

Abstract
We have investigated in rat pheochromacytoma PC12 cells the activation of the mitogen-activated protein kinases ERK1 and ERK2 by the mitochondrial uncoupler carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP). This treatment slowly decreases ATP levels to 30% of control, whereas the internal calcium level rises very rapidly to 250% of control, derived from internal stores. Tyrosine phosphorylation of ERK1 and ERK2 increases gradually, starting after 5 min of treatment, to reach a maximum at 30 min; the kinase activity reaches 250% when measured after 1 hr of treatment. The drop in ATP levels is slower still. Comparison of the time courses of the rapid rise in cytosolic calcium with the slower increase in ERK1 and ERK2 activation suggests one or more intermediate stages in this pathway. Chelation of cytosolic calcium with dimethyl bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid abolished the FCCP-stimulated rise in internal calcium, as well as the tyrosine phosphorylation and the activation of the ERKs. Surprisingly, caffeine, which releases calcium from different internal stores, did not increase the tyrosine phosphorylation and did not activate the ERKs. The FCCP effect on calcium storage may be related to mitochondrial dysfunction in Alzheimer disease, which might result in ineffective buffering of cytosolic calcium that leads to mitogen-activated protein kinase activation and subsequent protein phosphorylations.
The neurodegenerative chain of events in Alzheimer disease (AD) are unknown. Recent evidence indicates that mitochondrial dysfunction, defects in energy metabolism, and excitotoxicity might be involved (1, 2). For example, both mitochondrial DNA deletions (3, 4) and defects in mitochondrial oxidative phosphorylation (5, 6) have been observed in the aged and the AD brain. Mitochondria are the site of ATP production and also play a role in intracellular calcium homeostasis because they can sequester large quantities of calcium (7). Effective buffering of cytosolic calcium is critical to neuronal survival because elevated cytosolic calcium would stimulate glutamate release, resulting in activation of N-methyl-d-aspartate receptors, which in turn would result in massive Ca2+ influx and cell damage and death. Several lines of evidence indicate that calcium homeostasis is altered in aging and AD (8–10). A reduction of Ca2+ storage in mitochondria has been detected in old rats (11). Calcium release from the inositol 1,4,5-trisphosphate-sensitive endoplasmic reticulum (ER) pool (12) and from an additional ionophore-sensitive pool (13) is enhanced in fibroblasts from AD patients. These results suggest that an age-associated increase in internal calcium concentration may be relevant to such neurodegenerative disorders. In fact, neurofibrillary tangle-like antigenic changes and β-amyloidosis can be elicited by glucose deprivation and calcium influx in cultured hippocampal neurons (14), skin fibroblasts (15), human neuroblastoma cells (16), and COS cells (17).
The molecular mechanism that links mitochondrial dysfunction to neuronal death is not clear. Previously, we demonstrated (18) that treatment of PC12 cells with the mitochondrial uncoupler carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) led to hyperphosphorylation of neurofilament (NF) and Tau in these cells, suggesting an activation of NF and/or Tau kinases. We undertook this study to investigate the mechanism involved. We examined the role of increased cytosolic calcium in the activation of ERK1 and ERK2 in PC12 cells. Our results indicate that FCCP treatment of PC12 cells leads to calcium release from mitochondrial stores, and also to phosphorylation/activation of the mitogen-activated protein (MAP) kinases ERK1 and ERK2. The possible implications of these effects in AD are discussed.
EXPERIMENTAL PROCEDURES
Cells and Materials.
Rat pheochromacytoma (PC12) cells were obtained from the American Type Culture Collection and subcultured in our laboratory as described (18). The cells were not treated with nerve growth factor to decrease the baseline MAP kinase activity, and were fed 1 day prior to extraction to obtain a lower baseline level of MAP kinase activity. Phosphotyrosine204-specific MAP kinase polyclone antibody (anti-Ptyr-ERK1+2) was obtained from New England Biolabs; polyclonal antibody to ERK1 and ERK2 (anti-ERK1+ERK2) and myelin basic protein (MBP) was from Upstate Biotechnology (Lake Placid, NY); poly-d-lysine, ATP disodium salt, peroxidase-conjugated anti-rabbit IgG, oligomycin, FCCP, and ionomycin were from Sigma; fura-2-AM, 5,5′-dimethyl-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA-AM), and the calcium calibration kit were from Molecular Probes; Immobilon-P transfer membrane was from Millipore; enhanced chemiluminescence kit was from Amersham; P81 phosphocellulose paper was from Whatman; protein G-Sepharose 4 was from Pharmacia; [γ-33P]ATP was from New England Nuclear.
Preparation of Cell Extracts.
PC12 cells were incubated with vehicle or various drugs in the cell culture medium at 37°C for the indicated times. Cells were washed and scraped into 500 μl of the lysis buffer (50 mM Tris⋅HCl, pH 7.4/270 mM sucrose/0.1 mM okadaic acid/1 mM EDTA/1 mM EGTA/1 mM Na3VO4/1 mM benzamidine/4 mg/ml leupeptin/0.1% 2-mercaptoethanol). The cells were then sonicated for 20 sec on ice and centrifuged for 10 min in a microfuge at 4°C. Total protein from the supernatant was assayed using a BCA kit (Pierce). For Western blots, an equal amount of SDS sample buffer containing 0.125 M Tris⋅HCl (pH 6.8), 4% SDS, 20% glycerol, 0.2 M DTT, and 0.02% bromophenol blue was added to the lysate, which was then boiled for 4 min.
Immunoblotting.
Proteins were resolved by SDS/12% PAGE and transferred onto Immobilon-P membranes for 2 hr at 4°C at 200 mA. The membrane was blocked with 5% bovine serum albumin in PBS overnight at 4°C and then incubated with anti-Ptyr-ERK1+2 at a dilution of 1:1,000 for 2 hr. Blots were then washed and incubated with anti-rabbit IgG at a dilution of 1:5,000. Blots were developed by using the enhanced chemiluminescence kit.
Immunoprecipitate ERKs Kinase Assay.
The assay was based on methods described by Alessi et al. (19). Antibody was titered according to the manufacturer’s suggested dilution, so that the assay was carried out in antibody excess. Briefly, cell extract (100 μg of total protein), anti-phospho-ERK1+2 (2 μl), and protein G-Sepharose (10 μl) were rocked gently at 4°C for 2 hr followed by centrifugation at 4°C for 5 min. The pellets were washed with the lysis buffer. Each washed pellet was resuspended in 20 μl of the kinase assay buffer containing 25 mM Hepes (pH 7.0), 2 mM MgCl2, 0.2 mM phenylmethylsulfonyl fluoride, and 1 mM DTT. The kinase assay mixture contained 20 μl of the immunocomplex suspended in kinase assay buffer, 5 μl of MBP (final concentration 18 μM), and 5 μl of [γ-33P]ATP (5 μCi, 1.0 mM ATP per assay). The incubation was carried out at 30°C for 15 min, and the reaction was terminated by spotting 14 μl of the supernatant onto each of two 2.4-cm circular P81 phosphocellulose paper circles, which were then washed with 0.5% phosphoric acid. The radioactivity was detected in a liquid scintillation counter (Beckman LS7500) using 5 ml of Liquiscint (National Diagnostics).
Measurement of Cytosolic Calcium.
PC12 cells, seeded at 6 × 104 cells per 35 mm dish, were cultured for 4 days on 24-mm round glass coverslips that had been coated with 10 mg/ml poly-d-lysine. To load the dye, cells were incubated with 3 μM of fura-2-AM in Tyrode’s solution containing 10 mM Hepes (pH 7.4), 150 mM NaCl, 3 mM KCl, 2 mM CaCl2, and 10 mM d-glucose with an osmolarity of 320 mOsm, at room temperature for 30 min. After rinsing with the Tyrode’s solution, the loaded cells were equilibrated at room temperature for 30 min in the cell culture medium containing 0.4 mM calcium. In some cases, this medium was replaced by calcium-free Tyrode’s solution. The coverslip with loaded cells was transferred to a coverglass holder (Medical System, Greenvale, NY), which was then filled with 1 ml of the cell culture medium. Cytosolic calcium was determined with a Nikon microscope optically linked to a model RM-M ratio fluorescence spectrometer (Photon Technology International, South Brunswick, NJ). The excitation maximum for the Ca2+-bound and the free dye is 340 and 380 nm, respectively. Fluorescence was detected in the ratio mode at the emission wavelength of 510 nm. To apply a drug, data acquisition was interrupted for 20–30 sec, and the cell medium was replaced with fresh medium containing the drug at a desired concentration. After each measurement, cytosolic calcium was calculated using felix software provided by the company, according to the formula of Grynkiewicz et al. (20):
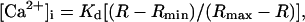 |
where Kd is the dissociation constant of the fura-2–Ca2+ complex and was assumed to be 274 nM at room temperature (21). R is the ratio of fluorescence when excited at 340 nm to that excited at 380 nm. To obtain Rmin (R at zero free calcium), fura-2 was placed in a calcium-free buffer (from the calcium calibration kit). Rmax (R when dye was saturated with calcium) value was determined in high calcium buffer (39.8 mM calcium, from the calcium calibration kit). This procedure yielded an Rmin of 0.14 and an Rmax of 1.13. The background fluorescence was subtracted from all measurements before calculation.
HPLC Analysis of the Levels of ATP.
PC12 cells were extracted with 0.4 M HClO4 for 10 min. The protein was spun down, dissolved in 1.0 ml 5% SDS/0.2 M NaOH, and quantified by the Lowry protein assay (22). Supernatants of the extracts were neutralized with 5 M KOH/2.1 M KH2PO4, and the insoluble KClO4 was discarded. A Waters HPLC apparatus was used to measure ATP levels. A254 was recorded and integrated with a Hewlett–Packard Model 3395 integrator. Chromatography was done with a Waters Nova-Pak C18 column at a flow rate of 1.3 ml/min. The loading buffer (A) was 35 mM KH2PO4/6 mM tetrabutylammonium hydrogen sulfate (pH 6.0) (23), and the elution buffer (B) consisted of 85% loading buffer/15% acetonitrile. After injection, initial conditions of 100% buffer A were held for 4 min. A linear gradient (Waters 660 slope no. 6) to 100% buffer B was then run in 10 min. Final conditions were held for 16 min, and returned to initial conditions in 3 min. Peaks were identified and quantified by analysis of ATP standards dissolved in the same buffer as the neutralized extracts.
Statistical Methods and Data Analysis.
Data are expressed as mean ± SD. Statistical significance was tested by the paired Student’s t test using an instat program (GraphPad, San Diego). A two-tailed value of P < 0.05 is taken as significant.
RESULTS
FCCP Decreases the Levels of ATP in PC12 Cells.
We first measured the total cellular ATP concentration following FCCP treatment of PC12 cells. Fig. 1 shows a time-dependent decrease in the ATP level after treatment with 30 μM FCCP, which differed significantly from the control values only after 2 hr of treatment (38% of control, P < 0.002; Fig. 1). Although at 30 min (P > 0.08) and 1 hr (P > 0.12) the ATP levels after FCCP treatment were not significantly lower, a decreasing trend over time is evident.
Figure 1.

Effects of FCCP on total ATP concentrations in PC12 cells. PC12 cells were treated with 0.10% EtOH (control) or 30 μM FCCP, and total ATP concentration in the cell extract was measured by HPLC. Data show the means and SD values of four separate experiments.
FCCP Raises Intracellular Calcium ([Ca2+]i) in PC12 Cells.
As an uncoupler of oxidative phosphorylation, FCCP collapses the mitochondrial membrane potential, thus releasing calcium. To measure the time course of changes in cytosolic calcium caused by FCCP, PC12 cells were loaded with fura-2-AM, and cytosolic calcium was measured for up to 1 hr after the drug addition. Fig. 2 shows a typical time course of the rapid rise in calcium produced by FCCP. In the example shown, immediately after addition of 30 μM FCCP, the cytosolic-free calcium level increased from about 80 nM to 185 nM followed by a plateau level at 152 ± 0.4 nM calcium (calculated between 10 and 20 min, 190% of control). Although there was some variation between cells in the shape of the immediate peak of the calcium profile, in general, FCCP caused a long elevated plateau phase (see Table 1 for summary and statistics). Oligomycin, an ATP synthase inhibitor, does not alter mitochondrial membrane potential and thus does not change calcium transport across the mitochondrial membrane (24). To determine the origin of the FCCP-stimulated increase in [Ca2+]i, we measured the FCCP effect either in calcium-free Tyrode’s solution or in culture medium containing 0.4 mM calcium. The FCCP-stimulated increase of cytosolic calcium in both media (Table 1) was similar, indicating that FCCP releases calcium from internal stores. We also observed increased cytosolic calcium levels with increasing concentrations (from 3.25 to 60 μM) of FCCP (data not shown).
Figure 2.

Time course of changes in cytosolic calcium following FCCP treatment of PC12 cells. Fura-2-loaded PC12 cells in cell culture medium were monitored for cytosolic calcium. At 5 min, the medium was replaced by a fresh medium containing vehicle (0.05% EtOH) or FCCP (30 μM). Data are representative of four separate measurements.
Table 1.
Summary of [Ca2+]i in PC12 cells treated with different drugs
| Drug | [Ca2+]e, mM | [Ca2+]i, nM* | % |
|---|---|---|---|
| Control | 0 | 71 ± 24 (12) | 100 |
| Control | 0.4 | 62 ± 13 (25) | 100 |
| FCCP, 30 μM | 0 | 153 ± 25 (4) | 215 |
| FCCP, 30 μM | 0.4 | 150 ± 33 (4) | 241 |
| Oligomycin, 50 μM | 0 | 79 ± 18 (4) | 111 |
| Oligomycin, 50 μM | 0.4 | 62 ± 8 (4) | 100 |
| Ionomycin, 10 μM | 0.4 | 130 ± 19 (4) | 172 |
| Caffeine, 10 mM | 0.4 | 147 ± 7 (3) | 210 |
| BAPTA, 10 μM† | 0.4 | 51 ± 12 (9) | 82 |
| BAPTA, 10 μM | |||
| FCCP, +30 μM‡ | 0.4 | 80 ± 16 (9) | 129 |
PC12 cells on glass coverslips were loaded with fura-2-AM, and [Ca2+]i was measured as described in Experimental Procedures.
Intracellular calcium concentration obtained at the plateau between 20 and 30 min after drug addition and calculated as the means ± SD; the number of experiments is indicated in parentheses.
Data averaged between 5 and 10 min after BAPTA addition.
BAPTA (10 μM) + 30 μM FCCP were added 15 min after addition of 10 μM BAPTA-AM, and measurements were averaged between 5 and 15 min later.
FCCP Induces Tyrosine Phosphorylation of MAP Kinases (ERK1 and ERK2).
Previously, we have found that FCCP treatment of PC12 cells correlates with an increase of phosphoepitopes on NF and Tau in vivo (18). To link the FCCP effect to the Tau hyperphosphorylation, we tested whether FCCP affects the activity of ERKs, which are expected to participate in Tau hyperphosphorylation. It is known that tyrosine phosphorylation is required for the activation of ERKs (25). Therefore, we first examined FCCP-treated cell extracts by Western blotting, using an antibody against tyrosine204-phosphorylated ERK1 and ERK2 (anti-Ptyr-ERK1+2). Fig. 3B shows that 5 min after FCCP treatment of PC12 cells, phosphorylated ERK1 and ERK2 were increased and reached a steady level at 30–60 min. For comparison, oligomycin (50 μM) had no effect (Fig. 3A). The concentration dependence of phosphorylation of ERKs by FCCP (Fig. 3A) indicates that the effect is FCCP specific.
Figure 3.

Immunoblots showing the dose response (A) and the time course (B) of FCCP-stimulated tyrosine phosphorylation of ERK1 and ERK2. (A) PC12 cells in culture medium were incubated with vehicle (0.05% EtOH, C), FCCP (7.5–60 μM, as indicated, F), or oligomycin (50 μM, oligo) at 37°C for 1 hr, or (B) for the indicated times at 30 μM. Tyrosine phosphorylation of ERK1 and ERK2 were analyzed by immunoblotting using anti-Ptyr-ERK1+ERK2. The results are representative of three separate experiments.
FCCP Increases the Activity of ERK1 and ERK2.
To investigate the correlation between tyrosine phosphorylation and the activity of ERKs stimulated by FCCP, we next performed an immunoprecipitate kinase assay to measure ERK1 and ERK2 activity specifically. The PC12 cell extracts were incubated with anti-Ptyr-ERK1+2. The immunoprecipitates were subjected to a standard MAP kinase assay by incubating with [γ-33P]ATP, using MBP as a substrate. Fig. 4 illustrates a more than 2-fold increase in ERK1 and ERK2 activity (240% of control, n = 5, P < 0.003) following FCCP treatment for 1 hr. Again, 50 μM oligomycin did not have the effect (78% of control, n = 3, P > 0.5). Ionomycin at 10 μM stimulated a smaller increase of the ERK1 and ERK2 activity than did FCCP (214% of control, n = 4, P < 0.01). Interestingly, 10 mM of caffeine had no effect on the activation of the ERKs (102%, n = 5, P > 0.8) even though the cytosolic calcium was raised 2-fold (Table 1; see Discussion).
Figure 4.

Effects of FCCP treatments on ERK1 and ERK2 kinase activity in PC12 cells. PC12 cells in the cell culture medium were treated with vehicle (0.05% EtOH, ctrl), oligomycin (50 μM, oligo), FCCP (30 μM), ionomycin (10 μM, iono), or caffeine (10 mM, caff) for 1 hr. ERK1 and ERK2 kinase activity was measured in phospho-ERK1+2-specific immunoprecipitates of the cell extracts using MBP as a substrate. Data are the means and SD values of at least three separate experiments (see Results).
Chelation of Calcium by BAPTA Blocks the FCCP-Stimulated Activation/Phosphorylation of ERKs.
If the activation of ERK1 and ERK2 by FCCP is mediated by calcium, depletion of cytosolic calcium should block such an activation. Table 1 shows that the basal cytosolic calcium level was reduced by BAPTA-AM (10 μM) and FCCP no longer caused an increase. Fig. 5A illustrates a typical experiment showing the chelating capacity of BAPTA. Coincidentally, the FCCP-induced tyrosine phosphorylation of ERK1 and ERK2 was blocked in the presence of BAPTA (10 μM; Fig. 5B), as well as with a combination of 1 mM EGTA and 10 μM ionomycin (data not shown). Fig. 5B also shows that FCCP-induced phosphorylation of ERK1 and ERK2 was not affected by pretreatment of the cells with 0.5 mM EGTA, which only chelates extracellular calcium. As a control, ionomycin (10 μM) increased the phosphorylation of ERK1+2, but to a lesser extent than FCCP (Fig. 5B). Again, caffeine alone did not stimulate phosphorylation of the ERKs. Fig. 5C shows the kinase activity of ERK1 and ERK2 after chelation of cytosolic calcium with BAPTA. Pretreatment of PC12 cells with 10 μM BAPTA-AM, followed by addition of 30 μM FCCP, blocked the activation of ERK1 and ERK2 stimulated by FCCP (63% by FCCP + BAPTA compared 256% with FCCP alone, n = 3, P < 0.05). These results support the idea that the activation of ERKs by FCCP may be triggered by an increase in cytosolic calcium.
Figure 5.

Chelation of cytosolic calcium with BAPTA blocks FCCP-stimulated phosphorylation and activation of ERK1 and ERK2. (A) Fura-2-loaded PC12 cells were monitored for basal [Ca2+]i. Cell medium was replaced at 5 min with the same medium containing 10 μM BAPTA-AM. At 15 min after the BAPTA addition, 30 μM FCCP in the presence of BAPTA was added. Data are representative of nine separate measurements. (B) PC12 cells were treated with either 10 μM BAPTA-AM, 0.5 mM EGTA, 10 mM caffeine alone for 15 min, or followed by 30 μM FCCP treatment for 1 hr. As a control, ionomycin (10 μM) was incubated with the cells for 15 min. The blot is representative of three separate experiments. (C) PC12 cells were pretreated with 10 μM BAPTA for 15 min followed by the addition of 30 μM FCCP for 1 hr. ERK1 and ERK2 kinase activity was assayed in phospho-ERK1+2-specific immunoprecipitates. Data are the means and SD of three separate experiments.
DISCUSSION
The purpose of this study was to help elucidate the chain of pathophysiological events that lead to neuronal dysfunction and neuronal cell death in AD. A likely candidate kinase to produce hyperphosphorylated Tau that resembles Alzheimer Tau is the MAP kinase ERK2 (26), and perhaps also the related ERK1, but other protein kinases may well be involved. ERK2 protein and mRNA are well represented in the dentate granule and pyramidal cells throughout all hippocampal subfields and the adjacent temporal cortex, regions believed to be important for memory (27). Furthermore, neurons that contain tangles are immunoreactive to ERK1 and ERK2 (28). Previously, indirect evidence had suggested that ATP depletion by FCCP in PC12 cells activates a NF kinase that could lead to neurofibrillary tangle formation seen in AD brains (18). We assume that the sole effect of FCCP on cells is the uncoupling of oxidative phosphorylation and that this results in a reduction of the proton gradient across the internal mitochondrial membrane. In turn, this results in a rapid release of calcium from this store (29) and a slower drop in ATP levels. No other effects of FCCP have been described. Oligomycin, an ATP synthase inhibitor, also depletes ATP but does not affect calcium levels (24).
We found that the time course of ATP depletion by FCCP is slower (30 min to 2 hr, Fig. 1) than the time course for activation of ERK1 and ERK2 (5 min, Fig. 3B). In contrast, the increase of cytosolic calcium by FCCP is immediate (a matter of seconds, Fig. 2). ERK2 activation at 5 min is not likely to be due to an immediate slight drop in ATP concentration. No causal relationships have been demonstrated between fall in ATP level and activation of ERKs. It is more likely due to the immediate dramatic increase in [Ca2+]i. The lag time between the calcium signal and the activation of MAP kinase is consistent with a role of calcium as an upstream activator of MAP kinase (30, 31). Although calcium-stimulated activation of ERKs by depolarization of PC12 cells (32) has been observed, this study links FCCP-sensitive mitochondrial calcium release with ERK1 and ERK2 activation. The ERKs presented in this study are distinct from other subgroups of MAP kinases—p38 and SAPKs, the stress-activated protein kinases (also called JNKs, c-Jun N-terminal kinases), which are activated in response to heat shock and oxidative stress (33). The antibody we applied does not recognize SAPKs and p38 (tested by the manufacturers); SAPKs and p38 are apparently activated through a distinct pathway, which does not require calcium or activation of MAP kinase kinase (34). However, the present results do not exclude the possibility that other subgroups of MAP kinases are also activated by FCCP treatment.
In this study, the observed increase in cytosolic calcium by FCCP treatment was similar in external medium that either did or did not contain calcium (Table 1). This indicates that FCCP does not cause an influx of calcium from the extracellular solution. Apparently, the release of calcium from internal stores by FCCP is sufficient to activate the MAP kinases. Chelation of intracellular calcium with BAPTA prevented both the FCCP-stimulated rise of cytosolic calcium and the FCCP-induced activation of ERKs, confirming the mediating role of calcium for FCCP-induced ERK activation. This was further confirmed by the combined treatment of external EGTA (1 mM) and of ionomycin (10 μM) in a calcium-free medium, which also prevented both the rise in calcium and the activation of ERKs. Although EGTA does not enter the cell, the ionophore ionomycin allows the diffusion of intracellular calcium down its concentration gradient out of the cell. Thus, the combination of EGTA and ionomycin would effectively deplete the intracellular calcium.
The FCCP-induced calcium release appears to be mitochondrial specific (29), while ionomycin and caffeine release calcium from different calcium pools. The slightly lower effect of ionomycin on the increase of cytosolic calcium (Table 1) may be due to its binding to serum present in the medium (ref. 35; and see Experimental Procedures). The same explanation applies to the lower magnitude of the ionomycin effect on the activation of ERK1 and ERK2 compared with that of FCCP. Surprisingly, caffeine, a ryanodine receptor agonist that releases calcium from the ER pool, increased cytosolic calcium as much as FCCP, but did not stimulate the tyrosine phosphorylation and the activation of ERK1 and ERK2. We offer three possible explanations: (i) MAP kinase, or its upstream activator, e.g., PYK2 (31), may respond differently to the increase in cytosolic calcium from different calcium pools. Interestingly, the ratios of the immunoreactive phospho-ERK1 and phospho-ERK2 induced by FCCP and ionomycin were different. Intensity of phospho-ERK1 was higher than that of phospho-ERK2 in the FCCP-treated cell extracts, whereas the opposite was true in the ionomycin treatment. This suggests that the activation of ERKs by calcium signaling may be source specific. The ability of calcium signals from different sources to trigger different responses (36) would add considerable flexibility to the cell-signaling system. (ii) Caffeine affects other cellular protein and kinases that may attenuate its effect on ERKs. Such multiple effects cannot be explained by the levels of cytosolic calcium alone. For example, caffeine only activates the protein kinase C β-isoform, although the activation of other protein kinase C isoforms are also calcium dependent (37). (iii) The action of FCCP may be less specific than that of ionomycin. Application of FCCP, apart from depolarizing the mitochondrial membrane, alters pH in mitochondria and/or cytosol, or decreases the ATP levels. Our present data do not allow us to decide between these possibilities.
Taken together, the results from this study indicate that the MAP kinases, ERK1 and ERK2, are activated by FCCP treatment of PC12 cells. The activation is apparently initiated by a sustained elevation of cytosolic [Ca2+]i from internal FCCP-sensitive calcium stores. Activation of ERK could have a number of important consequences in neurons (38, 39). These effects suggest that activated ERKs could mediate calcium-induced changes in cytoskeletal dynamics and gene expression that may underlie neuronal dysfunction and disorder in AD.
Acknowledgments
We thank Peggy Russell for supplying PC12 cells. We thank Ray Rabindran for important and useful discussions. This work was supported by the Markey Foundation (Y.L.), the John and Dorothy Wilson Fund (V.M.I.), and the Massachusetts Institute of Technology’s Undergraduate Research Opportunities Program (J.D.B.).
ABBREVIATIONS
- MAP
mitogen-activated protein
- AD
Alzheimer disease
- BAPTA
5,5′-dimethyl-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- BAPTA-AM
BAPTA acetoxymethyl ester
- [Ca2+]i
cytosolic free calcium concentration
- ER
endoplasmic reticulum
- FCCP
carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone
- MBP
myelin basic protein
- NF
neurofilament
- Ptyr-ERK1+2
tyrosine204-phosphorylated MAP kinases ERK1 and ERK2
References
- 1.Beal M F. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 2.Wallace D C. Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 3.Hutchin T, Cortopassi G. Proc Natl Acad Sci USA. 1995;92:6892–6895. doi: 10.1073/pnas.92.15.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanchard B J, Park T, Fripp W J, Lerman L S, Ingram V M. NeuroReport. 1993;4:799–802. doi: 10.1097/00001756-199306000-00051. [DOI] [PubMed] [Google Scholar]
- 5.Davis R E, Miller S, Herrnstadt C, Ghosh S S, Fahy E, Shinobu L A, Galasko D, Thal L J, Beal M F, Howell N, Parker W D., Jr Proc Natl Acad Sci USA. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Parker W D, Parks J K. Neurology. 1995;45:482–486. doi: 10.1212/wnl.45.3.482. [DOI] [PubMed] [Google Scholar]
- 7.Ernster L, Schatz G. J Cell Biol. 1981;91:227–255. doi: 10.1083/jcb.91.3.227s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCoy K R, Mullins R D, Newcomb T G. Neurobiol Aging. 1993;14:447–456. doi: 10.1016/0197-4580(93)90103-i. [DOI] [PubMed] [Google Scholar]
- 9.Peterson C, Goldman J E. Proc Natl Acad Sci USA. 1986;83:2758–2762. doi: 10.1073/pnas.83.8.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khachaturian Z S. Ann NY Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Serrano A, Blanco P, Satrustegui J. J Biol Chem. 1992;267:4672–4679. [PubMed] [Google Scholar]
- 12.Ito E, Oka K, Etcheberrigaray R, Nelson T J, McPhie D L, Tofel-Grehl B, Gibson G, Alkon D L. Proc Natl Acad Sci USA. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson G E, Lourdes H Z, Szolosi S, Tofel-Grehl B. Biochim Biophys Acta. 1996;1316:71–77. doi: 10.1016/0925-4439(96)00002-6. [DOI] [PubMed] [Google Scholar]
- 14.Cheng B, Mattson M. Exp Neurobiol. 1992;117:114–123. doi: 10.1016/0014-4886(92)90120-f. [DOI] [PubMed] [Google Scholar]
- 15.Blass J P, Baker A C, Ko L-W, Black R S. Arch Neurol. 1990;47:864–869. doi: 10.1001/archneur.1990.00530080046009. [DOI] [PubMed] [Google Scholar]
- 16.Shea T B, Klinger E P, Cressman C M. NeuroReport. 1995;6:1437–1440. doi: 10.1097/00001756-199507100-00019. [DOI] [PubMed] [Google Scholar]
- 17.Gabuzda D, Busciglio J, Chen L B, Matsudaira P, Yankner B A. J Biol Chem. 1994;269:13623–13628. [PubMed] [Google Scholar]
- 18.Bush M L, Miyashiro J S, Ingram V M. Proc Natl Acad Sci USA. 1995;92:1861–1865. doi: 10.1073/pnas.92.6.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alessi D R, Cohen P, Ashworth A, Cowley S, Leevers S J, Marshall C J. Methods Enzymol. 1995;255:279–289. doi: 10.1016/s0076-6879(95)55031-3. [DOI] [PubMed] [Google Scholar]
- 20.Grynkiewicz G, Poenie M, Tsien R Y. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 21.Shuttleworth T J, Thompson J L. J Biol Chem. 1991;266:1410–1414. [PubMed] [Google Scholar]
- 22.Robyt J, White B. Biochemical Techniques: Theory and Practice. Belmont, CA: Brooks/Cole; 1987. pp. 235–236. [Google Scholar]
- 23.Ally A, Park G. J Chromatogr. 1992;575:19–27. doi: 10.1016/0378-4347(92)80499-g. [DOI] [PubMed] [Google Scholar]
- 24.Budd S L, Nicholls D G. J Neurochem. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- 25.Anderson N G, Maller J L, Tonks N K, Sturgill T W. Nature (London) 1990;343:651–652. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- 26.Roder H M, Eden P A, Ingram V M. Biochem Biophys Res Commun. 1993;193:639–647. doi: 10.1006/bbrc.1993.1672. [DOI] [PubMed] [Google Scholar]
- 27.Hyman B T, Reiter J, Moss M, Rosene D, Pandya D. Neurosci Lett. 1994;166:113–116. doi: 10.1016/0304-3940(94)90853-2. [DOI] [PubMed] [Google Scholar]
- 28.Hyman B T, Elvhage T E, Reiter J. Am J Pathol. 1994;144:565–572. [PMC free article] [PubMed] [Google Scholar]
- 29.Friel D D, Tsien R W. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosen L B, Ginty D D, Weber M J, Greenberg M E. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 31.Lev S, Martinez R, Canoll P, Peles E, Musacchlo J M, Plowman G D, Rudy B, Schlessinger J. Nature (London) 1995;376:737–744. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 32.Rusanescu G, Qi H, Thomas S M, Brugge J S, Halegoua S. Neuron. 1995;15:1415–1425. doi: 10.1016/0896-6273(95)90019-5. [DOI] [PubMed] [Google Scholar]
- 33.Pombo C M, Bonventre J V, Avruch J, Woodgett J R, Kyriakis J M, Force T. J Biol Chem. 1994;269:26546–26551. [PubMed] [Google Scholar]
- 34.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 35.Kao J P Y. Methods Cell Biol. 1994;40:171. doi: 10.1016/s0091-679x(08)61114-0. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh A, Greenberg M E. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 37.Coulson R, Proch P S, Olsson R A, Chalfant C E, Cooper D R. Am J Physiol. 1996;270:263–274. doi: 10.1152/ajprenal.1996.270.2.F263. [DOI] [PubMed] [Google Scholar]
- 38.Drewes G, Lichtenberg-Kraag B, Döring F, Mandelkow E-M, Biernat J, Goris J, Dorée M, Mandelkow E. EMBO J. 1992;11:2131–2138. doi: 10.1002/j.1460-2075.1992.tb05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis R J. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]