

Abstract
Chronic hydrocephalus (CH) is a neurological disease characterized by increased cerebrospinal fluid volume and pressure that is often associated with impaired cognitive function. By and large, CH is a complex and heterogeneous CSF disorder where the exact site of brain insult is uncertain. Several mechanisms including neural compression, fiber stretch, and local or global hypoxia have been implicated in the underlying pathophysiology of CH. Specifically, the hippocampus, which plays a significant role in memory processing and is in direct contact with expanding CSF ventricles, may be involved. Using our model of chronic hydrocephalus, we quantified the density of VEGFR-2+ neurons, glial, endothelial cells, and blood vessels in hippocampal regions CA1, CA2-3, dentate gyrus and hilus using immunohistochemical and stereological methods. Density and %VEGFR-2+ cell populations were estimated for CH animals (2–3 weeks vs 12–16 weeks) and Surgical Controls (SC).
Overall, we found approximately 6–8 fold increase in the cellular density of VEGFR-2+ and more than double BVd in the hippocampus of CH compared with SC. There were no significant regional differences in VEGFR-2+ cellular and BV density expression in the CH group. VEGFR-2+ and BV densities were significantly related to changes in CSF volume (p≤0.05), and not ICP. The %VEGFR-2+ was significantly greater in CH than SC (p≤0.05), and was significantly correlated with BV density (p≤0.05).
These results showed that CH elicited a profound increase in VEGFR-2+ in hippocampus that corresponded to increased BV density. It was unclear whether increased VEGFR-2+ and BV expression was related to focal compression alone or in combination with global ischemia/hypoxia conditions as previously described. These findings suggest that VEGFR-2 may play an adaptive role in angiogenesis after CH-induced hypoxia. Modulation of VEGF/VEGFR-2+ may be important in developing treatments for hypoxic conditions including hydrocephalus and other forms of cerebral ischemia.
Keywords: canine, cerebrovascular, aquaductal stenosis, cerebrospinal fluid, circulation, ischemia, neurons, glia, endothelial cells, angiogenesis
Introduction
Chronic hydrocephalus (CH) is a neurological disease characterized by increased cerebrospinal fluid (CSF) volume and/or intracranial pressure (ICP). If left untreated, CH can result in cognitive impairment for memory, attention, information processing, executive function, and visuospatial and visuoconstructural skills similar to subcortical type dementia (Donnet et al., 2004, Devito et al., 2005, Thomas et al., 2005),(Iddon et al., 1999, Klinge et al., 2002c, Klinge et al., 2002d, Klinge et al., 2005, Relkin et al., 2005). While the exact pathophysiology is not clearly understood, cognitive deficits observed in CH may be related to direct compression of brain tissue and blood vessels, fiber stretching, or reduced cerebral blood flow(Graff-Radford et al., 1987, Mamo et al., 1987, Larsson et al., 1994, Tanaka et al., 1997, Chang et al., 1999, Owler and Pickard, 2001, Klinge et al., 2002b, Klinge et al., 2002c, Mori et al., 2002, Mataro et al., 2003, Momjian et al., 2004, Owler et al., 2004b). Damage specifically to the prefrontal cortex, thalamus, hippocampal formation and the fiber pathways connecting them may be involved in CH- induced memory impairment. Due to their proximity to CSF ventricular spaces namely the frontal and inferior horns of the lateral ventricles and the third ventricle, these areas as part of the Papez circuit may be particularly vulnerable to ventriculomegaly. The hippocampus, specifically the CA1 region, is also known to be susceptible to ischemic insult as seen in various forms of cerebral ischemia and stroke. Thus, it may be possible to compared and distinguish the degree and specific pattern of ischemic injury in CH from other forms of neurologic diseases and cerebral ischemia.
Experimental evidence suggests that angiogenesis occurs in CH as an adaptive response to hypoxia(Fukuhara et al., 2001, Luciano et al., 2001). Angiogenesis is a complex and tightly regulated process that involves endothelial cell division, migration and proliferation in the formation of new blood vessels(Plate, 1999, Dvorak, 2005). It is known that VEGF, a heparin-binding homodimeric glycoprotein with optimal bioavailability and potency effects, is an angiogenic factor involved in the fetal and adult brain(Rosenstein et al., 1998, Veikkola and Alitalo, 1999, Zhang et al., 2000, Rosenstein and Krum, 2004, Dvorak, 2005). Studies reported that the specific tyrosine kinase receptor VEGFR-2+, also known as KDR/Flk-1, becomes expressed in response to ischemia and VEGF signaling(Neufeld et al., 1999, Ferrara and Gerber, 2001). As a result, there is a direct relationship between VEGF/ VEGFR-2+ expression and angiogenesis. However, it is not known whether the degree of hypoxia is directly related to the angiogenic response.
In this study, we employed an experimental model of chronic obstructive hydrocephalus previously developed and studied in our laboratory(Johnson et al., 1999, Fukuhara et al., 2001, Luciano et al., 2001, Dombrowski et al., 2006) to investigate the degree of hypoxia insult in the hippocampus after chronic hydrocephalus (CH) induction. Specifically, we used histological and immunocytochemical staining, and stereologic counting methods to estimate the density of VEGFR-2+ neurons, glial and endothelial cells, and blood vessels in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar region in CH and surgical control animals.
MATERIALS and METHODS
Animals
Sixteen (n=16) young adult, male hounds (canis familiaris) approximately 8–9 months of age, weighing 25–30kg were used in this study. Animals were divided into two groups: Chronic Hydrocephalus (CH, n=11) and Surgical Controls (SC, n=5). CH animals were further subdivided into Short Term (ST; 14–33 days) and Long Term (LT; 90–180 days) according to duration of condition. Animals were obtained from licensed suppliers and quarantined for a minimum of seven days before entering into the study. All animals were maintained in the Cleveland Clinic Foundation fully accredited Animal Care Facility under the rules and regulations of the Guide for the Care and Use of Laboratory Animals.
Surgical Induction of Chronic Hydrocephalus (CH)
The surgical procedure used to induce CH was originally developed in our laboratory(Johnson et al., 1999) and has been studied extensively(Fukuhara et al., 2001, Luciano et al., 2001, Dombrowski et al., 2006). Pre-surgical medications included Dilantin to prevent post-operative seizures, dexamethasone to reduce inflammation, glycopyrrolate to reduce respiratory secretions, and Gentamicin and Cefazolin to prevent infection. In brief, animals were induced with sodium pentothal, connected to a ventilator, and maintained under general gas anesthesia (1% isoflurane). Animals were then placed in a prone position in a stereotaxic headframe. Under sterile conditions, skin, fascia and muscles of the posterior neck were retracted along the midline from the external occipital pertuberance to the first cervical vertebrae. Next, a sub-occipital craniectomy was performed to allow visualization of the cerebellar vermis and brainstem. A small opening was made in the dura where a flexible, silicon catheter was inserted into the CSF fourth ventricle and cyanoacrylic gel (0.3–0.6mL) was injected. The catheter was then cut below the level of dura, and left in position. The dura, muscle, fascia and skin layers were sutured in a layered fashion.
Surgical control animals were generated using the same procedures as those described for experimental CH animals with the exception of saline injected into the fourth ventricle as substitute for cyanoacrylic gel.
Post-Operative Care
All animals (CH and SC) received Dexamethasone, Gentamicin and Cefazolin immediately post-operatively. The animals also received analgesics for the management of pain, and oral antibiotics to prevent infection. Routine post-operative care was provided for pain, hydration and infection, and included regular examination of vitals, pupillary reflex, and leg responsiveness. Dextrose feeding (i.v.) was initiated in two animals that prolonged food/water abstinence occurred. Medical treatment for increased intracranial pressure (ICP) included mannitol, acetazolamide, and decadron. Due to acute fluctuations in intracranial pressure and fluid retention, post-operative orders for i.v. fluids were limited to a rate of 25mL/hr until the animal is able to drink independently. Body temperature, respiratory rate, heart rate, urine production, activity, fluid intake, and IV fluid intake were recorded every hour.
MRI and Volumetric Analysis
Magnetic resonance images were collected prior to CH induction (i.e., baseline) and again at sacrifice for CH and SC groups. The baseline and sacrifice MRI were used to evaluate the anatomical severity and progression of hydrocephalus. Routine spin-echo magnetic resonance images were acquired using a 1.5 T Siemens Vision Magnetom and archived onto optical disk for subsequent volumetric analyses. Separate measures for brain and ventricular volume were obtained by manually tracing their contours on approximately 60–80 sections in the coronal plane of 1mm thickness from digital images using a commercially available image analysis system (Microbrightfield™, Colchester, VT). Though some reports have cautioned against the use of mongrel dogs for the purpose of hydrocephalus research because of the high incidence of spontaneous ventriculomegaly in mixed strains, baseline MRI was used to identify any animals with congenital hydrocephalus and reject them from the study.
Intraoperative Intracranial Pressure (ICP) Measurement
Intraoperative ICP measures were obtained for both CH and SC animals at baseline and prior to sacrifice as previously described(Johnson et al., 1999, Fukuhara et al., 2001, Luciano et al., 2001, Dombrowski et al., 2006). In brief, animals were placed in a prone position in a stereotaxic headframe, and a central arterial line was established for monitoring and control of hemodynamic conditions (PaO2, 95–100mmHg; PaCO2, 35–45mmHg; pH=7.4).
A midline incision was made along the scalp, enabling the retraction of the right temporalis muscle to expose the parietal bone posterior to the coronal suture. Using a small twist-drill, a 5mm frontal burr hole craniotomy was made above the dorsolateral frontal cortex of the right hemisphere. The dura was opened and an ICP microsensor probe (Camino, #110–4BT; Integra NeuroSciences) was inserted approximately 2mm below the cortical surface. Approximately 30–45 minutes was allowed for stabilization before ICP baseline measurements were obtained.
Histology / Immunocytochemistry
For sacrifice, animals were deeply anesthetized with sodium pentobarbital in combination with inhaled isoflurane and perfused via bilateral catheterization of the carotid arteries with 0.1M PBS followed by 4% paraformaldehyde in PBS. Brains were then removed, post-fixed for 24 hours in 4% PFA, and cryoprotective solution consisting of 30% sucrose for study of gross pathology, frozen sectioning, and routine histology (H&E, cresyl violet) and VEGFR-2+ immunohistochemistry.
Brains were then cut in serial coronal sections on a freezing microtone at 40um and subjected to immunocytochemical staining for VEGFR-2+ cellular expression and silver staining for blood vessels was performed. Creysl violet was used as a counterstain to identifying architectonic boundaries and normal cellular (neuronal and glial) characteristics including cell size and shape, and nuclear staining intensities. Neurons were relatively large with pale cytoplasm and a clearly identified nucleolus, while glia was usually smaller with intensely stained nucleus. No distinction was made between pyramidal cells and interneurons. All glia, including astrocytes, oligodendrocytes and microglia, were placed in one category. Endothelial cells were smaller in size compared to glial cells, and intensely stained nucleus and sickle shaped in transverse sections.
Immunocytochemical staining for VEGFR-2+ was performed on free floating sections, rinsed thoroughly in 0.1%M PBS, and pre-treatment with 3.0% H2O2 and 10% methanol in PBS to reduce staining from endogenous peroxidase. Tissue was then incubated in 3.0% normal rabbit serum with 0.3% Triton X100 for 60 minutes followed by an overnight incubation at 4°C with VEGFR-2+ (AF644, Flk-1/VEGFR-2, R&D Systems, Minneapolis, MN, U.S.A.) diluted at 1:50. Sections were rinsed and treated with a biotinylated anti-goat IgG at a dilution of 1:200. Tissue sections were then rinsed and treated for 60 minutes in avidin-biotin complex (ABC Elite Kit, PK-6105, Vector Labs, Burlingame, CA, U.S.A.) then treated with 3,3-diaminobenzidine tetrahydrochloride (DAB, Sigma) for approximately 30 seconds. Sections were rinsed, mounted on glass slides, and allowed to dry overnight. Sections were then counterstained using creysl violet, cover slipped in preparation for microscopic analysis and stereologic counting methods. Several sections were processed using immunoflourescent methods for the purpose of showing VEGFR-2 localization in neurons and glial cells using antibodies NeuN (monoclonal, mouse anti-neuronal nuclei, #MAB377, Chemicon/Millipore) and GFAP (polyclonal, anti-glial fibrillary acidic protein, #G9269 Sigma-Aldrich) and coverslipped using vectashield mounting medium with dapi (#H1200, Vector Labs).
Immunohistological control experiments were performed to distinguish specific VEGFR-2 staining (Fig. 1, 2A) from non-specific binding of the VEGFR-2 antibody through direct omission of either the primary antibody (Fig. 2B), or anti-goat IgG secondary (Fig. 2C).
Figure 1.

Photomicrographs showing VEGFR-2+ immunohistochemical expression in neurons (A), glial cells (B) and endothelial cells (C). Example of increased VEGFR-2+ expression in CH (D, F) compared to SC (E, G), and high magnification (insert) showing staining specifically in CA2/3 region. Scale bar equals 10μm.
Figure 2.
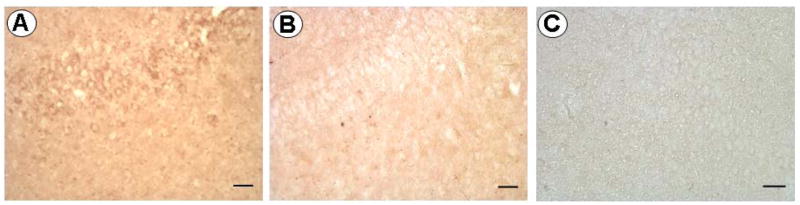
Photomicrographs showing immunohistochemical staining for VEGFR-2 (A) and in control experiments with the primary antibody omitted (B), and with secondary anti-goat IgG omitted (C) in order to discern non-specific staining. Results of control experiments reveal little if any non-specific staining.
Blood vessel staining
Tissue sections containing the ventral hippocampus were impregnated with silver (FD NeuroSilver Kit I, FD NeuroTechnologies, Inc., Baltimore, MD) to visualize changes in the vascular tree. After mounting, the tissue was dehydrated in absolute alcohol, cleared in xylene, and cover slipped using Cytoseal-60 mounting medium (Richard-Allan Scientific, Kalamazoo, MI).
Section and region sampling for stereologic counting
Starting from anterior to posterior, five randomly selected 40μm thick sections cut in the coronal plane were sampled from each animal. Stereologic counting methods were performed on a total of eighty tissue sections, five sections from each of eleven different animals (CH=11; SC=5). Counts were performed separately for VEGFR-2+ immunocytochemical cell staining and blood vessel silver staining. Stereologic procedures were performed by two independent investigators; inter- and intra-investigator reliability was >90%.
The anterior first-third of the ventral hippocampus was selected as the region of interest. This region was further subdivided into four distinct subfields: CA1, CA2/3 (amalgamation of CA2 and CA3), dentate cell layer, and hilar region. Cytoarchitectonic boundaries of these hippocampal subdivisions were identified in creysl violet stained sections and delineated according to the canine atlas of Dua-Sharma, S., Jacobs, H. L., & Sharma, K. N. (1970)(Sushil Dua-Sharma, 1970.) and a previous anatomical study(Salazar et al., 1990).
Stereological methods
Unbiased estimation of numerical density of neurons, glial cells and endothelial cells in the canine hippocampus were made by sampling the hippocampus in a systematic random manner with optical dissectors, an extension of the dissector principle. According to the principles of stereology, counts were obtained using approximately 25–30 optical dissectors (i.e., counting frames) with dimensions (75μm × 100μm × 40 μm) for each region of interest in each section, five sections per animal. Boundaries of the hippocampus and subdivisions were traced manually at low (2.5X) magnification under a light microscope with a camera attached to a computer with the aid imaging capture program. Cells were point counted by optical dissector at 100X magnification under oil immersion. Cellular density was estimated using a method adapted from previous studies(West, 1993, Dombrowski et al., 2001). Density of blood vessels in the canine hippocampus was performed using stereologic methods previously reported(Lokkegaard et al., 2001). Blood vessels were counted at 40X magnification from 40–45 randomly placed dissectors (75μm × 100μm × 40 μm) per region per section. Methods used to estimate blood vessel density did not distinguish between large (diameter >5μm) and small, i.e., capillary vessels. Quantitative differences in cellular density (cells/mm3) for VEGFR-2+ and blood vessel density were performed using a semi-automated stereological cell counting system (StereoInvestigator, v5.04.5; Microbrightfield; Williston, VT).
Statistical Analysis
The results of this study are expressed as mean ± the standard error of the mean for VEGFR-2+ and blood vessel density, total cell density, and percent (%) VEGFR-2+ density. Statistical comparisons were made across groups (CH-ST, CH-LT, and SC), hippocampal subdivisions (CA1, CA2/3, DG, and HL), and cell types (neurons, glial cells, and endothelial cells) using analysis of variance (ANOVA) followed by subsequent two-tailed unpaired t-tests. Correlations were made between blood vessel density, VEGFR-2+ cellular densities, and CSF volume and pressure. Statistical significance was accepted at the probability level less than 0.05.
RESULTS
In this study, we report the density and percent population of VEGFR-2+ neurons, glial and endothelial cells in CA1, CA2/3, dentate gyrus, and hilar regions of the canine hippocampus in both experimental and surgical control animals (Fig. 1A–C). We were able to clearly differentiate cell types and blood vessels based on morphological criteria and staining intensity (Figs. 1 and 4). Under microscopic visualization, CH animals appeared to have more VEGFR-2+ cells and blood vessels than surgical control animals (Figs. 1 and 4). Double labeling immunoflourescence investigation confirmed VEGFR-2 positive staining in neurons (Fig. 3A) and astrocytes (Fig. 3B). Quantitative stereologic counting methods to estimate neuronal, glial and endothelial cell and blood vessel densities confirm these differences.
Figure 4.
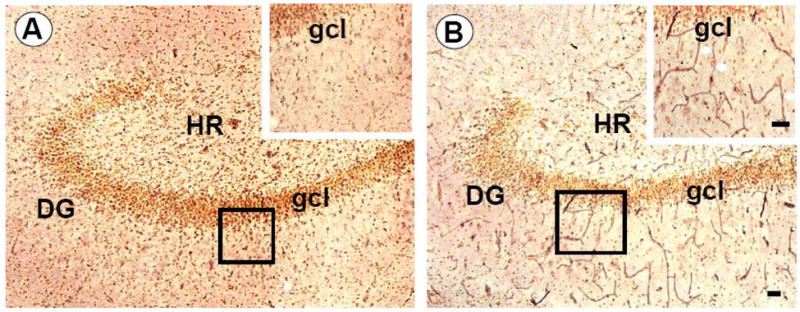
Photomicrographs showing differences in blood vessel density in control (A) and chronic hydrocephalus (B) canine hippocampus. Inserts illustrates increased BVd in CH compared to SC at high magnification silver staining in the dentate gyrus and granule cell layer. Scale bars equal 25μm.
Figure 3.
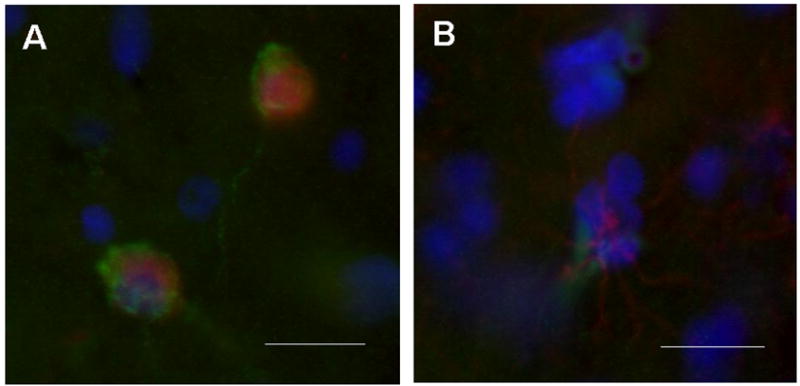
Photomicrographs showing immunoflourescent double labeling methods used to determine VEGFR-2 expression in neurons (A) and glial cells (B). VEGFR-2 staining (green) colocalizes with NeuN (red) for neuron identification in (A) and with GFAP (red) for astrocytes in (B). Sections counterstained with dapi (blue) for architectonic differentiation. These findings confirming that VEGFR-2 may be found in neurons as well as glial cells. Bars equal 10μm.
Confirmation of Chronic Hydrocephalus (CH)
Overall, baseline CSF total volume (0.22–1.9cc, mean 0.92cc) increased approximately 200% in CH animals and 14% in SC animals (Fig. 5). There was no significant difference in CSF volume between ST and LT CH groups. Baseline ICP, which ranged from 5.9–18.4 mmHg (mean 9.39mmHg), did not increase significantly in either the CH or SC groups, and there was no significant difference in ICP between groups.
Figure 5.
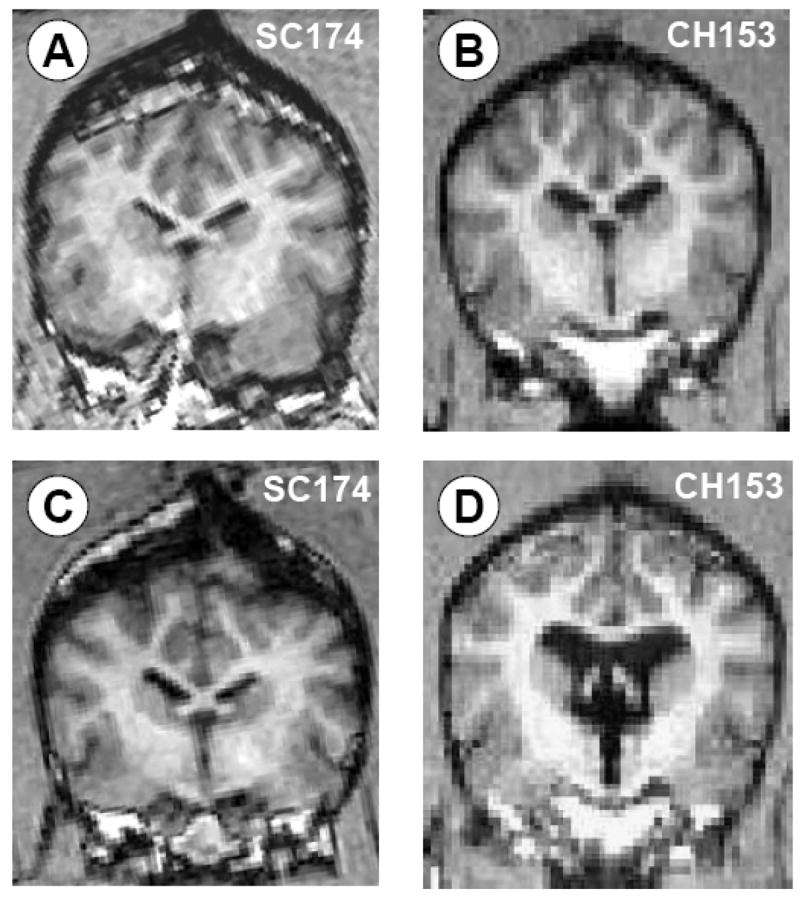
MRI confirmation of CSF ventricular volume change in SC (A,C) versus CH (B,D) animals. Overall, baseline CSF volume increased approximately 200% in CH animals compared with surgical controls as measured via 3D MRI volumetrics.
There was no incidence of mortality or morbidity in any animal for either the experimental or control groups. Recovered animals showed persistent ataxia, but were normally responsive and ambulatory. In CH-induced animals, gross morphological changes were observed including ventricular distension, sulcal widening, gyral flattening, cortical compression and distortion. Autopsy performed in each animal revealed no evidence of intracerebral or intraventricular bleeding, which corroborated with MRI data. Routine histological analysis showed no signs of intracerebral bleeding or anomalous pathological condition.
Density and Percentage (%) VEGFR-2+ Neurons, Glial and Endothelial Cells
Table 1 shows neuronal (Nd), glial (Gd) and endothelial cell (ECd) density and percentage (%) of VEGFR-2+ of neurons, glial and endothelial cells in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar region for CH (ST and LT) and SC groups. Overall, the density and percentage of VEGFR-2+ cells for CH animals was significantly higher than SC animals for all hippocampal areas investigated (Figs. 6 and 7). Specifically, VEGFR-2+ Nd and %N was approximately 3–6 times greater in the CH (ST and LT) groups (7,569±1,506 to 22,733±3,758 N/mm3; 50–81%) compared with the SC group (2,664±816 to 5,386±2015 N/mm3; 12–26%). Similarly, VEGFR-2+ Gd and %G was approximately 3–6 times greater in the CH (ST and LT) groups (5,885±1,460 to 21,454±6890 G/mm3; 13–64%) compared with the SC group (3,034±617 to 4,865±937 G/mm3; 5–9%). To a lesser extent, VEGFR-2+ ECd and %EC was 3–6 times greater in the CH (ST and LT) groups (2,237±414 to 5,197±1157 EC/mm3; 51–76%) compared with the SC group (963±193 to 1,287±284 EC/mm3; 10–18%). Statistically, there was no difference in VEGFR-2+ density or percentage between ST and LT hydrocephalus groups for any hippocampal region and/or cell type.
TABLE 1.
Vascular endothelial growth factor receptor 2 density (VEGFR-2+d) and percent (%) total population, and Blood Vessel density (BVd)
| VEGFR-2+d | (%) | VEGFR-2+d | (%) | VEGFR-2+d | (%) | |
|---|---|---|---|---|---|---|
| NEURONS | ||||||
| CH-ST | CH-LT | SC | ||||
| CA1 | 17581±3545 | (74±8.1) | 21557±4234 | (77±6.1) | 3580±1495 | (14±5.5) |
| CA2/3 | 20422±3422 | (80±3.1) | 20911±3107 | (81±2.5) | 5386±2015 | (26±7.7) |
| DG | 22733±3758 | (72±9.5) | 22630±5307 | (76±5.7) | 3875±1476 | (12±4.9) |
| Hilar | 7569±1506 | (50±9.5) | 10744±1597 | (59±9.9) | 2664±816 | (16±2.8) |
| GLIAL CELLS | ||||||
| CH-ST | CH-LT | SC | ||||
| CA1 | 14923±2898 | (62±12) | 16838±5003 | (59±10) | 4357±1315 | (9±4.2) |
| CA2/3 | 16903±2697 | (61±11) | 21454±6890 | (58±13) | 4865±937 | (7±2.2) |
| DG | 20798±2992 | (64±11) | 18335±4697 | (57±14) | 3819±1668 | (4±2.7) |
| Hilar | 5885±1460 | (13±1.1) | 5889±438 | (15±1) | 3034±617 | (5±1) |
| ENDOTHELIAL CELLS | ||||||
| CH-ST | CH-LT | SC | ||||
| CA1 | 4499±876 | (67±13) | 2237±414 | (68±15) | 972±301 | (12±5.8) |
| CA2/3 | 4238±644 | (76±12) | 3213±395 | (67±11) | 963±193 | (18±6.2) |
| DG | 3913±631 | (68±13) | 3622±455 | (62±15) | 1084±439 | (10±6.5) |
| Hilar | 5197±1157 | (54±7) | 3944±578 | (51±5.7) | 1287±284 | (15±2.5) |
| BLOOD VESSEL DENSITY | ||||||
| CH-ST | CH-LT | SC | ||||
| CA1 | 752±143 | 812±71 | 474±37 | |||
| CA2/3 | 735±120 | 765±39 | 483±44 | |||
| DG | 800±117 | 718±89 | 419±49 | |||
| Hilar | 1070±135 | 895±90 | 380±51 | |||
The density and percentage of VEGFR-2 neurons, glial and endothelial cells and blood vessel density in hippocampal regions CA1, CA2/3, dentate gyrus, and hilar regions in Chronic Hydrocephalus (Short Term, CH-ST; Long Term, CH-LT) and Surgical Control (SC) animals.
Figure 6.
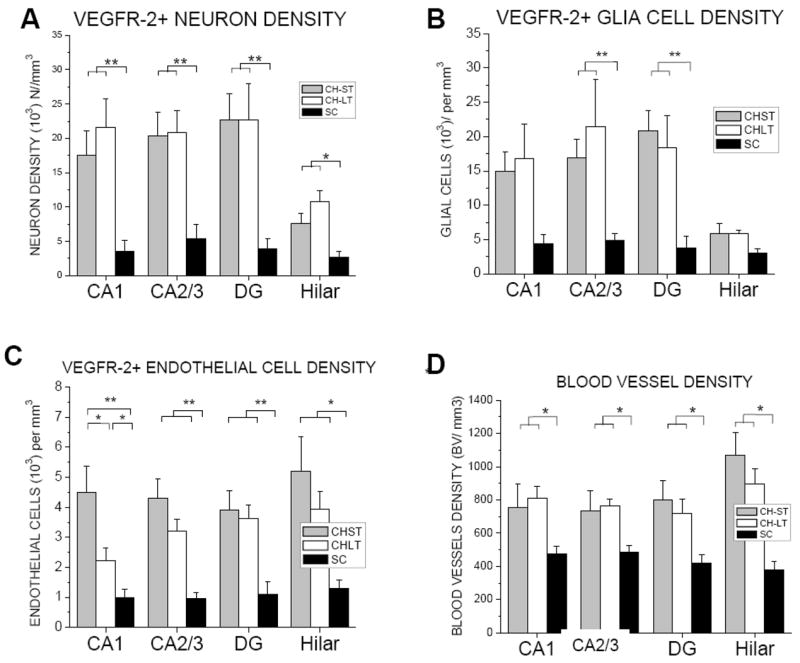
Density (cells per mm3) of VEGFR-2+ neurons (A), glial cells, (B) endothelial cells (C), and blood vessels (D) in CH (Short Term, ST; Long Term, LT) and SC in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar regions. * p≤0.05; **p≤0.01
Figure 7.
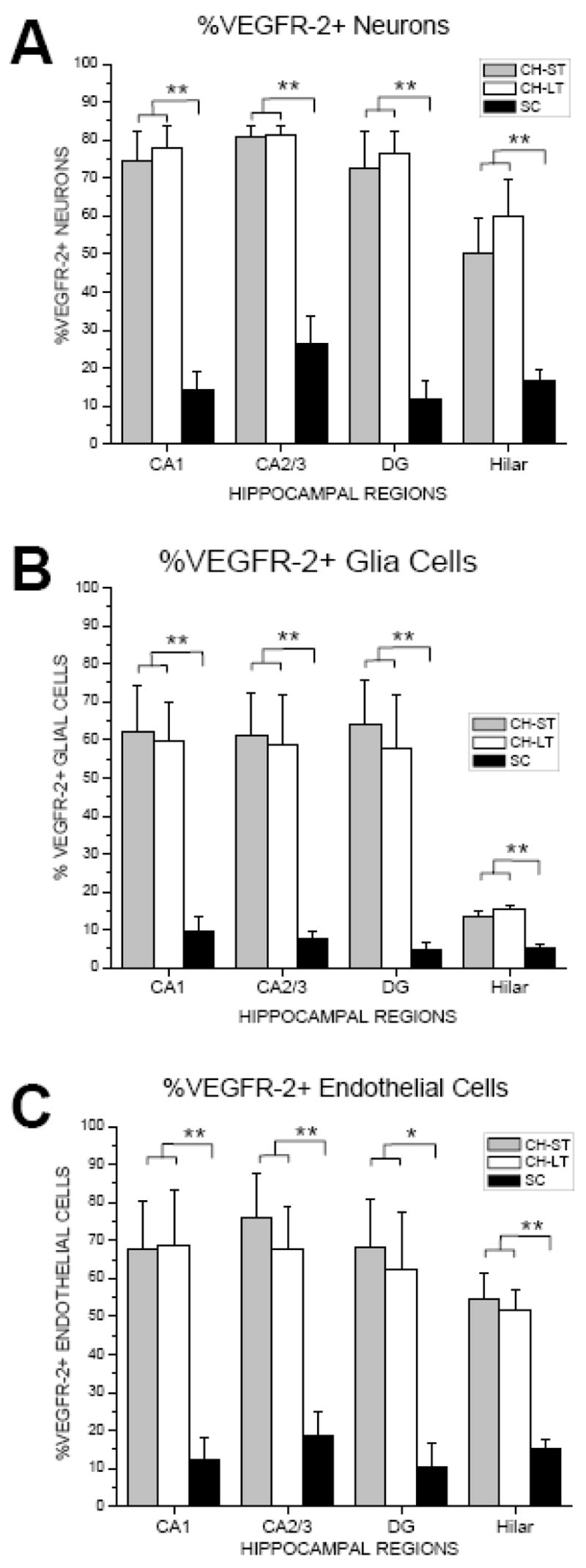
Bar graph illustrating percentage (%) VEGFR-2+ cells per total number of neurons (A), glial cells (B) and endothelial cells (C) in CH (ST, LT) and SC in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar regions.
* p≤0.05; **p≤0.01
Regionally, VEGFR-2+ density was similar between the four hippocampal regions investigated with the exception of Nd and Gd in the hilar region being were approximately 3 times lower than CA1, CA2/3 and DG in the CH-ST group (Table 1; Fig. 6; p≤0.01). By comparison, %VEGFR-2+ glial cells in the hilar region was approximately 5 times lower compared with CA1, CA2/3 and DG in both ST and LT CH groups (p≤0.001; p≤0.05, respectively).
While slight differences in the density and percentage of VEGFR-2+ between cell types were observed in control animals, larger differences were found in hydrocephalus animals (Table 1, Figs. 6 and 7). Specifically, the density of VEGFR-2+ neurons and glial cells, which were similar, were approximately 3-fold greater than EC density in all hippocampal areas with the exception of the hilar region where the opposite was noted (Table 1; Fig. 6). Comparatively, %VEGFR-2+ neurons was slightly greater than the percentage of glial and endothelial cell populations in all hippocampal areas with the exception of %VEGFR-2+ glial cells in the hilar region being approximately 4-fold less.
Blood Vessel density (BVd)
Table 1 and Figure 6D shows the density of blood vessels (BVd) for hippocampal areas CA1, CA2/3, dentate gyrus, and hilar region for CH (ST and LT) and SC groups. Overall, BVd in CH (ST and LT) animals (752 to 1070 BV/mm3) was approximately twice that of SC animals (380 to 483 BV/mm3) for all hippocampal areas investigated (p≤0.01). BVd was not significantly different between ST and LT CH groups. Regionally, there were no significant differences between hippocampal areas for either the CH (p=0.15) or SC (p=0.23) groups.
Relationship between VEGFR-2+, BVd and Hydrocephalus
Figure 8 shows the correlation between %VEGFR-2+ neurons, glial and endothelial cells and blood vessel density for hippocampal areas CA1, CA2/3, dentate gyrus, and hilar regions in hydrocephalus and control animals. As BVd increased in the hippocampus, the %VEGFR-2+ neurons (p=0.02, R=0.78) and endothelial cells (p=0.01, R=0.81) increased in the different groups.
Figure 8.
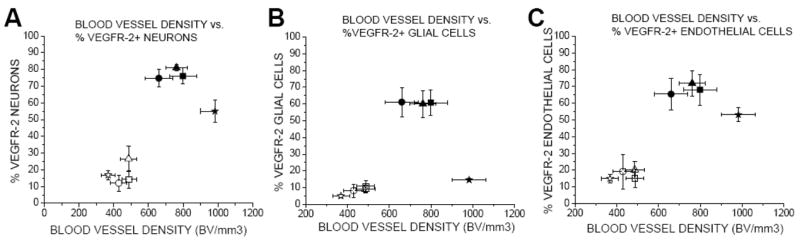
Two-dimensional box graph illustrating the relationship between %VEGFR-2+ and blood vessel density in CH (ST, LT) and SC in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar regions.
Change in CSF ventricular volume, and not ICP, was directly correlated with %VEGFR-2+ cellular expression and BVd in the hippocampus (Figs. 9 and 10). Specifically, when SC and CH data was combined, CSF ventricular volume change was significantly correlated with %VEGFR-2+ in CA1 (p=0.0001; R=0.65), CA2/3 (p=0.001; R=0.66) and DG (p=0.0004; R=0.63), but not in the hilar region. However, within the CH group alone, we found no correlation between %VEGFR-2+ and CSF volume change (Fig. 8). Finally, Figure 10 illustrates a significant correlation between BVd and CSF ventricular volume change in CA1 (p=0.002; R=0.77), CA2/3 (p=0.0002; R=0.85) and DG (p=0.001; R=0.78).
Figure 9.
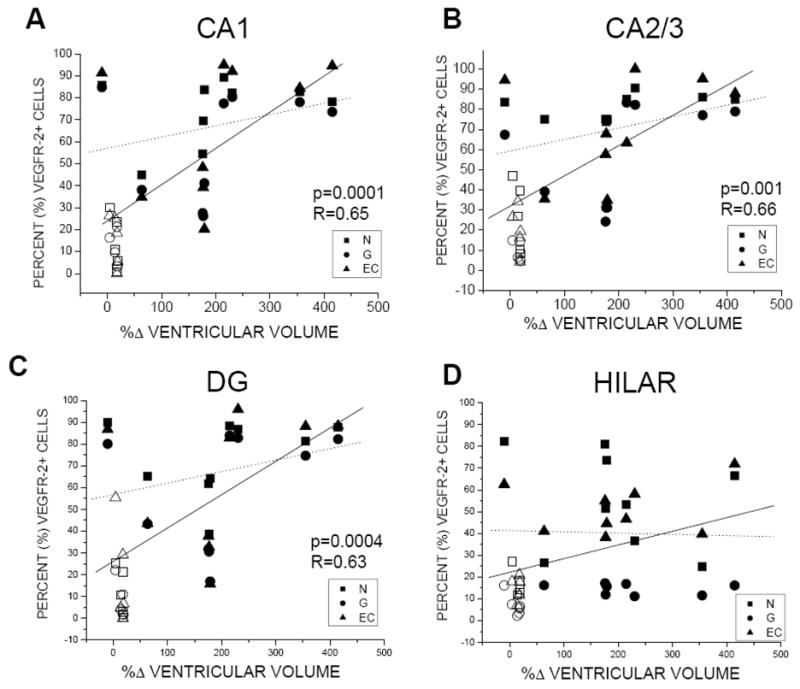
Relationship between %VEGFR-2+ and percent change (%Δ) CSF ventricular volume for CH and SC combined (solid line), and for CH only (dashed line) in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar region. Statistical significance (p≤0.001) was reported for CA1, CA2/3 and DG in CH + SC combined group (A,B,C).
Figure 10.
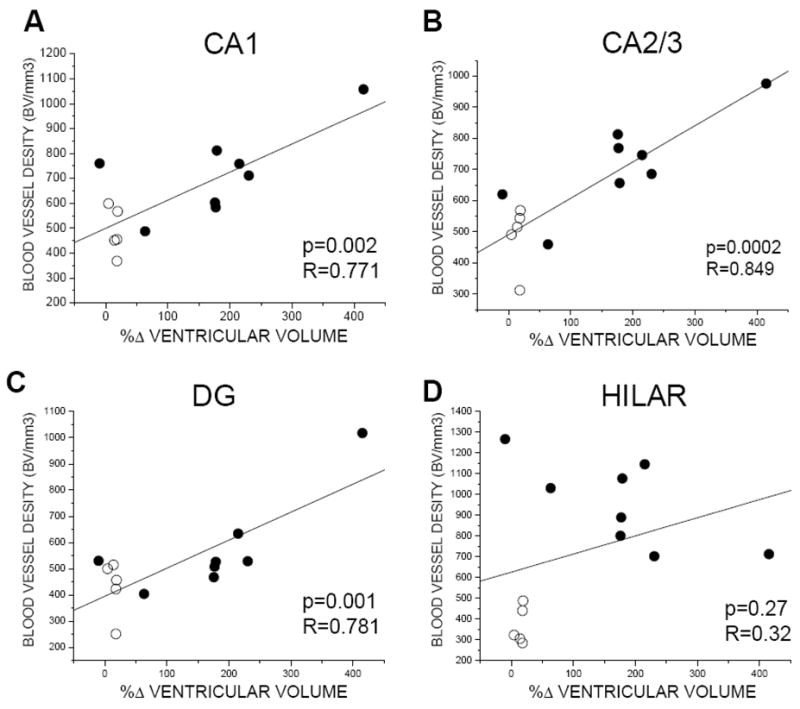
Relationship between BV density and CSF ventricular volume change (%Δ) for CH (ST, LT) and SC in hippocampal areas CA1, CA2/3, dentate gyrus, and hilar regions. Only when CH and SC groups were combined was there a significant correlation between ventriculomegaly and BV density.
DISCUSSION
This investigation found quantitative differences in the density of blood vessels and VEGFR-2+ + neurons, glia and endothelial cells in CA1, CA2/3, dentate gyrus, and hilar regions of the hippocampus in an experimental model of chronic obstructive hydrocephalus compared to surgical control animals. Overall, VEGFR-2+ expressions were similar across different regions and cell types, and were directly related to blood vessel density and ventriculomegaly. The current findings confirm and extend earlier studies from our laboratory showing blood vessel proliferation in occipital cortex and caudate nucleus(Luciano et al., 2001) that can be attributed to decreased cerebral blood flow(Dombrowski et al., 2006) and oxygen delivery(Fukuhara et al., 2001, Dombrowski et al., 2006) several weeks after the induction of hydrocephalus. Taken together, these findings support the idea that CH is a chronic ischemic condition which initiates an angiogenic response mediated through VEGFR-2.
Regional pattern of VEGFR-2 expression in hippocampus
We reported no significant regional differences in VEGFR-2+ and blood vessel density across hippocampal areas CA1, CA2/3 and dentate after hydrocephalus. Regional differences may have been expected based on findings from global ischemia and stroke models that showed a dissimilar spatial pattern of damage after ischemic insult, particularly in CA1 which seems to be most affected in low blood flow states.(1990, Hu et al., 2000a, Hu et al., 2000b, Sugino et al., 2000, Gu et al., 2001a, Gu et al., 2001b, Choi et al., 2006) In a rat model of kaolin-induced hydrocephalus, Klinge et al., 2003 showed a global increase in neuronal nitric oxide synthase, a selective marker for acute cerebral ischemia, in cerebral cortex and CA1 that corresponded to prolonged decreases in blood flow.(Klinge et al., 2003) It is possible that basic structural and functional differences in blood vessel anatomy in different hippocampal subfields may explain these findings.(Cervos-Navarro and Diemer, 1991, Patt et al., 1997)
In addition, the relative proximity of the hippocampal region to expanding CSF ventricular space may also influence VEGFR-2+ density. The medial temporal region, specifically the paraventricular areas of hippocampal cortex and CA1-3 may be susceptible to focal compression effects caused by ventriculomegaly. However, our results showed similar VEGFR-2+ and blood vessel densities across CA1, CA2/3, and dentate. The hilar region was the exception where the increase in neuronal and glial VEGFR-2+ density was not as profound as in the other regions, and where endothelial cell VEGFR-2+ density and blood vessel density was greater than CA1-3 and dentate. We showed that except for the hilar region, %VEGFR-2+ expression were directly related to ventriculomegaly. Thus, homogeneity in VEGFR-2+ expression may represent a suprathreshold stimulation of the hippocampal region, either through physical or hypoxic mechanisms, or both.
Specific VEGFR-2+ cell-type expression
Different hypoxia or ischemic conditions can induce different VEGF expression profiles and cellular localization (Cobbs et al., 1998) (Hayashi et al., 1997, Patt et al., 1997, Lennmyr et al., 1998, Patt et al., 1998, Kuo et al., 1999, Lee et al., 1999, Plate, 1999, Plate et al., 1999). For example, in rats sub-lethal chronic hypoxia conditions have been shown to elicit VEGF expression in astrocytes in vivo (Ogunshola et al., 2000) as well as in vitro (Ment et al., 1997, Rosenstein et al., 1998, Chow et al., 2001). By comparison, in a rat model of MCAO, transient hypoxia resulted in an early onset and rapid disappearance of VEGF mRNA expression in neurons(Hayashi et al., 1997). In clinical studies for stroke and tumor, and other chronic neurologic diseases such as Parkinson’s and Alzheimer’s disease the ischemic condition and VEGF expression is prolonged(Kalaria et al., 1998, Issa et al., 1999, Yasuhara et al., 2005). Regardless of pathology, under hypoxic conditions the normal temporal-spatial expression of VEGF-induced angiogenesis is disturbed that is usually maintained by neurons, and then controlled via glial-endothelial cell interactions. However, our findings showed relatively similar VEGFR-2+ expressions among different cell types in hippocampus after CH induction. Specifically, we reported the total proportion of VEGFR-2+ neurons (50–81%), glial cells (15–64%) and endothelial cells (51–75%) to be approximately 3–6 times greater in hydrocephalic animals compared with 10–15% in control animals. These results suggest that CH elicited an extraordinary high and nearly equivalent VEGFR-2+ response in neurons, glial and endothelial cell types. This may reflect differences in the degree and/or the duration of hypoxia, specifically as it relates to hydrocephalus insult. As shown here for CH, such a profound expression of VEGF in all cell types suggests a severe and extended hypoxic event. The degree or level of cellular response to hypoxia shown here for CH was similar to earlier studies in stroke, tumor, and cerebral ischemia which reported a significant increase in VEGF expression (Issa et al., 1999) (for reviews see (Rosenstein et al., 1998, Ferrara and Gerber, 2001, Rosenstein and Krum, 2004)). Though it is well known that VEGF plays a significant role in angiogenesis and neuroprotection, it is not evident from our results which cell types may be involved in adaptive or protective responses after CH-induced ischemic injury.
Early versus late stage chronic hydrocephalus
There is evidence from experimental studies that the expression of VEGF and VEGFR-2 is closely associated with onset and cessation of ischemia/hypoxia. In experimental stroke models, it has reported that VEGFR-2 expression increases as early as 6 hours after middle cerebral artery occlusion with peak activity observed after 5–7 days, and where VEGF expression returned to near normal levels after 3 weeks. We report similar VEGFR-2+ and blood vessel density expression at the early (2 week) and late (12+ week) post-CH induction time points. This suggests that the onset of VEGF expression most likely occurred earlier than two weeks and persisted longer than late stage (>12 weeks) hydrocephalus. This also suggests that adaptive processes involving VEGFR-2 induced angiogenesis may be active as early as two weeks. However, due to similar blood vessel density findings early versus late, it is unclear whether VEGFR-2 continues to have a role in blood vessel proliferation late (>12 weeks) in CH. Compared with MCAO stroke models, CH-induced VEGFR-2+ expression remained high even after 12+ weeks. These findings suggest that CH can result in a significant increase in VEGFR-2+ expression that is similar to permanent cerebral ischemia such as in stroke (Marti and Risau, 1998, Issa et al., 1999, Marti and Risau, 1999, Marti et al., 2000, Marti, 2005), and may correspond to an extended ischemic/hypoxic state.
VEGFR-2+ expression related to CH severity
Clinically the degree or severity of CH symptoms can vary from patient to patient and are often directly associated with ventriculomegaly and/or increased ICP. Since increased ICP may be transient, changes in CSF ventricular volume may be a better measure for hydrocephalus severity. Our results showed a direct relationship between %VEGFR-2+ and blood vessel density, and CSF ventricular enlargement, but not ICP. This finding most likely corresponds to the persistence of ventriculomegaly and transient ICP changes in our experimental model that mimics the same clinical condition. The exact mechanisms underlying these findings are unclear. First, albeit transient we cannot ignore possible damage sustained during increased ICP. Next, we cannot rule out local effects relating to focal compression or afferent/efferent fiber stretching to the hippocampal region that could contribute to ischemic damage observed. Finally, we must consider other direct (i.e., damage to brainstem cardio-respiratory nuclei) or indirect (i.e., neurohumoral) systemic factors that could affect cardiac function leading to reduced CBF and eventual global ischemia as described in an earlier study from our laboratory.(Dombrowski et al., 2006)
The role of VEGFR-2+ in CH: angiogenesis and neuroprotection
The increase in cellular VEGFR-2+ density and blood vessel density may not be coincidental. There is considerable evidence that VEGFR-2 play a significant role in angiogenesis. Consequently, we found increased VEGFR-2+ expression that corresponded directly to increased blood vessel density in hippocampus after CH-induced hypoxia. Thus, we confirm earlier reports that show a close relationship between VEGF/VEGFR-2 expression and angiogenesis. Newly formed blood vessels, however, have fewer and more immature endothelial cells that may have different structural and functional properties such as a permeable blood brain barrier(Rosenstein et al., 1998, Fischer et al., 2002, Schoch et al., 2002, Valable et al., 2005).
In addition, there is experimental evidence suggesting that VEGF plays an important role in neurodegeneration by impairing neural tissue perfusion (for review see (Storkebaum and Carmeliet, 2004, Storkebaum et al., 2004)), or is a critical factor in neurogenesis(Jin et al., 2002, Louissaint et al., 2002, Fabel et al., 2003), and/or neuroprotection (for review see (Storkebaum et al., 2004)). It is not known whether changes in VEGFR-2 density as shown here reflect protective or destructive mechanism in the hippocampus after CH induction. However, earlier studies reported axonal and neuropile degeneration but no cell loss in the hippocampus and cortex of hydrocephalus animal models(Ding et al., 2001, Del Bigio et al., 2003). Therefore, the increased expression of VEGFR-2+ we found may indicate certain adaptive mechanisms for recovery as seen in other forms of cerebral ischemia (Cobbs et al., 1998, Jin et al., 2000) (Hayashi et al., 1997) (Lennmyr et al., 1998) (Pichiule et al., 1999, Pichiule et al., 2003)
Alternative explanation for increased VEGFR-2 expression and angiogenesis
While previous work from our laboratory showing decreased cerebral blood flow (Dombrowski et al., 2006), decreased tissue and CSF oxygen saturation (Fukuhara et al., 2001), and specific changes in cerebrovascular adaptation (Luciano et al., 2001) support an argument for this model of chronic obstructive hydrocephalus being similar to other ischemic/hypoxic conditions including stroke, there still may be other explanations for increased VEGFR-2 and blood vessel density. Studies have shown increased VEGF and VEGFR-2 expression relating to direct cellular/axonal injury, chronic physiological stress, and, in part, after inflammatory response. Wang et al. (2005) showed that expressions of VEGF and VEGFR-2 in the hippocampus of ICR mice were upregulated after entorhinal deafferentation (Wang et al., 2005)(Wang, WY, 2005 Neuroscience). Heine et al. (2005) reported increased VEGF and VEGFR-2 expressions in GFAP positive astrocy(Hara and Okayasu, 2004)tes in the hilar region and to a lesser extent in the granule cell layer of the rat hippocampus after chronic stress (Heine et al., 2005)(Heine, VM 2005 Eur J Neuroscience). In addition several growth factors and pro-inflammatory mediators including TGF-alpha and beta, IGF-1, FGF, PDGF, and keratinocyte growth factor have been shown to be directly involved in the up regulation of VEGF mRNA (for review see Ferrara, 2004, 2005)(Ferrara, 2004, Ferrara, 2005). Croll et al. (2004) and later Kasselman et al. (2007), who demonstrated that inhibition of the inflammatory mediators significantly attenuated VEGF induced pathological angiogenesis (Kasselman et al, J Vas Res. 2007)(Croll et al., 2004, Kasselman et al., 2007). Gallo et al. (2002) reported that overexpression of COX-2 and iNOS may contribute to VEGF-induced angiogenesis head and neck squamous cell carcinomas(Gallo et al., 2002). Similarly, Hara et al. (2004) reported COX-2 expression significantly correlated with iNOS and VEGF modulation in human astrocytic gliomas(Hara and Okayasu, 2004). Many growth factors including FGF-1 and 2, HGF, PDGF, Ang-1 and 2, TGF-alpha and beta, IL-8, leptin and prostaglandins are also able to generate new blood vessels. TGF-alpha, HGF and FGF-2 act at least in part by regulating VEGF expression and others have secondary roles in vessel formation and differentiation(Dvorak, 2005). Thus, it is possible that increased VEGFR-2 expression and blood vessel density may be the result of several factors unrelated to hypoxia/ischemia. The fact that ventriculomegaly may result in cellular injury relating directly to white matter fiber stretch may be a plausible explanation. However, the current results showing equal distributions of both VEGFR-2 and blood vessel density across different hippocampal areas most likely reflects a global phenomena and not regional specific changes.
Causes of CH-induced hypoxia
Clinical and experimental studies have shown that chronic hydrocephalus is associated with decreased cerebral blood flow and oxygen delivery to the brain(Graff-Radford et al., 1987, Mamo et al., 1987, Larsson et al., 1994, Tanaka et al., 1997, Chang et al., 1999, Owler and Pickard, 2001, Klinge et al., 2002a, Klinge et al., 2002b, Mori et al., 2002, Mataro et al., 2003, Momjian et al., 2004, Owler et al., 2004a) (for review see Owler, 2001 (Owler and Pickard, 2001)). Experimental studies from our laboratory(Fukuhara et al., 2001, Luciano et al., 2001, Dombrowski et al., 2006) and others(da Silva et al., 1995, Richards et al., 1995, Klinge et al., 2003) report similar hypoxia or low blood flow conditions in experimental models of hydrocephalus. It is well known that VEGF is up regulated in hypoxic conditions where blood flow is reduced. Our results correspond showing an increase in VEGFR-2+ density in an experimental model of CH that most likely reflects the low CBF state in CH. Though we did not directly measure blood flow to the hippocampal region, earlier results from our laboratory suggest that global ischemia in CH may be a response to reduced CBF as described earlier(Dombrowski et al., 2006).
VEGF, hippocampus, and memory
Chronic hydrocephalus is often associated with memory impairment, specifically subcortical type dementia(Iddon et al., 1999, Klinge et al., 2002c, Klinge et al., 2002d, Klinge et al., 2005, Relkin et al., 2005). However, the exact pathophysiology underlying cognitive impairment in CH is not well understood. Damage specifically to the hippocampal formation and the fornix/fimbria pathways have been implicated in memory processes including consolidation, storage and retrieval. Due to their proximity to CSF ventricular spaces namely the frontal and inferior horns of the lateral ventricles and the third ventricle, these areas as part of the Papez circuit may be particularly vulnerable to ventriculomegaly. Findings from earlier studies reporting little, if any cell loss in the hippocampal region along with our results showing increased VEGFR-2+ expression in CH suggests that while the hippocampus may be spared, it may be functionally incapacitated. Studies have found that VEGF can act as a neurotrophic factor and promote neurogenic effects in brain(Jin et al., 2002, Zhu et al., 2003, Sun et al., 2006, Wang et al., 2007). Cao et al. (2004) reported that inhibition of VEGF expression completely blocked neurogenesis, while VEGF gene transfer in hippocampus resulted in neurogenesis and cognitive improvement, thus showing that hippocampal VEGF expression may respond to environmental stimuli. Therefore, it is possible that VEGF may play an important role in cellular adaptation but also plasticity in the hippocampus.
CONCLUSIONS
Overall, chronic hydrocephalus may be considered a chronic ischemic condition which initiates an angiogenic response mediated through VEGFR-2 (Fig. 11). Specifically, hydrocephalus can result in brain injury through increased CSF volume and/or pressure. As a result, a hypoxic response can occur through either decreased cerebral perfusion pressure caused by increased ICP, or via reduced cardiac output and cerebral blood flow relating to brain and blood vessel compression caused by ventriculomegaly. Hypoxia, in turn, activates VEGF and VEGFR-2 mechanisms in neurons, glial and endothelial cells that can be involved in adaptive processes including angiogenesis (Fig. 11).
Figure 11.
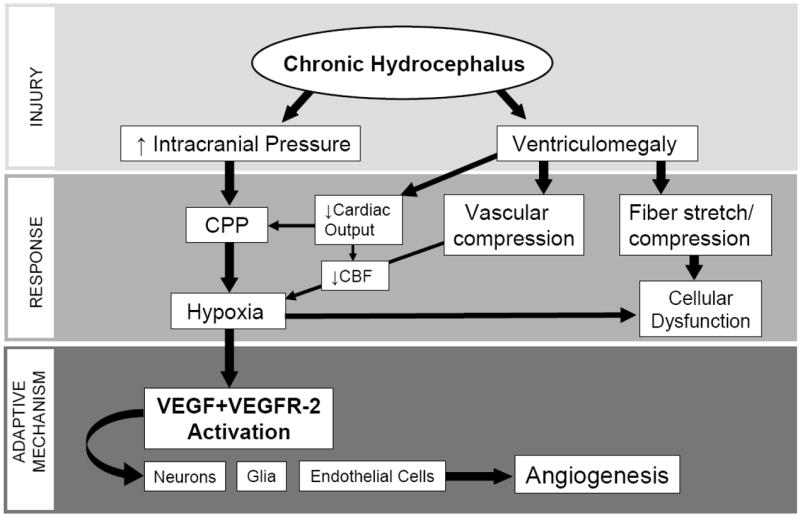
Flow diagram postulating the injury, response and adaptive mechanisms relating to CH. CH can result in brain injury through increased CSF volume and/or pressure. A hypoxic response can occur via increased ICP through decreased cerebral perfusion pressure or via ventriculomegaly through brain and blood vessel compression, decreased cerebral blood flow and cardiac output. Hypoxia directly activates VEGF and VEGFR-2 mechanisms in neurons, glial and endothelial cells that can be involved in adaptive processes including angiogenesis.
The findings of VEGFR-2 expression and BVd reported in this study suggest that: (1) chronic hydrocephalus may be considered a significant and prolonged ischemic condition; (2) different hippocampal (CA1, CA2/3, and DG) regions may be equally vulnerable to CH-induced ischemic injury; (3) the extent of neuronal, glial and endothelial cell VEGFR-2 response to hypoxia is similar, and may be unrelated to cell loss in the hippocampus; (4) increased BVd may be directly related to increased VEGFR-2 expression; (5) the exact role of VEGF activation is uncertain, but may be involved in an adaptive protective response to chronic hypoxia; (6) the relationship to ventriculomegaly suggests a specific “hydrocephalus effect” but the exact mechanism, i.e., local compression, remains uncertain. Finally, treatment options specifically those targeting VEGF activation may be considered as adjunct management for hydrocephalus and other cerebral ischemic conditions including stroke.
Acknowledgments
This work was supported by NIH-RO1 NS041553.
Abbreviations
- BV
Blood Vessel
- CH
Chronic Hydrocephalus
- CSF
Cerebrospinal Fluid
- DG
Dentate Gyrus
- ECd
Endothelial Cell Density
- ICP
Intracranial Pressure
- gc
granule cell layer
- Gd
Glial Cell Density
- HR
Hilar Region
- LT
Long Term
- Nd
Neuron Density
- SC
Surgical Control
- ST
Short Term
- VEGF
Vascular Endothelial Growth Factor
- VEGFR-2
Vascular Endothelial Growth Factor Receptor 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Crit Rev Neurobiol. 1991;6:149–182. [PubMed] [Google Scholar]
- Chang CC, Kuwana N, Noji M, Tanabe Y, Koike Y, Ikegami T. Cerebral blood flow in patients with normal pressure hydrocephalus. NuclMedCommun. 1999;20:167–169. doi: 10.1097/00006231-199902000-00009. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim HY, Cha JH, Lee MY. Ischemic preconditioning-induced activation of ERK1/2 in the rat hippocampus. NeurosciLett. 2006;409:187–191. doi: 10.1016/j.neulet.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Chow J, Ogunshola O, Fan SY, Li Y, Ment LR, Madri JA. Astrocyte-derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Brain Res Dev Brain Res. 2001;130:123–132. doi: 10.1016/s0165-3806(01)00220-6. [DOI] [PubMed] [Google Scholar]
- Cobbs CS, Chen J, Greenberg DA, Graham SH. Vascular endothelial growth factor expression in transient focal cerebral ischemia in the rat. Neurosci Lett. 1998;249:79–82. doi: 10.1016/s0304-3940(98)00377-2. [DOI] [PubMed] [Google Scholar]
- Croll SD, Ransohoff RM, Cai N, Zhang Q, Martin FJ, Wei T, Kasselman LJ, Kintner J, Murphy AJ, Yancopoulos GD, Wiegand SJ. VEGF-mediated inflammation precedes angiogenesis in adult brain. Exp Neurol. 2004;187:388–402. doi: 10.1016/j.expneurol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- da Silva MC, Michowicz S, Drake JM, Chumas PD, Tuor UI. Reduced local cerebral blood flow in periventricular white matter in experimental neonatal hydrocephalus-restoration with CSF shunting. JCerebBloodFlowMetab. 1995;15:1057–1065. doi: 10.1038/jcbfm.1995.132. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Wilson MJ, Enno T. Chronic hydrocephalus in rats and humans: white matter loss and behavior changes. Ann Neurol. 2003;53:337–346. doi: 10.1002/ana.10453. [DOI] [PubMed] [Google Scholar]
- Devito EE, Pickard JD, Salmond CH, Iddon JL, Loveday C, Sahakian BJ. The neuropsychology of normal pressure hydrocephalus (NPH) Br J Neurosurg. 2005;19:217–224. doi: 10.1080/02688690500201838. [DOI] [PubMed] [Google Scholar]
- Ding Y, McAllister JP, Yao B, Yan N, Canady AI. Neuron tolerance during hydrocephalus. Neuroscience. 2001;106:659–667. doi: 10.1016/s0306-4522(01)00166-x. [DOI] [PubMed] [Google Scholar]
- Dombrowski SM, Hilgetag CC, Barbas H. Quantitative architecture distinguishes prefrontal cortical systems in the rhesus monkey. CerebCortex. 2001;11:975–988. doi: 10.1093/cercor/11.10.975. [DOI] [PubMed] [Google Scholar]
- Dombrowski SM, Schenk S, Leichliter A, Leibson Z, Fukamachi K, Luciano MG. Chronic hydrocephalus-induced changes in cerebral blood flow: mediation through cardiac effects. JCerebBloodFlowMetab. 2006 doi: 10.1038/sj.jcbfm.9600282. [DOI] [PubMed] [Google Scholar]
- Donnet A, Schmitt A, Dufour H, Giorgi R, Grisoli F. Differential patterns of cognitive impairment in patients with aqueductal stenosis and normal pressure hydrocephalus. Acta Neurochir (Wien) 2004;146:1301–1308. doi: 10.1007/s00701-004-0384-3. discussion 1308. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Angiogenesis: update 2005. JThrombHaemost. 2005;3:1835–1842. doi: 10.1111/j.1538-7836.2005.01361.x. [DOI] [PubMed] [Google Scholar]
- Fabel K, Fabel K, Tam B, Kaufer D, Baiker A, Simmons N, Kuo CJ, Palmer TD. VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur J Neurosci. 2003;18:2803–2812. doi: 10.1111/j.1460-9568.2003.03041.x. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. Exs. 2005:209–231. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP. The role of vascular endothelial growth factor in angiogenesis. ActaHaematol. 2001;106:148–156. doi: 10.1159/000046610. [DOI] [PubMed] [Google Scholar]
- Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63:70–80. doi: 10.1006/mvre.2001.2367. [DOI] [PubMed] [Google Scholar]
- Fukuhara T, Luciano MG, Brant CL, Klauscie J. Effects of ventriculoperitoneal shunt removal on cerebral oxygenation and brain compliance in chronic obstructive hydrocephalus. JNeurosurg. 2001;94:573–581. doi: 10.3171/jns.2001.94.4.0573. [DOI] [PubMed] [Google Scholar]
- Gallo O, Masini E, Bianchi B, Bruschini L, Paglierani M, Franchi A. Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Hum Pathol. 2002;33:708–714. doi: 10.1053/hupa.2002.125376. [DOI] [PubMed] [Google Scholar]
- Graff-Radford NR, Rezai K, Godersky JC, Eslinger P, Damasio H, Kirchner PT. Regional cerebral blood flow in normal pressure hydrocephalus. JNeurolNeurosurg Psychiatry. 1987;50:1589–1596. doi: 10.1136/jnnp.50.12.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Zhang G. Extracellular signal-regulated kinase 1/2 activation in hippocampus after cerebral ischemia may not interfere with postischemic cell death. Brain Res. 2001a;901:79–84. doi: 10.1016/s0006-8993(01)02275-2. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Zhang G. Extracellular signal-regulated kinase and c-Jun N-terminal protein kinase in ischemic tolerance. Neuroreport. 2001b;12:3487–3491. doi: 10.1097/00001756-200111160-00023. [DOI] [PubMed] [Google Scholar]
- Hara A, Okayasu I. Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: correlation with angiogenesis and prognostic significance. Acta Neuropathol (Berl) 2004;108:43–48. doi: 10.1007/s00401-004-0860-0. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. doi: 10.1161/01.str.28.10.2039. [DOI] [PubMed] [Google Scholar]
- Heine VM, Zareno J, Maslam S, Joels M, Lucassen PJ. Chronic stress in the adult dentate gyrus reduces cell proliferation near the vasculature and VEGF and Flk-1 protein expression. Eur J Neurosci. 2005;21:1304–1314. doi: 10.1111/j.1460-9568.2005.03951.x. [DOI] [PubMed] [Google Scholar]
- Hu BR, Liu CL, Park DJ. Alteration of MAP kinase pathways after transient forebrain ischemia. J Cereb Blood Flow Metab. 2000a;20:1089–1095. doi: 10.1097/00004647-200007000-00008. [DOI] [PubMed] [Google Scholar]
- Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000b;20:3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iddon JL, Pickard JD, Cross JJ, Griffiths PD, Czosnyka M, Sahakian BJ. Specific patterns of cognitive impairment in patients with idiopathic normal pressure hydrocephalus and Alzheimer’s disease: a pilot study. JNeurolNeurosurg Psychiatry. 1999;67:723–732. doi: 10.1136/jnnp.67.6.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa R, Krupinski J, Bujny T, Kumar S, Kaluza J, Kumar P. Vascular endothelial growth factor and its receptor, KDR, in human brain tissue after ischemic stroke. LabInvest. 1999;79:417–425. [PubMed] [Google Scholar]
- Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience. 2000;99:577–585. doi: 10.1016/s0306-4522(00)00207-4. [DOI] [PubMed] [Google Scholar]
- Johnson MJ, Ayzman I, Wood AS, Tkach JA, Klauschie J, Skarupa DJ, McAllister JP, Luciano MG. Development and characterization of an adult model of obstructive hydrocephalus. JNeurosciMethods. 1999;91:55–65. doi: 10.1016/s0165-0270(99)00072-2. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain ResMolBrain Res. 1998;62:101–105. doi: 10.1016/s0169-328x(98)00190-9. [DOI] [PubMed] [Google Scholar]
- Kasselman LJ, Kintner J, Sideris A, Pasnikowski E, Krellman JW, Shah S, Rudge JS, Yancopoulos GD, Wiegand SJ, Croll SD. Dexamethasone treatment and ICAM-1 deficiency impair VEGF-induced angiogenesis in adult brain. J Vasc Res. 2007;44:283–291. doi: 10.1159/000101450. [DOI] [PubMed] [Google Scholar]
- Klinge P, Berding G, Brinker T, Schuhmann M, Weckesser E, Knapp WH, Samii M. The role of cerebral blood flow and cerebrovascular reserve capacity in the diagnosis of chronic hydrocephalus--a PET-study on 60 patients. ActaNeurochirSuppl. 2002a;81:39–41. 39–41. doi: 10.1007/978-3-7091-6738-0_10. [DOI] [PubMed] [Google Scholar]
- Klinge P, Berding G, Brinker T, Weckesser E, Knapp WH, Samii M. Regional cerebral blood flow profiles of shunt-responder in idiopathic chronic hydrocephalus--a 15-O-water PET-study. ActaNeurochirSuppl. 2002b;81:47–9. 47–49. doi: 10.1007/978-3-7091-6738-0_12. [DOI] [PubMed] [Google Scholar]
- Klinge P, Marmarou A, Bergsneider M, Relkin N, Black PM. Outcome of shunting in idiopathic normal-pressure hydrocephalus and the value of outcome assessment in shunted patients. Neurosurgery. 2005;57:S40–52. doi: 10.1227/01.neu.0000168187.01077.2f. [DOI] [PubMed] [Google Scholar]
- Klinge P, Ruckert N, Schuhmann M, Berding G, Brinker T, Knapp WH, Samii M. Neuropsychological sequels to changes in global cerebral blood flow and cerebrovascular reserve capacity after shunt treatment in chronic hydrocephalus--a quantitative PET-study. ActaNeurochirSuppl. 2002c;81:55–7. 55–57. doi: 10.1007/978-3-7091-6738-0_14. [DOI] [PubMed] [Google Scholar]
- Klinge P, Ruckert N, Schuhmann M, Dorner L, Brinker T, Samii M. Neuropsychological testing to improve surgical management of patients with chronic hydrocephalus after shunt treatment. ActaNeurochirSuppl. 2002d;81:51–3. 51–53. doi: 10.1007/978-3-7091-6738-0_13. [DOI] [PubMed] [Google Scholar]
- Klinge PM, Samii A, Muhlendyck A, Visnyei K, Meyer GJ, Walter GF, Silverberg GD, Brinker T. Cerebral hypoperfusion and delayed hippocampal response after induction of adult kaolin hydrocephalus. Stroke. 2003;34:193–199. doi: 10.1161/01.str.0000048820.17198.15. [DOI] [PubMed] [Google Scholar]
- Kuo NT, Benhayon D, Przybylski RJ, Martin RJ, LaManna JC. Prolonged hypoxia increases vascular endothelial growth factor mRNA and protein in adult mouse brain. J Appl Physiol. 1999;86:260–264. doi: 10.1152/jappl.1999.86.1.260. [DOI] [PubMed] [Google Scholar]
- Larsson A, Bergh AC, Bilting M, Arlig A, Jacobsson L, Stephensen H, Wikkelso C. Regional cerebral blood flow in normal pressure hydrocephalus: diagnostic and prognostic aspects. EurJNuclMed. 1994;21:118–123. doi: 10.1007/BF00175758. [DOI] [PubMed] [Google Scholar]
- Lee MY, Ju WK, Cha JH, Son BC, Chun MH, Kang JK, Park CK. Expression of vascular endothelial growth factor mRNA following transient forebrain ischemia in rats. Neurosci Lett. 1999;265:107–110. doi: 10.1016/s0304-3940(99)00219-0. [DOI] [PubMed] [Google Scholar]
- Lennmyr F, Ata KA, Funa K, Olsson Y, Terent A. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–882. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- Lokkegaard A, Nyengaard JR, West MJ. Stereological estimates of number and length of capillaries in subdivisions of the human hippocampal region. Hippocampus. 2001;11:726–740. doi: 10.1002/hipo.1088. [DOI] [PubMed] [Google Scholar]
- Louissaint A, Jr, Rao S, Leventhal C, Goldman SA. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron. 2002;34:945–960. doi: 10.1016/s0896-6273(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Luciano MG, Skarupa DJ, Booth AM, Wood AS, Brant CL, Gdowski MJ. Cerebrovascular adaptation in chronic hydrocephalus. JCerebBlood Flow Metab. 2001;21:285–294. doi: 10.1097/00004647-200103000-00012. [DOI] [PubMed] [Google Scholar]
- Mamo HL, Meric PC, Ponsin JC, Rey AC, Luft AG, Seylaz JA. Cerebral blood flow in normal pressure hydrocephalus. Stroke. 1987;18:1074–1080. doi: 10.1161/01.str.18.6.1074. [DOI] [PubMed] [Google Scholar]
- Marti HH. Angiogenesis--a self-adapting principle in hypoxia. Exs. 2005:163–180. doi: 10.1007/3-7643-7311-3_12. [DOI] [PubMed] [Google Scholar]
- Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci U S A. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti HH, Risau W. Angiogenesis in ischemic disease. Thromb Haemost. 1999;82(Suppl 1):44–52. [PubMed] [Google Scholar]
- Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. doi: 10.1016/S0002-9440(10)64964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataro M, Poca MA, Salgado-Pineda P, Castell-Conesa J, Sahuquillo J, Diez-Castro MJ, Aguade-Bruix S, Vendrell P, del Mar MM, Junque C. Postsurgical cerebral perfusion changes in idiopathic normal pressure hydrocephalus: a statistical parametric mapping study of SPECT images. JNuclMed. 2003;44:1884–1889. [PubMed] [Google Scholar]
- Ment LR, Stewart WB, Fronc R, Seashore C, Mahooti S, Scaramuzzino D, Madri JA. Vascular endothelial growth factor mediates reactive angiogenesis in the postnatal developing brain. Brain Res Dev Brain Res. 1997;100:52–61. doi: 10.1016/s0165-3806(97)00012-6. [DOI] [PubMed] [Google Scholar]
- Momjian S, Owler BK, Czosnyka Z, Czosnyka M, Pena A, Pickard JD. Pattern of white matter regional cerebral blood flow and autoregulation in normal pressure hydrocephalus. Brain. 2004;127:965–972. doi: 10.1093/brain/awh131. [DOI] [PubMed] [Google Scholar]
- Mori K, Maeda M, Asegawa S, Iwata J. Quantitative local cerebral blood flow change after cerebrospinal fluid removal in patients with normal pressure hydrocephalus measured by a double injection method with N-isopropyl-p-[(123)I] iodoamphetamine. ActaNeurochir(Wien) 2002;144:255–262. doi: 10.1007/s007010200033. [DOI] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. Faseb J. 1999;13:9–22. [PubMed] [Google Scholar]
- Ogunshola OO, Stewart WB, Mihalcik V, Solli T, Madri JA, Ment LR. Neuronal VEGF expression correlates with angiogenesis in postnatal developing rat brain. Brain Res Dev Brain Res. 2000;119:139–153. doi: 10.1016/s0165-3806(99)00125-x. [DOI] [PubMed] [Google Scholar]
- Owler BK, Momjian S, Czosnyka Z, Czosnyka M, Pena A, Harris NG, Smielewski P, Fryer T, Donovan T, Coles J, Carpenter A, Pickard JD. Normal pressure hydrocephalus and cerebral blood flow: a PET study of baseline values. JCerebBloodFlowMetab. 2004a;24:17–23. doi: 10.1097/01.WCB.0000093326.88757.49. [DOI] [PubMed] [Google Scholar]
- Owler BK, Pena A, Momjian S, Czosnyka Z, Czosnyka M, Harris NG, Smielewski P, Fryer T, Donvan T, Carpenter A, Pickard JD. Changes in cerebral blood flow during cerebrospinal fluid pressure manipulation in patients with normal pressure hydrocephalus: a methodological study. JCerebBloodFlowMetab. 2004b;24:579–587. doi: 10.1097/00004647-200405000-00012. [DOI] [PubMed] [Google Scholar]
- Owler BK, Pickard JD. Normal pressure hydrocephalus and cerebral blood flow: a review. Acta NeurolScand. 2001;104:325–342. doi: 10.1034/j.1600-0404.2001.00092.x. [DOI] [PubMed] [Google Scholar]
- Patt S, Danner S, Theallier-Janko A, Breier G, Hottenrott G, Plate KH, Cervos-Navarro J. Upregulation of vascular endothelial growth factor in severe chronic brain hypoxia of the rat. Neurosci Lett. 1998;252:199–202. doi: 10.1016/s0304-3940(98)00582-5. [DOI] [PubMed] [Google Scholar]
- Patt S, Sampaolo S, Theallier-Janko A, Tschairkin I, Cervos-Navarro J. Cerebral angiogenesis triggered by severe chronic hypoxia displays regional differences. J Cereb Blood Flow Metab. 1997;17:801–806. doi: 10.1097/00004647-199707000-00010. [DOI] [PubMed] [Google Scholar]
- Pichiule P, Agani F, Chavez JC, Xu K, LaManna JC. HIF-1 alpha and VEGF expression after transient global cerebral ischemia. Adv Exp Med Biol. 2003;530:611–617. doi: 10.1007/978-1-4615-0075-9_60. [DOI] [PubMed] [Google Scholar]
- Pichiule P, Chavez JC, Xu K, LaManna JC. Vascular endothelial growth factor upregulation in transient global ischemia induced by cardiac arrest and resuscitation in rat brain. Brain Res Mol Brain Res. 1999;74:83–90. doi: 10.1016/s0169-328x(99)00261-2. [DOI] [PubMed] [Google Scholar]
- Plate KH. Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol. 1999;58:313–320. doi: 10.1097/00005072-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Plate KH, Beck H, Danner S, Allegrini PR, Wiessner C. Cell type specific upregulation of vascular endothelial growth factor in an MCA-occlusion model of cerebral infarct. J Neuropathol Exp Neurol. 1999;58:654–666. doi: 10.1097/00005072-199906000-00010. [DOI] [PubMed] [Google Scholar]
- Relkin N, Marmarou A, Klinge P, Bergsneider M, Black PM. Diagnosing idiopathic normal-pressure hydrocephalus. Neurosurgery. 2005;57:S4–16. doi: 10.1227/01.neu.0000168185.29659.c5. discussion ii–v. [DOI] [PubMed] [Google Scholar]
- Richards HK, Bucknall RM, Jones HC, Pickard JD. Uncoupling of LCBF and LCGU in two different models of hydrocephalus: a review. ChildsNervSyst. 1995;11:288–292. doi: 10.1007/BF00301762. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Krum JM. New roles for VEGF in nervous tissue--beyond blood vessels. Exp Neurol. 2004;187:246–253. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Rosenstein JM, Mani N, Silverman WF, Krum JM. Patterns of brain angiogenesis after vascular endothelial growth factor administration in vitro and in vivo. Proc Natl Acad Sci U S A. 1998;95:7086–7091. doi: 10.1073/pnas.95.12.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar I, Fernandez Troconiz P, Cifuentes JM, Ruiz Pesini P. Macroscopical and microscopical anatomy of the hippocampus in the dog. Biol Struct Morphog. 1990;3:45–52. [PubMed] [Google Scholar]
- Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125:2549–2557. doi: 10.1093/brain/awf257. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Carmeliet P. VEGF: a critical player in neurodegeneration. J Clin Invest. 2004;113:14–18. doi: 10.1172/JCI200420682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26:943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- Sugino T, Nozaki K, Hashimoto N. Activation of mitogen-activated protein kinases in gerbil hippocampus with ischemic tolerance induced by 3-nitropropionic acid. Neurosci Lett. 2000;278:101–104. doi: 10.1016/s0304-3940(99)00906-4. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA. Vascular endothelial growth factor-B (VEGFB) stimulates neurogenesis: evidence from knockout mice and growth factor administration. Dev Biol. 2006;289:329–335. doi: 10.1016/j.ydbio.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Sushil Dua-Sharma KNS, Jacobs Harry L. The Canine Brain in Stereotaxic Coordinates: Full Sections in Frontal, Sagittal and Horizontal Planes. MIT Press; Cambridge, MA: 1970. [Google Scholar]
- Tanaka A, Kimura M, Nakayama Y, Yoshinaga S, Tomonaga M. Cerebral blood flow and autoregulation in normal pressure hydrocephalus. Neurosurgery. 1997;40:1161–1165. doi: 10.1097/00006123-199706000-00009. [DOI] [PubMed] [Google Scholar]
- Thomas G, McGirt MJ, Woodworth G, Heidler J, Rigamonti D, Hillis AE, Williams MA. Baseline neuropsychological profile and cognitive response to cerebrospinal fluid shunting for idiopathic normal pressure hydrocephalus. Dement Geriatr Cogn Disord. 2005;20:163–168. doi: 10.1159/000087092. [DOI] [PubMed] [Google Scholar]
- Valable S, Montaner J, Bellail A, Berezowski V, Brillault J, Cecchelli R, Divoux D, Mackenzie ET, Bernaudin M, Roussel S, Petit E. VEGF-induced BBB permeability is associated with an MMP-9 activity increase in cerebral ischemia: both effects decreased by Ang-1. J Cereb Blood Flow Metab. 2005;25:1491–1504. doi: 10.1038/sj.jcbfm.9600148. [DOI] [PubMed] [Google Scholar]
- Veikkola T, Alitalo K. VEGFs, receptors and angiogenesis. Semin Cancer Biol. 1999;9:211–220. doi: 10.1006/scbi.1998.0091. [DOI] [PubMed] [Google Scholar]
- Wang WY, Dong JH, Liu X, Wang Y, Ying GX, Ni ZM, Zhou CF. Vascular endothelial growth factor and its receptor Flk-1 are expressed in the hippocampus following entorhinal deafferentation. Neuroscience. 2005;134:1167–1178. doi: 10.1016/j.neuroscience.2005.04.064. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007;27:304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. NeurobiolAging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Shingo T, Muraoka K, Kameda M, Agari T, Wen Ji Y, Hayase H, Hamada H, Borlongan CV, Date I. Neurorescue effects of VEGF on a rat model of Parkinson’s disease. Brain Res. 2005;1053:10–18. doi: 10.1016/j.brainres.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Jin K, Mao XO, Greenberg DA. Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. Faseb J. 2003;17:186–193. doi: 10.1096/fj.02-0515com. [DOI] [PubMed] [Google Scholar]