inhibitor) in the reaction mixture and compared influence of substrate and P450 reduction on the relative affinities of their interaction with TBCYP51 (spectral responses of substrate-bound and Na2S2O4-reduced TBCYP51 to the azole binding are shown in Figure 1, insert). No trace of metabolism or decomposition during the reaction time was detected for either azole. Excess of substrate, however, was found to result in a 13-fold higher increase in the calculated Kd upon titration of TBCYP51 with 10 than upon titration with 4 (Table 1). If the substrate was added after the azole, it did not cause spectral changes when TBCYP51 was bound with 4 but produced time-dependent type 1 response in the case of 10. A negative effect of P450 reduction on the interaction with 10 was also stronger, 4-fold increase in the apparent Kd versus only 1.5-fold increase in the case of compound 4. Again, if TBCYP51 was reduced after the titration with the azoles and then exposed to CO, the rate and efficiency of the CO-complex formation [43] indicated that the heme-coordinated imidazole nitrogen of compound 10 is much more easily replaced by CO from the iron coordination sphere.
Table 1.
Modification of azole binding properties upon substrate binding or heme reduction and replacement of the bound azoles by substrate or carbon monoxide.
| Selected azoles |
Apparent Kd, μM | Replacement by substrate (% of high-spin form) |
Replacement by CO (vs. no inhibitor), %** |
|||
|---|---|---|---|---|---|---|
| Oxidized, no substrate |
Oxidized, plus substrate* |
Reduced, no substrate |
Rate (per minute) |
Maximum | ||
|
4 10 |
0.05 ± 0.003 0.05 ± 0.002 |
0.17 ±0.02 (3↑) 2.2 ± 0.44 (44↑) |
0.07 ± 0.01 (1.6↑) 0.26 ± 0.02 (4↑) |
− + (34 ± 0.7) |
6 ± 0.7 13 ± 0.8 |
52 ± 7 95 ± 6 |
Obtusifoliol, 20μM (azole titration range 0.2-10 μM)
the spectra are shown in Supplemental Data, Figure S2. The results of two experiments are presented as mean ± standard error.
One explanation of the data is that the affinity of azole-CYP51 interaction in the reaction mixture can be altered, the expression of the differences depending on the inhibitor structure. The number of contacts between the azole molecule and the enzyme substrate binding cavity might be decreased as a result of protein conformational dynamics upon substrate recognition, interaction with the reductase, electron transfer and subsequent reduction so that inhibitors such as 10 can be more easily released from the active center. An alternative explanation can be connected with the fact that binding of some inhibitors alone is known to cause conformational changes in P450s [44]. By tightening the complex, such changes might impede protein dynamics and thus make the inhibitory effect much stronger, for example as with 4. To summarize, being a useful tool in the primary search for binding ligands, spectral response of substrate-free oxidized P450 may not truly reflect the relative affinity of the interaction under the reaction conditions. Prediction of the inhibitory potency relying solely on binding parameters or IC50 values determined as the influence on the initial rate of reaction (especially without considering P450 concentration) can be misleading. Long-term inhibitory effect appears to be most informative for identification of strongest CYP51 inhibitors.
C. Azole structure/activity relationship
The two strongest TB and TC CYP51 inhibitors (2 and 4 Figure 2 A) differ in composition of the substituents at the δ-position relative to the imidazole yet both contain a phenyl ring at the α-position. The inhibitory effect of the levorotatory enantiomer of 2 (compound 3) is slightly weaker but the I/E2 ratios after extended incubation with CYP51 still remain less than 1. On the contrary, in all inhibitors which revealed sharp time-dependent increase in the I/E2 values (5-10) the phenyl group is present at the β-position. Because structurally the β-phenyl compounds differ only in the composition on the δ-substituents, influence of these differences on the apparent binding affinity can be easily seen. While a short 5-member heterocycle of 5 is clearly insufficient to provide efficient binding, bulkier constituents (6-10) produce better Kd values. When this part of the molecule consists of two aromatic rings with rigid connection (6, 7), the calculated affinities remain moderate but when the connection is flexible (especially 9 and 10) the Kds become very low, the best values (0.05 μM) being as good as those obtained for compound 4. Perhaps flexibility of this part of azole molecule leads to its easier entry into the CYP51 substrate binding cavities, but similar to the other β-phenyl azoles, it still lacks restraining interactions necessary to prevent the inhibitors from being replaced by the substrate upon catalysis. On the other hand, compounds 1 and 11, having β-substituents more bulky than a single phenyl ring, produce long-term inhibitory effects stronger than 5-10 (Figure 2A). In addition, these two azoles demonstrate some selectivity. Compound 1 (two phenyl rings at the β-position) is a little more favorable for TBCYP51, while for TCCYP51 the longer arm of the benzyl group (so that benzene and imidazole rings remain separated by two C-atoms) of 11 is preferable.
D. Modification of a basic structure
Because any drug development process can face problems such as cytotoxicity, cell permeability, solubility, rapid turnover in plasma, insufficient life-time, etc., it is important to be able to predict the most promising directions for further modifications of a lead structure. To verify the importance of the phenyl group location at the α- versus the β-position, the β-phenyl isomer of 4 (compound 12, Figure 2B) was tested and in correspondence with our predictions from compounds 6-10, regardless of its strong apparent binding affinity, showed a rapid decrease in the inhibitory effect over time. Influence of alteration in the composition of the α-substituent was investigated using an additional set of eight derivatives of compound 4. The inhibitory effects were compared as inhibition of CYP51 activity at 1:1 molar ratio azole/P450 after 5 and 60 min reaction and quite significant differences in their potencies were observed (Supplemental Data, Figure S3). Two of these compounds (17 and 19, Figure 2B), revealed inhibitory effects as strong as 4 and were included in further studies.
Correlation between CYP51 inhibition and antiparasitic effect
A. Cellular responses in the mammalian and insect life stages of TB
Only two of four clinically available drugs (melarsoprol and eflornithine (Supplemental Data, Figure S1) can treat late stages of sleeping sickness. However, eflornithine is only efficient for TB gambiense, and melarsoprol is extremely toxic, causing severe, often fatal encephalopathy, and has up to 30% resistance in parts of central Africa. Suramin and pentamidine do not penetrate the blood/brain barrier and can treat only the first, hemolymphatic stage of the disease, which sometimes passes unnoticed because of non-specific symptoms. Recently quite promising data on use of nitroimidazoles as possible drug candidates for African trypanosomiasis have been reported [46], yet the target for these compounds has not been determined. Though major sterol components identified in bloodstream TB most likely originate from host cholesterol [27], it is not excluded that the parasite might still require functional sterols, for which hormonal level of action can be below routine sterol detection limits. We aimed to clarify this issue by comparison of the effects of potent TBCYP51 inhibitors in bloodstream and procyclic (known to synthesize endogenous sterols [27, 47]) cells of TB.
The Alamar Blue method was adjusted to monitor TB cell growth. While the procyclic form, though growing slowly at low concentrations provided very good fluorescent signal starting from plating density of 106 cells/ml (Figure 4A) for the bloodstream TB originally cultured in the commonly used HMI-9 medium [48], the signal/noise ratios remained below 2 and rapidly dropped over time with increase in the cell densities. Using absorbance spectroscopy we have observed that several components of the HMI-9 themselves cause fast Alamar Blue reduction, and that the bloodstream TB can further convert the reduced (fluorescent) dye into its colorless hydroxy form [49]. To overcome this problem several different cell culture media were tested and RPMI 1640 found to combine both requirements, support bloodstream TB growth and maintain the dye in the oxidized non fluorescent form, was used in further experiments.
Figure 4. Growth and inhibition of TB.

A. Sensitivity of Alamar Blue assay to monitor viable TB cells: the ratios between fluorescence emission in growing cells (signal) and in the growth media (noise) at increasing plating densities of procyclic and bloodstream TB. B. Cellular responses to the CYP51 inhibitors (1, 5, 10, 20, 30 and 50 μM) BS, bloodstream TB, PC, procyclic TB, HL60, human leukemia cell line. Effects of the compounds on the Alamar Blue absorbance spectra are shown in Supplemental Figure S4.
All the azoles demonstrated clear dose-dependent inhibitory effect on both procyclic and bloodstream TB (Figure 4B). Among them, ED50 above 10 μM for bloodstream TB was obtained only for ketoconazole (35 and 37% responses for procyclic and bloodstream TB, respectively, with less than 10% negative influence on human HL60). At increasing ketoconazole concentration, inhibition of human cells becomes more pronounced, selectivity index human/bloodstream TB being 1.8 (Table 2). This low selectivity is not surprising for ketoconazole since the drug is known to inhibit growth of cancer cells and has been used for a long time in cancer chemotherapy [50, 51]. Though stronger effect of 10 in bloodstream versus procyclic TB (8 and 20 μM, respectively) may reflect slower sterol flow (which means lower concentration of the CYP51 substrate) in the mammalian form of the parasite, most likely a large portion of sensitivity of the TB cells to 10 might result from general cytotoxicity of the compound unless the effect on HL60 is cancer cell specific. The best antitrypanosomal activity is provided by the four strongest CYP51 inhibitors, compounds 2, 4, 17, and 19. At 10 μM concentration these azoles cause more than 90% growth inhibition of bloodstream TB, three of them produce ED50 below 2 μM. The observed direct correlation between the potency of the tested azoles as CYP51 inhibitors and their antiparasitic effect both in insect and mammalian stages of the TB life cycle indicates that CYP51 reaction is essential in the bloodstream form of the parasite and thus supports the notion [10] that sparking sterols may play an important role in Trypanosomatidae. High selectivity of compounds 2, 4, 17 and 19 for trypanosomal cells makes them good candidates for further testing as potential antitrypanosomal agents.
Table 2.
Sensitivities of trypanosomal cells to CYP51 inhibitors. The values were calculated from the doseresponse curves.
| CYP51 inhibitor |
TB testing system | TC testing system | ||||||
|---|---|---|---|---|---|---|---|---|
| ED50, μM* | Selectivity index ** |
Trypomastigotes | Amastigotes | |||||
| Procyclic TB |
Bloodstream TB |
Human HL60 |
ED50, μM |
Inhibition at 1 μM, % |
ED95, μM |
Inhibition at 1 μM, % |
||
|
K*** 2 4 10 17 19 |
18 7 4 20 1 5 |
16 1.3 2.5 8 1.3 1.5 |
29 >50 >50 4 20 22 |
1.8 >38 >20 0.5 15 15 |
nt**** <1 <1 8 nt <1 |
- 78 72 7 - 92 |
- <1 <1 20 - <1 |
- 99 97 43 - 99 |
the dose causing a 50% cell growth inhibition, ED50< 10 μM being proposed as a threshold to consider a compound as a potential antisleeping sickness drug [45]
ED50(HL60)\/ED50(bloodstream TB)
ketoconazole
not tested.
B. Effect on TC: bloodstream trypomastigotes and intracellular amastigotes
Both clinical drugs currently available for treatment of TC infections (benzinidazole and nifutrimox (Supplemental Data, Figure S1)) are effective only for the early stages, intracellular forms of TC being more difficult targets. Because human stages of TC are known to synthesize endogenous sterols with the major component being the final sterol product of the fungal pathway, ergosterol, azole derivatives were tested as potential anti-TC agents. Some of them were found effective and are considered for clinical trials [18-20]. However, specific inhibition of the target enzyme has never been investigated.
Potent TCCYP51 inhibitors produce profound antiparasitic effect on TC (Figure 5, Table 2). The ability of trypomastigotes to infect heart cells after exposure to 1 μM concentration of 2, 4, and 19 was decreased by 78, 72 and 92% respectively (Figure 5 A, lower panel). Only for compound 10 (short-term TCCYP51 inhibitor) 8 μM concentration was required to reach 50% inhibition. Most remarkably, the compounds strongly inhibit multiplication of amastigotes within cardiomyoblasts, ED95 being less than 1 μM for 2, 4, and 19 (inhibition at 1 μM 99, 97 and 99%, respectively) as seen in Figure 5A, upper panel, and microscopically observed in Figure 5B, suggesting their potential applicability to be used for treatment of currently incurable late stages of the disease. Alterations in the sterol composition of TC cells upon treatment with compound 2 (sharp decrease in the ergosterol formation and accumulation of the C14 methylated precursors 24-methylenedihydrolanosterol and lanosterol (Figure 5C) provide direct evidence that the mode of action of the compound is connected with CYP51 inhibition.
Figure 5. Exposure of T. cruzi trypomastigotes to CYP51 inhibitors dramatically inhibits T. cruzi multiplication within cardiomyocytes and the percentage of cellular infection.

A. Potent inhibition of both T. cruzi intracellular multiplication and infection in cardiomyoblasts by CYP51 inhibitors. Trypomatigotes were pre-treated with several concentrations of CYP51 inhibitors 2, 4, 10 and 19 or mock-treated, exposed to cardiomyoblasts monolayers and T. cruzi multiplication was evaluated at 72 h by determining the number of T. cruzi/cell (upper insert) and the percent of infection (lower insert). The data represent the mean ± standard deviation (SD) of results from triplicate samples. SD did not exceed 10% of the mean. B. Microscopic observation of the inhibition of T. cruzi multiplication by 1 μM CYP51 inhibitors within cardiomyoblasts at 72 hr. T. cruzi pre-treated with control DMSO showed high levels of parasite multiplication, whereas cells exposed to trypanosomes pre-incubated with 1 μM azoles showed dramatic decrease in the number of intracellular parasites. C. TLC analysis of sterol composition of TC cells. Sterol standards: 24-methylenedihydrolanosterol (M), lanosterol (L), obtusifoliol (O), cholesterol (C) and ergosterol (E), 5 nmoles each (lane 1). Sterols from 10 mg of cell pellet of untreated epimastigotes (lane 2) and epimastigotes incubated for 120 hours with compound 2 (lane 3). The results of GC-MS analysis of TC sterols are provided in Supplemental data, Table S2.
Significance
Because TB species contain only two CYP genes, CYP51 and a putative P450 of unknown function (http://www.tigr.org), antiparasitic effect of potent CYP51 inhibitors in TB must be directly connected with disturbances in sterol 14α-demethylation. Comparable sensitivities of bloodstream and procyclic TB to the TBCYP51 inhibitors implies that the CYP51 gene is active in the mammalian stages of the parasite life cycle and thus support the notion [29] that, similar to plants/algae, biologically active (functional) sterols of endogenous nature must be essential in Trypanosomatidae. This finding suggests that CYP51 inhibitors should be included in testing as potential antisleeping sickness agents and that the nitroimidazoles, recently found to be potent in killing bloodstream TB [46], might actually target TBCYP51. Supported by the profound effect of the most potent TCCYP51 inhibitors both in trypomastigote and intracellular human stages of TC, the work offers a novel lead structure with a defined region most promising for possible further modifications for antitrypanosomal therapy. Taking into account high amino acid sequence identity between the CYP51s from Trypanosoma and Leishmania (74-78%) there is a high probability that the same compounds will be effective against leishmaniasis, which might be of vital importance for visceral forms of the disease as many azoles are known to penetrate the blood/brain barrier [52]. The approach undertaken in this study including large-scale spectroscopic screening for binding ligands and quantification of the inhibitory potencies in the reconstituted enzyme reaction followed by testing the best compounds in the parasite cells is promising to follow for the discovery of new, rationally designed antiprotozoan drugs and perhaps novel antifungal agents as well.
Experimental Procedures
Overexpression of TB and TC CYP51 in E. coli, purification to electrophoretic homogeneity and reconstitution of enzymatic activities using 3H-labeled sterol substrates were performed as previously described [31, 32]. Experimental azole derivatives and their further modifications were a generous gift from Novartis Research Institute (Vienna, Austria); the Novartis numbers for the compounds are: 1, SDZ-89443(−), 2, SDZ-284692(+); 3, SDZ-284693(−); 4, SDZ-285604(−); 5, SDZ-287113; 6, VAB251(R); 7, VAB309(R); 8, SDZ-287115; 9, VAB636(R); 10, VAB635(R); 11, SDZ-285936; 12, SDZ-285428(S); 17, SDZ-284531; 19, SDZ-284797(+); 20, SDZ-284798(−).
Spectroscopic titration of TB and TCCYP51 with azoles
Azole-induced spectral changes in TC and TBCYP51 (2μM) were monitored in 2 ml tandem cuvettes in the wavelength range 350-500 nm using a Shimadzu UV-2401PC spectrophotometer. The azoles were dissolved in DMSO (1mM) and added in the range 0.2-20 μM. Maximal spectral response per nanomole of P450 (ΔA426-410 for oxidized substrate free, ΔA426-390 for oxidized CYP51 in presence of substrate, and ΔA424-445 for reduced, substrate free CYP51 and apparent Kd were determined from the equilibrium titration curves (Figure 1, insert) by plotting absorbance changes against the concentration of free ligand and fitting the data to a rectangular hyperbola using SigmaPlot Statistics [53]. Replacement of the azoles by sterol substrate and by carbon monoxide after the titration experiments was monitored as type 1 spectral response and as CO binding spectra, respectively.
Inhibition of CYP51 activity
Equal volumes of the reaction mixture containing 1 μM P450, 2 μM TB cytochrome P450 reductase, 50 μM obtusifoliol for TBCYP51 and 24-methylenedihydrolanosterol for TCCYP51 were added to the tubes with the azole inhibitors (final concentration 1-100 μM) and the reaction was initiated with the addition of NADPH. For time-course measurements aliquots of the reaction mixture were taken over time. The sterols were extracted with ethyl acetate and analyzed by reverse phase HPLC equipped with β-RAM radioactivity flow detector using Nova-Pak C18 column. Reverse phase HPLC using 0.05M disodium hydrogen orthophosphate/acetonitrile (50:50 v/v) adjusted to pH 6.0 with glacial acetic acid as the mobile phase and UV detection at 230 nm was employed to confirm stability of the azoles in the solution during the reaction. CO-spectra of the control (no inhibitor) sample were taken to confirm stability of the P450s. The inhibitory potencies of azoles were compared upon time course measurements as I/E2 (molar ratio inhibitor/enzyme at which the substrate conversion decreases 2-fold) and as percentage of inhibition at molar ratio 1:1.
Cell culture and growth inhibition assay for TB
The procyclic forms of the TB cell line 29-13 were grown in SDM-79 medium (JRH Biosciences, Lenox, KS) supplemented with 10% heat-inactivated fetal bovine serum at 27°C [54]. Bloodstream forms of the TB cell line SM427 [48] and human myeloid leukemia cells HL60 (DSZM, Braunschweig, Germany) were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum at 37°C. Both cultures were maintained in a humidified atmosphere containing 5% CO2. Cell counting was done using a Neubauer haemocytometer. The cells were seeded at various densities (105 − 2×106/ml) in 24-well plates and incubated for 48 hours. The cell viability indicator Alamar Blue (Biosource, USA) was added to final concentration of 10 %, and after 2 hours of additional incubation 200 μl aliquots were placed into 96-well flat bottom microtiter plates (Costar, USA), and fluorescence was measured at 520 nm excitation wavelength and 590 nm emission wavelength using a FLUOstar Optima plate reader (BMG Lab Technologies, Germany). Cell viability was monitored as emission in the cells versus emission in the growth media (signal to noise ratio). Plating cell densities of 106 for the procyclic forms and 5×105 for the bloodstream forms of TB and for human HL60 (signal to noise ratios, 12, 6 and 4.5, respectively) were selected for further measurements. Reduction of Alamar Blue in the cells, absence of media-induced reduction or further conversion into the hydro-form were confirmed by absorbance spectroscopy in the range of 400-700 nm (the spectra are shown as Supplemental Figure S4). To evaluate the growth inhibition effect, the cells were incubated in the presence of CYP51 inhibitors (0-50 μM), the anti-sleeping sickness drug suramin (bloodstream TB) and NaN3 (procyclic TB). Wells containing cells, medium and 1% DMSO alone served as controls. The concentrations causing 50% growth inhibition (ED50, 50% effective dose) were calculated from dose-response curves. The selectivity of the inhibitors for the parasite cells was estimated as ratio ED50 (human HL60)/ED50 (bloodstream TB) [45].
Growth and inhibition of TC
Trypomastigotes (106 organisms) were pre-exposed to the CYP51 inhibitors 2, 4, 10 and 19 dissolved in DMSO/DMEM at several concentrations (1-50 μM) or to control DMSO/DMEM for 30 min. The parasites were then exposed in triplicate to rat cardiomyocyte monolayers at the ratio 10 parasites/cell in Lab Tech chambers for 2h as described [55]. After removing the unbound parasites, monolayers were incubated with DMEM supplemented with 10% FBS for 72 hr to allow parasite intracellular multiplication and the number of T. cruzi per 200 cells and the percent of infection were microscopically determined in Giemsa stained monolayers [55].
TLC and GC-MS analysis of TC sterols
Epimastigotes (plating density 107 cells/ml) were cultured in brain heart infusion supplemented with hemin and 10% calf serum for 120 hours without an inhibitor and in the presence of compound 2. The inhibitor was added daily to maintain 1 μM concentration for the first 3 days and 2 μM concentration for the last 2 days. The cell pellet was washed with Hanks' balanced salt solution without phenol red to remove excess of cholesterol from the serum and saponified using 10% KOH in 98% aqueous methanol at reflux temperature for 1 hour. The neutral lipids obtained by dilution with water and extraction with hexane were subjected to silica gel TLC plates (0.25 μm, Whatman, Germany) for 30 min in hexane:ethyl acetate (8:2) as developing solvent and monitored in an iodine chamber. Individual sterols were identified using a GC-MS apparatus with Hewlett-Packard LS 6500 gas chromatograph interfaced to a 5973 mass spectrometer as described previously [29].
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (GM067871 to M.R.W, GM63477 to W.D.N, SC1GM 081168 and GM 008037 to F.V.), from the American Heart Association (0535121N to G.I.L), and from Welch Foundation (D-1276 to W.D.N.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S. The trypanosomiases. Lancet. 2003;362:1469–1480. doi: 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- 2.Chagas' disease- an epidemic that can no longer be ignored. Lancet. 2006;368:619. doi: 10.1016/S0140-6736(06)69217-9. Editorial. [DOI] [PubMed] [Google Scholar]
- 3.Newsome AL, McGhee CR. Trypanosoma cruzi in Triatomes from an urban and domestic setting in Middle Tennessee. J. Ten. Acad. Sci. 2006;81:62–65. [Google Scholar]
- 4.Diaz JH. Chagas disease in the United States: a cause for concern in Louisiana? J La State Med. Soc. 2007;159:25–29. [PubMed] [Google Scholar]
- 5.Morel CM, Lazdins J. Chagas disease. Nat .Rev. Microbiol. 2003;1:14–15. doi: 10.1038/nrmicro735. [DOI] [PubMed] [Google Scholar]
- 6.Luscher A, de Koning HP, Maser P. Chemotherapeutic strategies against Trypanosoma brucei: drug targets vs. drug targeting. Curr. Pharm. Des. 2007;13:555–567. doi: 10.2174/138161207780162809. [DOI] [PubMed] [Google Scholar]
- 7.El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- 8.Croft SL, Barrett MP, Urbina JA. Chemotherapy of trypanosomiases and leishmaniasis. Trends in Parasitol. 2005;21:508–512. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 9.Nes WR, McKean MR. Biochemistry of steroids and other isopentenoids. University Park Press; Baltimore: 1977. Ch.4. [Google Scholar]
- 10.Roberts CW, McLeodm R, Rice DW, Ginger M, Chance ML, Goad LJ. Fatty acid and sterol metabolism: potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 2003;126:129–142. doi: 10.1016/s0166-6851(02)00280-3. [DOI] [PubMed] [Google Scholar]
- 11.Vanden Bossche H, Koymans L, Moereels H. P450 inhibitors of use in medical treatment: focus on mechanisms of action. Pharmacol. Ther. 1995;67:79–100. doi: 10.1016/0163-7258(95)00011-5. [DOI] [PubMed] [Google Scholar]
- 12.Gupta AK, Tomas E. New antifungal agents. Dermatol. Clin. 2003;3:565–576. doi: 10.1016/s0733-8635(03)00024-x. [DOI] [PubMed] [Google Scholar]
- 13.Docampo R, Moreno SN, Turrens JF, Katzin AM, Gonzalez-Cappa SM, Stoppani AO. Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1981;3:169–180. doi: 10.1016/0166-6851(81)90047-5. [DOI] [PubMed] [Google Scholar]
- 14.Lazardi K, Urbina JA, de Souza W. Ultrastructural alterations induced by two ergosterol biosynthesis inhibitors, ketoconazole and terbinafine, on epimastigotes and amastigotes of Trypanosoma (Schizotrypanum) cruzi. Antimicrob. Agents Chemother. 1990;34:2097–2105. doi: 10.1128/aac.34.11.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbina JA, Payares G, Molina J, Sanoja C, Liendo A, Lazardi K, Piras M, Piras R, Perez N, Wincker P, Ryley JF. Cure of short- and long-term experimental Chagas' disease using D0870. Science. 1996;273:969–971. doi: 10.1126/science.273.5277.969. [DOI] [PubMed] [Google Scholar]
- 16.Apt W, Aguilera X, Arribada A, Perez C, Miranda C, Sanchez G, Zulantay I, Cortes P, Rodriguez J, Juri D. Treatment of chronic Chagas' disease with itraconazole and allopurinol. Am. J. Trop. Med. Hyg. 1998;59:133–138. doi: 10.4269/ajtmh.1998.59.133. [DOI] [PubMed] [Google Scholar]
- 17.Araujo MS, Martins-Filho OA, Pereira ME, Brener Z. A combination of benznidazole and ketoconazole enhances efficacy of chemotherapy of experimental Chagas' disease. J. Antimicrob. Chemother. 2000;45:819–824. doi: 10.1093/jac/45.6.819. [DOI] [PubMed] [Google Scholar]
- 18.Molina J, Martins-Filho O, Brener Z, Romanha AJ, Loebenberg D, Urbina JA. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob. Agents. Chemother. 2000;44:150–155. doi: 10.1128/aac.44.1.150-155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urbina JA. Specific treatment of Chagas disease: current status and new developments. Curr. Opin. Infect. Dis. 2001;6:733–741. doi: 10.1097/00001432-200112000-00012. [DOI] [PubMed] [Google Scholar]
- 20.Buckner F, Yokoyama K, Lockman J, Aikenhead K, Ohkanda J, Sadilek M, Sebti S, Van Voorhis W, Hamilton A, Gelb MH. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15149–15153. doi: 10.1073/pnas.2535442100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urbina JA, Concepcion JL, Caldera A, Payares G, Sanoja C, Otomo T, Hiyoshi H. In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob. Agents Chemother. 2004;48:2379–2387. doi: 10.1128/AAC.48.7.2379-2387.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hucke O, Gelb MH, Verlinde CL, Buckner FS. The protein farnesyltransferase inhibitor Tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 2005;48:5415–5418. doi: 10.1021/jm050441z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gebre-Hiwot A, Frommel D. The in-vitro anti-leishmanial activity of inhibitors of ergosterol biosynthesis. J. Antimicrob. Chemother. 1993;32:837–842. doi: 10.1093/jac/32.6.837. [DOI] [PubMed] [Google Scholar]
- 24.Haughan PA, Chance ML, Goad LJ. Effects of an azasterol inhibitor of sterol 24-transmethylation on sterol biosynthesis and growth of Leishmania donovani promastigotes. Biochem. J. 1995;308:31–38. doi: 10.1042/bj3080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodrigues JCF, Urbina JA, de Souza W. Antiproliferative and ultrastructural effects of BPQOH, a specific inhibitor of squalene synthase, on Leishmania amazonensis. Exp. Parasitol. 2005;111:230–238. doi: 10.1016/j.exppara.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Coppens I, Baudhuin P, Opperdoes FR, Courtoy PJ. Receptors for the host low density lipoproteins on the hemoflagellate Trypanosoma brucei: purification and involvement in the growth of the parasite. Proc. Natl. Acad. Sci. U. S. A. 1988;85:6753–6757. doi: 10.1073/pnas.85.18.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coppens I, Courtoy PJ. The adaptative mechanisms of Trypanosoma brucei for sterol homeostasis in its different life-cycle environments. Annu. Rev. Microbiol. 2000;54:129–156. doi: 10.1146/annurev.micro.54.1.129. [DOI] [PubMed] [Google Scholar]
- 28.Lorente SO, Rodrigues JC, Jimenez C, Joyce-Menekse M, Rodrigues C, Croft SL, Yardley V, de Luca-Fradley K, Ruiz-Perez LM, Urbina J. Novel azasterols as potential agents for treatment of leishmaniasis and trypanosomiasis. Antimicrob. Agents. Chemother. 2004;48:2937–2950. doi: 10.1128/AAC.48.8.2937-2950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou W, Cross GA, Nes WD. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol synthesis and cell proliferation of Trypanosoma brucei. J. Lipid Res. 2007;48:665–673. doi: 10.1194/jlr.M600404-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta. 2007;1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lepesheva GI, Nes WD, Zhou W, Hill GC, Waterman MR. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry. 2004;43:0789–10799. doi: 10.1021/bi048967t. [DOI] [PubMed] [Google Scholar]
- 32.Lepesheva GI, Zaitseva NG, Nes WD, Zhou W, Arase M, Liu J, Hill GC, Waterman MR. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B' helix defines substrate preferences of sterol 14alpha-demethylase. J. Biol. Chem. 2006;281:3577–3585. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- 33.Lepesheva GI, Hargrove TY, Ott RD, Nes WD, Waterman MR. Biodiversity of CYP51 in trypanosomes. Biochem. Soc. Trans. 2006;34(Pt 6):1161–1164. doi: 10.1042/BST0341161. [DOI] [PubMed] [Google Scholar]
- 34.Ortiz de Montellano PR, Correia MA. Cytochrome P450: Structure, Mechanism, and Biochemistry. Plenum Publishing Corp.; New York: 1995. Ch. 3. [Google Scholar]
- 35.Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug, Metab. Dispos. 2002;30:314–318. doi: 10.1124/dmd.30.3.314. [DOI] [PubMed] [Google Scholar]
- 36.Marichal P, Koymans L, Willemsens S, Bellens D, Verhasselt P, Luyten W, Borgers M, Ramaekers FC, Odds FC, Bossche HV. Contribution of mutations in the cytochrome P450 14alpha-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology. 1999;145:2701–2713. doi: 10.1099/00221287-145-10-2701. [DOI] [PubMed] [Google Scholar]
- 37.Schiaffella F, Macchiarulo A, Milanese L, Vecchiarelli A, Fringuelli R. Novel ketoconazole analogues based on the replacement of 2,4-dichlorophenyl group with 1,4-benzothiazine moiety: design, synthesis, and microbiological evaluation. Bioorg. Med. Chem. 2006;14:5196–5203. doi: 10.1016/j.bmc.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Sun QY, Xu JM, Cao YB, Zhang WN, Wu QY, Zhang DZ, Zhang J, Zhao HQ, Jiang YY. Synthesis of novel triazole derivatives as inhibitors of cytochrome P450 14alpha-demethylase (CYP51) Eur. J. Med. Chem. 2007;42:1226–1233. doi: 10.1016/j.ejmech.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Munayyer HK, Mann PA, Chau AS, Yarosh-Tomaine T, Greene JR, Hare RS, Heimark L, Palermo RE, Loebenberg D, McNicholas PM. Posaconazole is a potent inhibitor of sterol 14alpha-demethylation in yeasts and molds. Antimicrob. Agents. Chemother. 2004;48:3690–3696. doi: 10.1128/AAC.48.10.3690-3696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogers TR. Antifungal drug resistance: limited data, dramatic impact? Int. J. Antimicrob. Agents. 2006;27:7–11. doi: 10.1016/j.ijantimicag.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Schuster I, Egger H, Nussbaumer P, Kroemer RT. Inhibitors of Vitamin D Hydroxylases: Structure-Activity Relationships. J. Cell Biochem. 2003;88:372–380. doi: 10.1002/jcb.10365. [DOI] [PubMed] [Google Scholar]
- 42.Schuster I, Egger H, Astecker N, Herzig G, Schussler M, Vorisek G. Selective inhibitors of CYP24: mechanistic tools to explore vitamin D metabolism in human keratinocytes. Steroids. 2001;66:451–462. doi: 10.1016/s0039-128x(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 43.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 44.Yano JK, Denton TT, Cerny MA, Zhang X, Johnson EF, Cashman JR. Synthetic inhibitors of cytochrome P-450 2A6: inhibitory activity, difference spectra, mechanism of inhibition, and protein cocrystallization. J. Med. Chem. 2006;49:6987–7001. doi: 10.1021/jm060519r. [DOI] [PubMed] [Google Scholar]
- 45.Hoet S, Opperdoes F, Brun R, Quetin-Leclercq J. Natural products active against African trypanosomes: a step towards new drugs. Nat. Prod. Rep. 2004;21:353–364. doi: 10.1039/b311021b. [DOI] [PubMed] [Google Scholar]
- 46.Torreelee E. Drugs Against Protozoan Parazites, Keystone Symposia on Molecular and Cellular Biology. Tahoe City, CA: 2007. Rediscovering nitro-imidazoles as promising drug candidates for human African Trypanosomiasis; p. 36. [Google Scholar]
- 47.Dixon H, Ginger CD, Williamson J. Trypanosome sterols and their metabolic origins. Comp. Biochem. Physiol. B. 1972;41:1–18. doi: 10.1016/0305-0491(72)90002-8. [DOI] [PubMed] [Google Scholar]
- 48.Carruthers VB, Cross GA. High-efficiency clonal growth of bloodstream- and insect-form Trypanosoma brucei on agarose plates. Proc. Natl. Acad. Sci. U.S.A. 1992;89:8818–8821. doi: 10.1073/pnas.89.18.8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang YJ, Jeng JH, Chen RJ, Tseng H, Chen LC, Liang YC, Lin CH, Chen CH, Chu JS, Ho WL, Ho YS. Ketoconazole potentiates the antitumor effects of nocodazole: In vivo therapy for human tumor xenografts in nude mice. Mol. Carcinog. 2002;34:199–210. doi: 10.1002/mc.10066. [DOI] [PubMed] [Google Scholar]
- 51.Dyrstad SW, Shah P, Rao K. Chemotherapy for prostate cancer. Curr. Pharm. Des. 2006;12:819–837. doi: 10.2174/138161206776056100. [DOI] [PubMed] [Google Scholar]
- 52.Goodpasture HC, Hershberger RE, Barnett AM, Peterie JD. Treatment of central nervous system fungal infection with ketoconazole. Arch. Intern. Med. 1985;145:879–880. [PubMed] [Google Scholar]
- 53.Lepesheva GI, Virus C, Waterman MR. Conservation in the CYP51 family. Role of the B' helix/BC loop and helices F and G in enzymatic function. Biochemistry. 2003;42:9091–9101. doi: 10.1021/bi034663f. [DOI] [PubMed] [Google Scholar]
- 54.Hirumi H, Hirumi K, Moloo SK, Shaw MK. Trypanosoma brucei brucei: in vitro production of metacyclic forms. J. Protozool. 1992;39:619–627. doi: 10.1111/j.1550-7408.1992.tb04861.x. [DOI] [PubMed] [Google Scholar]
- 55.Nde PN, Simmons KJ, Kleshchenko Y, Pratap S, Lima MF, Villalta F. Silencing of the laminin γ-1 gene blocks Trypanosoma cruzi infection. Infect. Immun. 2006;74:1643–1648. doi: 10.1128/IAI.74.3.1643-1648.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.