

Abstract
DBA/2 mice are much more susceptible to infection with Mycobacterium tuberculosis than major histocompatibility complex-compatible BALB/c mice. It is shown here that, although vaccination provided mice of both strains with a capacity to reduce the level of infection in their lungs, vaccinated DBA/2 mice remained much more susceptible in this organ than vaccinated BALB/c mice. Consequently, the former mice developed more lung pathology and died much earlier than the latter. On the other hand, colony-forming unit counts and histology suggest that vaccination provided mice of both strains with an increased and equal ability to express immunity in the liver and spleen, thereby indicating that they possessed equal systemic levels of vaccine-induced immunity at the time of M. tuberculosis challenge. The results indicate that inefficient expression of immunity in the lungs is likely to prove an obstacle to successful vaccination against tuberculosis in resistant and susceptible mouse strains, but more so in the latter strains.
INTRODUCTION
The increased incidence of tuberculosis in developed countries, coupled with the emergence of multi-drug-resistant strains of Mycobacterium tuberculosis, has drawn attention to the need to develop an efficacious anti-tuberculosis vaccine that is more protective than Bacillus Calmette–Guèrin (BCG), the attenuated vaccine strain of M. bovis. It is generally believed1,2 that this will be achieved by employing recombinant DNA technology to make vaccines that are capable of inducing higher levels of immunity than can be induced by BCG. This assumes, however, that BCG is a poor vaccine because it lacks sufficient immunogenicity, an assumption that may prove to be incorrect. It needs to be brought to mind, in this connection, that tuberculosis in mice3 and in 85% of humans4 is a progressive disease only of the lungs, in spite of the ability of M. tuberculosis to disseminate systemically and implant in other organs. This information, together with the knowledge that the disease can become systemic in immunocompromised mice3 and humans,5 indicates that confinement of the disease to the lungs of immunocompetent individuals is based on an acquired mechanism of immunity capable of being expressed efficiently in all organs except the lungs. It is possible that the inability of acquired immunity to protect the lungs is the result of mechanisms possessed by M. tuberculosis itself, which enable it to survive and induce lung pathology in the face of even high levels of systemic immunity. If this proved to be the case, it would seem pointless to attempt to develop vaccines that are more immunogenic than BCG.
This paper will show with major histocompatibility complex (MHC)-compatible DBA/2 and BALB/c mice, which are known to be genetically susceptible and genetically resistant to M. tuberculosis, respectively,6 that the lung is peculiarly susceptible to tuberculosis in the face of acquired systemic immunity. It will demonstrate that, although BCG vaccination protects mice of both strains equally in the liver and spleen, it leaves DBA/2 mice proportionally much more susceptible to tuberculosis in the lung.
MATERIALS AND METHODS
Mice
Male DBA/2 and BALB/c mice 8–10 weeks of age were purchased from the Jackson Laboratories (Bar Harbor, MN).
Bacteria and infection
The virulent H37Rv strain of M. tuberculosis (TMC# 102) was obtained from the Trudeau Institute Culture Collection (TMC) as a frozen (−70°) log-phase [2×108 colony-forming units (CFU) per ml] suspension culture3 in Proskauer and Beck medium (Difco Laboratories Inc., Detroit, MI) containing 0·01% Tween-80. For intravenous (i.v.) inoculation a culture was thawed, subjected to 5 seconds of ultrasound to break up aggregates, and diluted appropriately in phosphate-buffered saline containing 0·01% Tween-80. Each mouse received ≈105 CFU i.v., and infection was monitored by enumerating M. tuberculosis in lungs, livers and spleens at the times indicated in the Results. This involved plating serial, 10-fold dilutions of whole organ homogenates on nutrient agar, and counting colonies 2–3 weeks later, as described previously.3
Vaccination
Mice were vaccinated by inoculating them i.v. with 106 BCG Pasteur, and allowing infection to proceed for 40 days before treating them with chemotherapy to reduce the number of BCG in major organs to almost undetectable numbers. BCG Pasteur (TMC# 1101) was obtained as a frozen log-phase culture and prepared as described above for M. tuberculosis. Chemotherapy involved providing BCG-infected and control mice with drinking water containing 200mg of isoniazid and 100mg of rifampin (Sigma Chemical Company, St. Louis, MO) perml for 10 days. Chemotherapy was withdrawn 3 days before giving the M. tuberculosis challenge infection. At day 40 of the immunizing infection when chemotherapy was commenced the spleens of BALB/c and DBA/2 mice contained 4·5+0·32 and 4·8+0·41 logs of BCG CFU, respectively, as determined with five mice per group. Ten days later, at the completion of chemotherapy, the number of BCG was below the detection limits (102) of the plating assay.
Histology
Major organs were removed and fixed in neutral-buffered formalin, washed in water, dehydrated in graded ethanols and embedded in wax by standard procedures, or in glycol methacrylate (JB-4 embedding kit; Polysciences Inc., Warrington, PA) according to the supplier's instructions. Wax sections 5 or 10μm in thickness were stained for acid-fast bacilli with a modified basic fuchsin stain7 and counterstained with methylene blue, whereas plastic sections 2–3 μm in thickness were stained with 2% crystal violet containing 0·5% LOC High Suds detergent (Amway Corp., Ada, MI). Photomicrographs were taken with a Nikon Optiphot-2 photomicroscope.
RESULTS
Tuberculosis in vaccinated and unvaccinated mice
The inferior ability of DBA/2 mice, compared with BALB/c mice, to deal with M. tuberculosis infection in the lungs has already been documented.6 The results of an experiment designed to determine whether BCG vaccination enables DBA/2 mice to overcome this deficiency and make them as resistant as BALB/c mice in the lungs are shown in Fig. 1. It can be seen first that unvaccinated DBA/2 mice were as capable as unvaccinated BALB/c mice at controlling M. tuberculosis infection in the liver and spleen, and that vaccination provided mice of both strains with an increased and equal capacity to reduce the level of infection in these organs. Thus, whereas unvaccinated mice of both strains allowed M. tuberculosis to grow in the liver and spleen for about 10 days before bringing infection under control, vaccinated mice were able to retard or prevent any bacterial growth during the same time period. This resulted in 2–3 logs lower levels of infection in the livers and spleens of vaccinated mice from about day 10 of infection until about day 40. After this time infection reached higher levels in the livers and spleens of vaccinated DBA/2 mice than vaccinated BALB/c mice.
Figure 1.

Growth over 120 days of a 105M. tuberculosis i.v. challenge in the lungs livers and spleens of vaccinated (IMM) and unvaccinated (CONT) BALB/c and DBA/2 mice. Mice of both strains were equally capable of dealing with infection in their livers and spleens before vaccination, and were protected to the same extent in these organs during the first 40–80 days of infection. However, in the lungs, DBA/2 mice were at a significant disadvantage both before and after vaccination. This was the case, even though vaccination enabled mice of both strains to reduce the level of infection in the lungs by about 90% at day 40. Means±SEM of five mice per group.
The situation in the lungs was different, in that DBA/2 mice were less capable than BALB/c mice of controlling infection in this organ, both before and after vaccination. Thus, although vaccination provided both mouse strains with a capacity to mediate immunity earlier and to thereby reduce the level of infection in their lungs by about 1 log at day 40, this still left the lungs of vaccinated DBA/2 mice with about 1 log more bacilli than the lungs of vaccinated BALB/c mice at this stage of infection. Moreover, after day 40, infection was progressive in the lungs of unvaccinated and vaccinated DBA/2 mice, whereas it was caused to plateau in unvaccinated and vaccinated BALB/c mice. Consequently, by day 120, the lungs of vaccinated DBA/2 mice contained over 2·5 logs more bacilli than the lungs of vaccinated BALB/c mice, and about 1·5 logs more bacilli than the lungs of unvaccinated BALB/c mice. It will be noted that in unvaccinated and vaccinated mice of both strains, immunity was expressed later in the lungs than in the liver and spleen.
The consequence of these different levels of lung infection on host survival is shown in Fig. 2, where it can be seen that lower levels of lung infection in vaccinated mice resulted in longer survival. Thus, whereas control BALB/c mice died with a median survival time of 250 days, vaccinated BALB/c mice were all alive by day 300 when the experiment was terminated. The survival time of DBA/2 mice, however, was only slightly extended by vaccination, and all vaccinated DBA/2 mice died before unvaccinated BALB/c mice.
Figure 2.

Survival times of unvaccinated (CONT) and vaccinated (IMM) BALB/c and DBA/2 mice infected i.v. with 105M. tuberculosis. Results with 10 mice per group.
Effect of vaccination on the development of infection-induced lung histopathology
Microscopic examination of appropriately stained sections revealed that vaccination served to reduce the extensiveness of histopathology at sites of infection in the lungs of mice of both strains, but far less so in the case of DBA/2 mice. Thus in unvaccinated BALB/c mice at day 80 of infection the lungs contained relatively small discrete areas of infection-induced pathology (Fig. 3a) that were heavily populated by mononuclear cells (Fig. 3b,c). These lesions were mostly comprised of air sacs filled with foamy epithelioid macrophages (Figs 3b,c and 5a), some of which contained small numbers of acid-fast bacilli. Only a small proportion of the air sacs were populated with neutrophils. Therefore, these lesions can be considered to be of the compact epithelioid type.8 By contrast, most of the lung lesions in unvaccinated DBA/2 mice were of the acute necrotic type.8 Each of these lesions was larger than those in BALB/c mice (Fig. 4a) and populated by massive numbers of neutrophils (Figs 4b,c and 5b) most of which were in different stages of degeneration and contained large numbers of bacilli.
Figure 3.
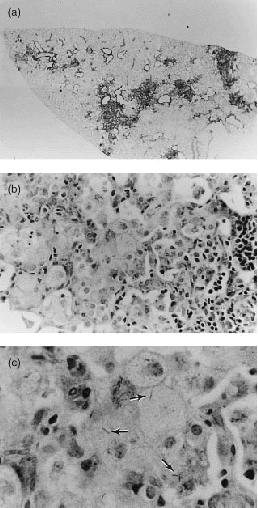
Histopathology of lungs of unvaccinated BALB/c mouse inoculated i.v. with 105 H37Rv 80 days earlier. (a) Low magnification of left lobe showing small discrete lesions; (b) higher magnification of a lesion showing mononuclear cells including epithelioid macrophages on the left side, and lymphoid cells on the right; (c) some epithelioid macrophages contain acid-fast bacteria (arrows). (a) ×5; (b) ×200; and (c) ×448.
Figure 5.
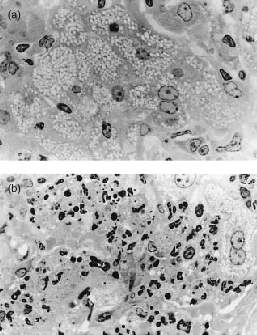
Histopathology of part of lesion in lung of (a) BALB/c and (b) DBA/2 mouse as seen in plastic sections. The ‘foaminess’ of the cytoplasm of epithelioid macrophages in the lesion of the BALB/c mouse is obvious, as is the dominance of degenerating neutrophils in the lesion of the DBA/2 mouse. (a) ×500; and (b) ×500.
Figure 4.
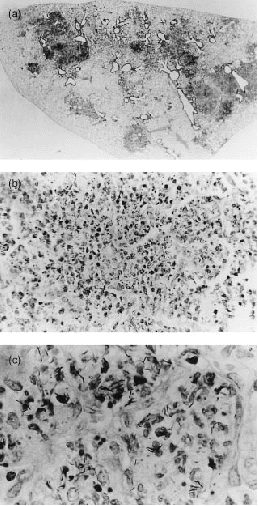
Histology of left lung lobe of unvaccinated DBA/2 mouse infected as in Fig. 1. (a) The lobe is swollen and individual lesions are extensive. (b) At higher magnification each lesion can be seen to be densely populated with neutrophils, many of which are necrotic. (c) At still higher magnification the lesion can be seen to contain a large number of acid-fast bacilli. (a) ×5; (b) ×200; and (c) ×448.
Vaccination provided BALB/c mice with the ability to reduce the size of lung tubercles and the number of acid-fast bacilli within them. However, the histopathology of the lesions was very similar to that seen in unvaccinated BALB/c mice. Likewise, vaccination did not prevent the development of lung pathology of the acute necrotizing type in DBA/2 mice. Thus although the lesions that developed in vaccinated DBA/2 mice appeared somewhat smaller than those that developed in unvaccinated DBA/2 mice, they were nevertheless dominated by neutrophils heavily infected with acid-fast bacilli. Because of the similarities between the lung pathology of vaccinated and unvaccinated mice, the lung pathology of the former mice is not shown.
Infection in the livers and spleens of control and vaccinated DBA/2 and BALB/c mice was confined to granulomas that were small in size and number, although smaller in number in vaccinated mice. In the spleens of vaccinated DBA/2 mice on day 80 of infection (Fig. 6) compact granulomas were mainly seen in the white pulp. In the livers of the same mice (Fig. 7) compact granulomas were sparsely distributed throughout the parenchyma. Because the histology of the livers and spleens of vaccinated BALB/c mice was essentially the same as that of vaccinated DBA/2 mice, it is not shown. Again, except for a somewhat larger number of granulomas, the histology of the livers and spleens of unvaccinated mice was very similar to that of vaccinated mice.
Figure 6.
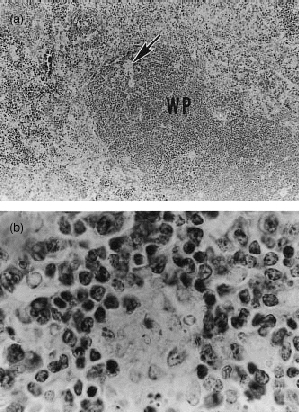
Low-power (a) and high-power (b) micrograph of spleen of DBA/2 mouse on day 80 of the challenge infection, as shown in Fig. 1. At this stage of infection the spleen showed a normal architecture. The only evidence of infection was the presence of a very small number of compact granulomas distributed mainly in the white pulp (WP). These were difficult to see (arrow) at low magnification (a), and showed little evidence of infection at higher power (b). (a) ×109·6; and (b) ×552.
Figure 7.
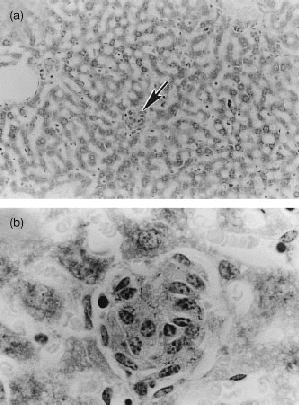
Low-power (a) and high-power (b) micrograph of section of liver of DBA/2 mouse infected for 80 days, as for Fig. 1. Granulomas (arrow) were small in number and size (a) and showed few acid-fast bacilli at high power (b). (a) ×56; and (b) ×552.
DISCUSSION
The results of this study of genetically susceptible DBA/2 mice and genetically resistant BALB/c mice show that vaccination failed to make the former mice as capable as the latter in defending against M. tuberculosis infection. Even though vaccination enabled mice of both strains to express immunity to infection earlier and to reduce the level of infection in their lungs by 1 log by day 40, the lungs of vaccinated DBA/2 mice still contained 1 log more M. tuberculosis than the lungs of vaccinated BALB/c at this time. Moreover, because the growth of M. tuberculosis was progressive in the lungs of vaccinated DBA/2 mice after day 40, but was caused to plateau in the lungs of vaccinated BALB/c mice, the lungs of the former mice contained much higher numbers of M. tuberculosis as infection progressed. Again, there was little difference between vaccinated and unvaccinated mice of either strain in the rate at which infection progressed in the lungs after day 40. It follows therefore that increased protection afforded by vaccination is confined to the first 40 or so days of infection. After this time lung infection in vaccinated mice resembled infection that would be expected to develop (R.J. North, unpublished results) in unvaccinated mice infected with a smaller inoculum of M. tuberculosis. There seems no reason to propose at this time that there was any difference between vaccinated and unvaccinated mice in terms of the quantity or quality of immunity they possessed after day 40. It is well established9,10 that immunity to M. tuberculosis infection depends predominantly on CD4Tcells of the T helper type 1 (Th1) type. These cells mediate immunity via the secretion of interferon-γ (IFN-γ) which serves to activate the anti-mycobacterial function of macrophages, the cells that need to ingest and destroy the pathogen.9,10
The possibility that the inferior ability of vaccinated DBA/2 mice to deal with M. tuberculosis infection in the lung was due to their having generated lower levels of systemic immunity in response to the BCG vaccine than BALB/c mice seems unlikely. This can be suggested on the grounds that even in the lungs, vaccination enabled mice of both strains to reduce the level of infection to about the same degree during the first 40 days of infection. In addition, vaccinated mice of both strains were equally capable of reducing the levels of infection in the liver and spleen. The higher levels of infection in the liver and spleens of vaccinated DBA/2 mice compared to vaccinated BALB/c mice at later stages of infection probably resulted from continuous seeding from more heavily infected lungs. It should be brought to mind that DBA/2 mice carry the resistance allele of the antimicrobial natural resistance gene, Nramp1, whereas BALB/c mice carry the susceptibility allele.11 Consequently, DBA/2 mice are capable of displaying more natural resistance to low-dose BCG infection than BALB/c mice during the first 20 days or so of infection, at least as measured in the spleen. It is known, however, that after 20 days both strains are equally capable of resolving infection in this organ.12 Because the 106 i.v. immunizing inoculum of BCG Pasteur used in this study was large enough to overcome the natural resistance difference between DBA/2 and BALB/c mice,13 and because the number of BCG in the spleens of mice of both strains was essentially the same on day 40 of the immunizing infection, there seems no reason to believe that DBA/2 mice generated lower levels of specific immunity in response to BCG vaccination than BALB/c mice.
If the foregoing argument is correct, it would suggest that a key obstacle to successful vaccination against M. tuberculosis infection in genetically resistant, as well as in genetically susceptible, mice is the inability of systemic immunity to protect the lungs against infection-induced pathology. In the case of BALB/c mice there is little doubt that eventual stabilization of infection in the lungs was the result of the expression of specific immunity, since it is known3 that infection is progressive in this and other organs in genetically resistant mice depleted of CD4 T cells. In spite of stabilization of infection, however, lung pathology of the compact epithelioid type continued to develop in vaccinated, as well as unvaccinated BALB/c mice. This resulted in death of unvaccinated BALB/c mice beginning on day 180. According to the results of a related ongoing study (E. Medina and R. J. North, manuscript in preparation), vaccinated BALB/c mice eventually also would have died from lung pathology. Progressive development of lung pathology of this type in the absence of progressive M. tuberculosis growth has been shown to occur in other genetically resistant mice.14 In vaccinated and unvaccinated DBA/2 mice pathology in the lungs was more extensive and of a different type, presumably because bacterial growth was not stabilized by specific immunity, but progressed at a reduced rate. A key finding with DBA/2 mice is that vaccination did not prevent lung pathology of the acute, necrotic type from developing. It is worth mentioning that M. tuberculosis-induced lung pathology described here for DBA/2 mice is similar to that described for mice of a resistant strain immunocompromised by CD4 T-cell depletion.3
In view of the results presented it can be suggested that the protective value of any subunit, recombinant, or DNA vaccine under experimental investigation needs to be judged in terms of its ability to reduce the level of M. tuberculosis infection and infection-induced pathology in the lungs, rather than in other organs. It also can be suggested that an efficacious vaccine will be one that protects the lungs of genetically susceptible, as well as the lungs of genetically resistant hosts. It is not apparent from the literature2,15 that any of the new vaccine preparations recently tested is significantly more protective in these regards than BCG.
Acknowledgments
We wish to express our thanks to Lynn Ryan and Ron LaCourse for their expert technical contribution. The research was supported by grants AI-40071 and AI-37844 from the National Institute of Allergy and Infections Diseases, United States Public Health Service; and a grant from the Mathers Charitable Trust.
REFERENCES
- 1.Kaufmann SHE. Vaccines against tuberculosis: the impact of modern technology. Scand J Infect Dis. 1990;76(Suppl.):54. [PubMed] [Google Scholar]
- 2.Orme IM. Progress in the development of new vaccines against tuberculosis. Int J Tuberc Lung Dis. 1997;1:95. [PubMed] [Google Scholar]
- 3.Dunn PL, North RJ. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect Immun. 1995;63:3428. doi: 10.1128/iai.63.9.3428-3437.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farer LS, Lowell AM, Meador MP. Extrapulmonary tuberculosis in the United States. Am J Epidemiol. 1979;109:205. doi: 10.1093/oxfordjournals.aje.a112675. [DOI] [PubMed] [Google Scholar]
- 5.Barnes PF, Bloch AB, Davidson PT, Snider DE. Tuberculosis in patients with human immunodeficiency virus infection. N Engl J Med. 1991;324:1644. doi: 10.1056/NEJM199106063242307. [DOI] [PubMed] [Google Scholar]
- 6.Medina E, North RJ. Evidence inconsistent with a role for the Bcg gene (Nramp1) in resistance of mice to infection with Mycobacterium tuberculosis. J Exp Med. 1996;183:1045. doi: 10.1084/jem.183.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis RC, Zabrowarny LA. Safer staining method for acid fast bacilli. J Clin Pathol. 1993;46:559. doi: 10.1136/jcp.46.6.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rich AR. The pathogenesis of tuberculosis. Springfield, IL: Charles C. Thomas; 1951. p. 823. [Google Scholar]
- 9.Orme IA, Andersen P, Boom WH. Tcell response to Mycobacterium tuberculosis. J Infect Dis. 1993;167:1481. doi: 10.1093/infdis/167.6.1481. [DOI] [PubMed] [Google Scholar]
- 10.Boom WH. The role of Tcell subsets in Mycobacterium tuberculosis infection. Infect Agents Dis. 1996;5:73. [PubMed] [Google Scholar]
- 11.Malo D, Vogan K, Vidal S, et al. Haplotype mapping and sequence analysis of the mouse Nramp gene predicts susceptibility to infection with intracellular parasites. Genomics. 1994;23:51. doi: 10.1006/geno.1994.1458. [DOI] [PubMed] [Google Scholar]
- 12.Forget A, Skamene E. Genetic regulation of early host defense mechanisms controlling mycobacterial infection. In: Skamene E, editor. Genetic Control of Host Resistance to Infection and Malignancy. New York: Alan R. Liss, Inc.; 1985. p. 265. [Google Scholar]
- 13.Orme IM, Collins FM. Demonstration of acquired resistance in Bcgr inbred mouse strains infected with a low dose of BCG Montreal. Clin Exp Immunol. 1984;56:81. [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn PL, North RJ. Persistent infection with virulent but not avirulent Mycobacterium tuberculosis in the lungs of mice causes progressive pathology. J Med Microbiol. 1996;45:103. doi: 10.1099/00222615-45-2-103. [DOI] [PubMed] [Google Scholar]
- 15.Lowrie DB, Silva CL, Tascon RE. DNA vaccines against tuberculosis. Immunol Cell Biol. 1997;75:591. doi: 10.1038/icb.1997.93. [DOI] [PubMed] [Google Scholar]