

Abstract
Sodium salicylate (NaSal) is a commonly used agent with a wide pharmacological spectrum. The objective of the present study was to investigate the effect of NaSal on anaphylaxis. NaSal (10−1 and 1 mm) significantly inhibited systemic anaphylaxis induced by compound 48/80 in rats. NaSal also significantly inhibited local anaphylaxis activated by anti-dinitrophenyl (DNP) immunoglobulin E (IgE). NaSal (10−1 and 1 mm) significantly inhibited histamine release from rat peritoneal mast cells (RPMC) activated by compound 48/80 or anti-DNP IgE. Northern-blot analysis demonstrated that a significantly reduced level of the mRNA of l-histidine decarboxylase was expressed in mast cells treated with NaSal, compared with that without NaSal. NaSal (10−2 and 10−1 mm) had a significant inhibitory effect on anti-DNP IgE-induced tumour necrosis factor-α secretion from RPMC. The level of cyclic AMP in RPMC, when NaSal (1 mm) was added, transiently and significantly increased about sixfold compared with that of basal cells. These results suggest a possible use of NaSal in managing mast cell-dependent anaphylaxis.
INTRODUCTION
Salicylate may have been the first anti-inflammatory preparation used by modern-day humans. Salicylate is known to have additional effects. For example, in cultured neuronal cells, salicylate protects neurotoxicity elicited by the excitatory amino acid glutamate,1 in cultured cardiac fibroblasts, it inhibits the inducible nitric oxide synthase,2 in HeLa cells, it activates human heat-shock transcription factor,3 and in plants, it activates pathogenesis-related genes in response to infection and injury.4,5 It is now well established that the mast cell triggers anaphylaxis in response to allergens by releasing chemical mediators.6 Among the preformed and newly synthesized inflammatory substances released on degranulation of mast cells, histamine is the best characterized and most potent vasoactive mediator implicated in the acute phase of immediate-type allergic reactions.7 Mast cell degranulation can be elicited by a number of positively charged substances, collectively known as the basic secretagogues of mast cells.8 Compound 48/80 and polymers of basic amino acids, such as substance P, are some of the most potent secretagogues of mast cells.9 Compound 48/80 is a mixture of polymers synthesized by condensing N-methyl-P-methoxyphenyl ethlyamine with formaldehyde. Compared with the natural process, a high concentration of compound 48/80 induces almost a 90% release of histamine from mast cells. Thus, an appropriate amount of compound 48/80 has been used as a direct and convenient reagent to study the mechanism of anaphylaxis.10 The secretory response of mast cells can also be induced by aggregation of their cell surface-specific receptors for immunoglobulin E (IgE) by the corresponding antigen.11–13 It has been established that the anti-IgE antibody induces passive cutaneous anaphylaxis (PCA) reactions as a typical model for the immediate hypersensitivity.14 Given the recent evidence that upon antigen stimulation mast cells are a potential source of various cytokines, including tumour necrosis factor-α (TNF-α),15–19 it is likely that they play a crucial role in allergic inflammation. Therefore, modulation of TNF-α secretion by mast cells should provide us with a useful therapeutic strategy for allergic disease.
In this paper, we showed that NaSal inhibited compound 48/80-induced systemic anaphylaxis, anti-dinitrophenyl (DNP) IgE antibody-induced PCA, and histamine and TNF-α secretion from rat peritoneal mast cells (RPMC). We also investigated the cyclic AMP content to clarify the mechanism by which the NaSal inhibited histamine release from RPMC.
MATERIALS AND METHODS
Materials
NaSal, compound 48/80, anti-DNP IgE, DNP-human serum albumin (HSA), substance P, metrizamide and all other reagents were purchased from Sigma Chemical Co. (St. Louis, MO). α-minimal essential medium (α-MEM) was purchased from Flow Laboratories (Irvine, CA). Fetal calf serum (FCS) was purchased from Life Technologies Inc. (Gaithersburg, MD). The original stock of Wistar rats was purchased from The Korean Research Institute of Chemical Technology (Taejeon, Chungnam, South Korea), and the animals were maintained at the College of Pharmacy, Wonkwang University. The animals were housed five to 10 per cage in a laminar air flow room maintained under a temperature of 22±1° and relative humidity of 55±10% throughout the study.
Systemic anaphylaxis
Rats were given an intraperitoneal (i.p.) injection of 8 mg/kg of the mast cell degranulator, compound 48/80. Compound 48/80 and NaSal were dissolved in saline and administered by i.p. injections, with each concentration of drug being given 10 min before the injection of compound 48/80. Mortality was monitored for 1 hr after induction of anaphylactic shock. Blood was taken by cardiac puncture from rats 30 min after compound 48/80 administration in each of the experimental groups.
PCA
An IgE-dependent cutaneous reaction was generated by sensitizing the skin with an intradermal injection of anti-DNP IgE followed 48 hr later with an injection of DNP-HSA into the rat's tail vein. The anti-DNP IgE and DNP-HSA were diluted in phosphate-buffered saline (PBS). The rats were injected intradermally with 0·5 μg (50 μl) of anti-DNP IgE into each of four dorsal skin sites that had been shaved 48 hr earlier. The sites were outlined with a water-insoluble red marker. Forty-eight hours later each rat received an injection of 1 mg (100 μl) of DNP-HSA in PBS containing 4% Evans blue (1:4) via the tail vein. Two hundred microliters of NaSal was administered i.p. 10 min before the challenge. Thirty minutes after the challenge, the rats were killed and the dorsal skin was removed for measurement of the pigment area. The amount of dye was then determined colorimetrically after extraction with 1 ml of 1·0 m KOH and 9 ml of a mixture of acetone and phosphoric acid (5:13) based on the method of Katayama et al.20 The absorbence intensity of the extract was measured at 620 nm in a spectrophotometer, and the amount of dye was calculated from the Evans blue measuring-line.
Preparation of plasma and histamine determination
The blood was centrifuged at 400 g for 10 min. The plasma was withdrawn and the histamine content was measured by the o-phthalaldehyde spectrofluorometric procedure of Shore et al.21 The fluorescence intensity was measured at 438 nm (excitation at 353 nm) in a spectrofluorometer.
RPMC
RPMC were isolated as previously described.22 In brief, rats were anaesthetized by ether and injected with 20 ml of Tyrode buffer B (137 mm NaCl, 5·6 mm glucose, 12 mm NaHCO3, 2·7 mm KCl, 0·3 mm NaH2PO4) containing 0·1% gelatin (Sigma Chemical Co.) into the peritoneal cavity, and the abdomen was gently massaged for about 90 seconds. The peritoneal cavity was carefully opened, and the fluid containing peritoneal cells was aspirated with a Pasteur pipette. Thereafter, the peritoneal cells were sedimented at 150 g for 10 min at room temperature and resuspended in Tyrode buffer B. Mast cells were separated from the major components of rat peritoneal cells, i.e. macrophages and small lymphocytes, according to the method described by Yurt et al.23 In brief, peritoneal cells suspended in 1 ml Tyrode buffer B were layered on 2 ml of 22·5% w/v metrizamide (density, 1·120 g/ml) and centrifuged at room temperature for 15 min at 400 g. The cells remaining at the buffer–metrizamide interface were aspirated and discarded. Mast cell preparations were about 95% pure as assessed by Toluidine blue staining. More than 97% of the cells were viable as judged by Trypan blue uptake.
Inhibition of histamine release
Purified mast cells were resuspended in Tyrode buffer A (10 mm HEPES, 130 mm NaCl, 5 mm KCl, 1·4 mm CaCl2, 1 mm MgCl2, 5·6 mm glucose) containing 0·1% bovine serum albumin (BSA; Sigma Chemical Co.) for the treatment of compound 48/80. Mast cell suspensions (2×105 cells/ml) (reaction volume: 1 ml) were preincubated for 10 min at 37° before the addition of compound 48/80 (5 μg/ml), and were also sensitized with 10 μg/ml anti-DNP IgE for 6 hr. The cells were preincubated with the NaSal at 37° for 10 min prior to the treat with compound 48/80 or the challenge with DNP-HSA (1 μg/ml). After 40 min the reaction was stopped by cooling the tubes in ice. The cells were separated from the released histamine by centrifugation at 400 g for 5 min at 4°. Residual histamine in the cells was released by disrupting the cells with perchloric acid and centrifugation at 400 g for 5 min at 4°.
Assay of histamine release
The inhibition percentage of histamine release was calculated using the following equation:
Molecular probes
The l-histidine decarboxylase (HDC) probe used a 32P-labelled, Pst I-digested cDNA insert from a mouse mastocytoma P-815 cells cDNA clone kindly provided Dr A. Ichikawa, Kyoto University, Kyoto, Japan.24
RNA extraction and Northern blotting
Mastocytoma P-815 cells were maintained as a suspension culture in α-MEM with 10% FCS. Total RNA was isolated by the modified LiCl–urea method,25 electrophoresed in 1·2% agarose–formaldehyde gels, and transferred on nylon membranes by capillary action in 20×SSC (1×SSC = 0·15 m NaCl, 0·015 m sodium citrate, pH 7·2). Baked filters were prehybridized at 42° in a buffer containing 50% formamide, 4×SSC, 0·5 mg/ml sheared salmon sperm DNA, and 1×Denhardt's solution. Hybridization proceeded at 42° for 15 hr in the same buffer containing 1×106 c.p.m./ml of an [α-32P]dCTP-labelled probe. The filters were then washed in 2×SSC/0·1% sodium dodecyl sulphate (SDS), 1×SSC/0·1% SDS, and 0·2× SSC/0·1% SDS at 55° for 20 min, dried, and examined by autoradiography.
Assay of TNF-α secretion
TNF-α secretion was measured by a modified enzyme-linked immunosorbent assay (ELISA) as described.26 The ELISA was sensitive to TNF concentrations in medium above 1 pg/ml. The ELISA was devised by coating 96-well plates with 6·25 ng/well of murine monoclonal antibody with specificity for murine TNF-α. Before use and between subsequent steps in the assay, coated plates were washed twice with PBS containing 0·05% Tween-20 (PBS–Tween) and twice with PBS alone. All reagents used in this assay were incubated for 1 hr at room temperature with coated wells. For the standard curve, recombinant TNF-α (rTNF-α; Genzyme, Munich, Germany) was added to serum previously determined to be negative for endogenous TNF-α. After exposure to medium, assay plates were sequentially exposed to rabbit anti-murine TNF-α antibody (Genzyme), phosphatase-conjugated goat anti-rabbit IgG, and P-nitrophenyl phosphate. Optical density readings were made within 10 min of addition of the substrate on a Titertek Multiscan (Flow Laboratories, North Ryde, Australia) with a 405-nm filter. Appropriate specificity controls were included.
Measurement of cyclic AMP level
The cyclic AMP level was measured according to the method of Peachell et al.27 In brief, purified mast cells were resuspended in prewarmed (37°) Tyrode buffer A. Typically, an aliquot of cells (105 cells) was added to an equivalent volume (50 μl) of prewarmed buffer containing the drug in an Eppendorf tube. The reaction was allowed to proceed for discrete time intervals, terminated by the addition of ice-cold acidified ethanol (0·9 ml of 86% ethanol/1 m HCl, 99:1) with brief vigorous vortexing and then snap frozen in liquid nitrogen. The sample was later thawed and vortexed, then the debris was sedimented in a centrifuge (400 g at 4°, for 5 min), and an aliquot (0·9 ml) of the supernatant was removed and evaporated to dryness under a reduced pressure. The dried sample was reconstituted in assay buffer (150–200 μl) and stored frozen. The cyclic AMP level was determined by enzyme immunoassay, using a commercial kit (Amersham International plc, Buckinghamshire, UK).
Statistical analysis
The data were expressed as mean±SEM. Student's t-test was used to make a statistical comparison between the groups. Results with P < 0·05 were considered statistically significant.
RESULTS
Effect of NaSal on compound 48/80-induced systemic anaphylaxis
To assess the contribution of NaSal in anaphylaxis, we first used the in vivo model of systemic anaphylaxis. We used compound 48/80 (8 mg/kg) as a systemic fatal anaphylaxis inducer. After the injection of compound 48/80 the rats were monitored for 1 hr, after which the mortality rate was determined. As shown in Table 1, i.p. injection of saline plus compound 48/80 as a control induced fatal shock in 100% of rats. When rats were pretreated with NaSal at concentrations ranging from 10−3 to 101 mm for 10 min, the mortality caused by compound 48/80 was reduced in a dose-dependent fashion (n = 10 rats per group). Especially, NaSal significantly inhibited compound 48/80-induced anaphylaxis with the concentrations of 10−1 and 1 mm. When NaSal was administered 5 min or 10 min after compound 48/80 injection (n = 10 rats per group), the mortality increased in a time-dependent fashion (Table 2).
Table 1.
Effect of NaSal on compound 48/80-induced systemic anaphylaxis
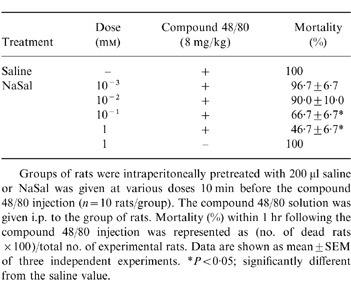
Groups of rats were intraperitoneally pretreated with 200 μl saline or NaSal was given at various doses 10 min before the compound 48/80 injection (n = 10 rats/group). The compound 48/80 solution was given i.p. to the group of rats. Mortality (%) within 1 hr following the compound 48/80 injection was represented as (no. of dead rats ×100)/total no. of experimental rats. Data are shown as mean±SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
Table 2.
Time-dependent effect of NaSal on compound 48/80-induced anaphylaxis
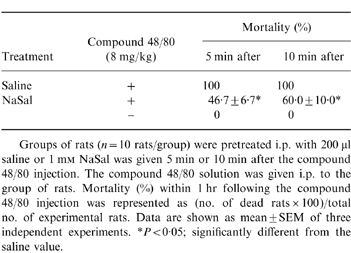
Groups of rats (n = 10 rats/group) were pretreated i.p. with 200 μl saline or 1 mM NaSal was given 5 min or 10 min after the compound 48/80 injection. The compound 48/80 solution was given i.p. to the group of rats. Mortality (%) within 1 hr following the compound 48/80 injection was represented as (no. of dead rats×100)/total no. of experimental rats. Data are shown as mean±SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
Effect of NaSal on compound 48/80-induced plasma histamine release
The ability of NaSal to influence compound 48/80-induced plasma histamine release was investigated. NaSal, at concentrations ranging from 10−3 to 101 mm, was given 10 min before the compound 48/80 injection (n = 10 rats per group). While serum levels of histamine were markedly elevated after the compound 48/80 injection in all groups of rats, the rats injected with NaSal (10−1 and 1 mm) showed a significant reduction in plasma histamine levels (Fig. 1). The actual concentration of histamine in control plasma was 0·21±0·18 ng/ml.
Figure 1.
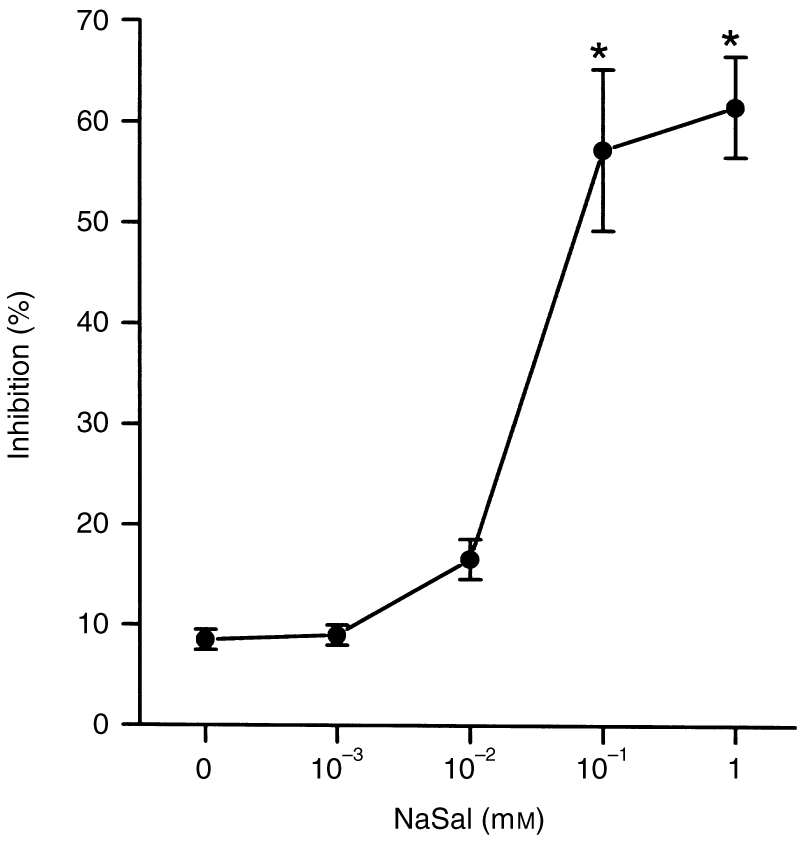
Effect of NaSal on compound 48/80-induced plasma histamine release. Groups of rats were pretreated i.p. with 200 μl NaSal. NaSal was given at various doses 10 min before i.p. compound 48/80 injection (n = 10 rats/group). The data represent the mean±SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
Effect of NaSal on anti-DNP IgE-induced PCA
Another way to test anaphylactic reactions is to induce PCA. As described in the experimental procedures, local extravasation is induced by a local injection of anti-DNP IgE followed by an antigenic challenge. As shown in Table 3, the i.p. administration of NaSal (10−1 and 1 mm) showed a marked inhibitory effect in PCA.
Table 3.
Effect of NaSal on the 48-hr PCA in rats
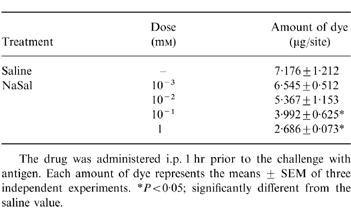
The drug was administered i.p. 1 hr prior to the challenge with antigen. Each amount of dye represents the means ± SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
Effect of NaSal on compound 48/80-induced or anti-DNP IgE-induced histamine release from RPMC
The inhibitory effects of NaSal on compound 48/80-induced or anti-DNP IgE-induced histamine release from RPMC following a 1-min preincubation are shown in Fig. 2. NaSal inhibited compound 48/80- induced or anti-DNP IgE-induced histamine release from RPMC in a dose-dependent fashion. Especially, NaSal significantly inhibited the compound 48/80-induced or IgE-mediated histamine release at the concentrations of 10−1 and 1 mm.
Figure 2.
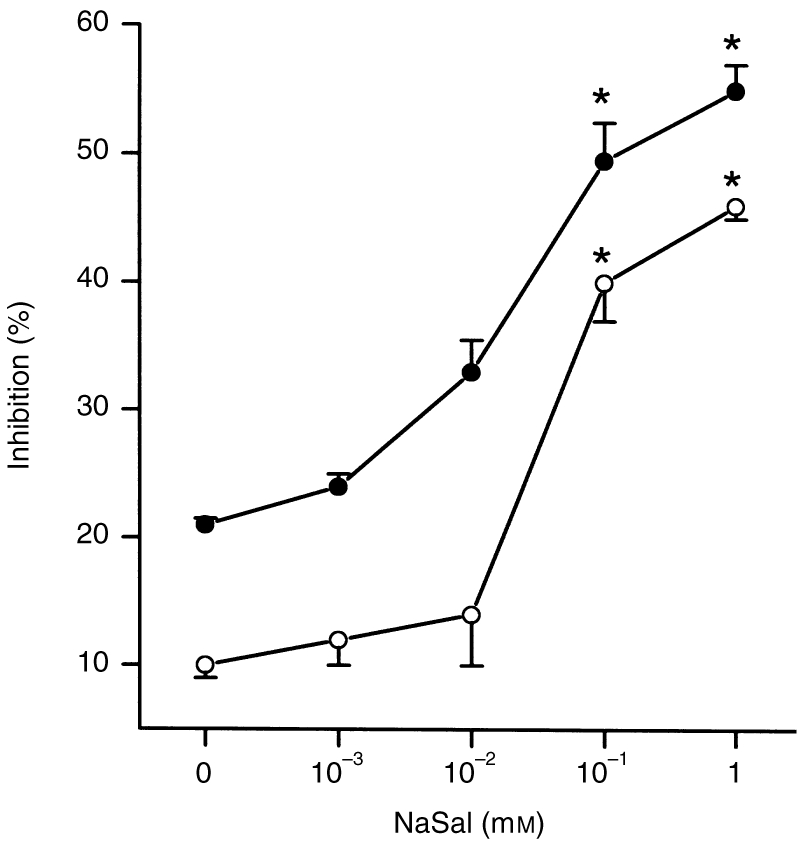
Effect of NaSal on compound 48/80-induced or IgE-mediated histamine release from RPMC. RPMC (2×105 cells/ml) were preincubated with the drug at 37° for 10 min prior to the incubation with compound 48/80 (○) or challenge with DNP-HSA (•) (n = 3 rats/group). The data represent the mean±SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
Effect of NaSal on substance P-induced HDC mRNA expression
Substance P appears to induce histamine release by interacting with the same site as compound 48/80.28 NaSal (10−1 and 1 mm) also significantly inhibited substance P-induced histamine release from RPMC (data not shown). HDC catalyzes the formation of histamine from its precursor, histidine, in a single step. Mastocytoma P-815 cells are the proper cell type for elucidating the mechanism underlying histamine formation in mast cells, because they synthesize HDC in response to various stimuli.24 Data in Fig. 3 show the Northern blot analysis. As shown in a previous study,24 the mRNA encoding the amino acid sequence of HDC in mastocytoma P-815 cells is 2·7 kilobases. NaSal inhibited the increase in substance P-induced accumulation of HDC mRNA on incubation for 6 hr. We did not detect the compound 48/80-induced accumulation of HDC mRNA in RPMC.
Figure 3.
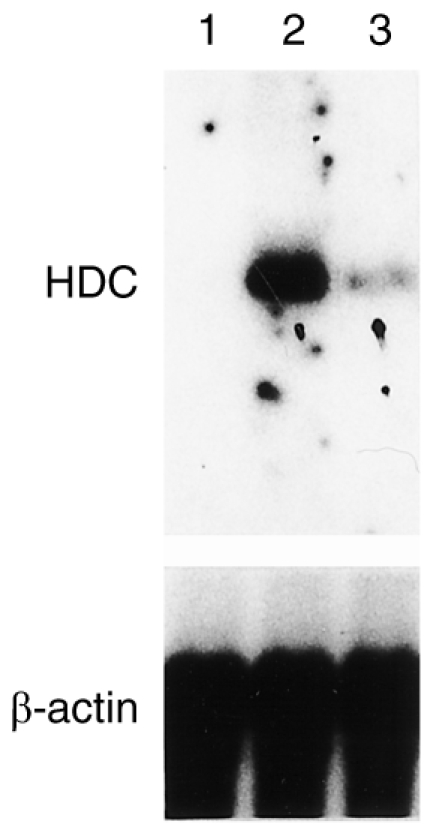
Effect of NaSal on HDC mRNA level in mastocytoma P-815 cells. Cells were incubated in the absence (lane 1) or presence (lane 2) of 100 nm substance P 100 nm, or in the presence of substance P plus 1 mm NaSal for 6 hr (lane 3) at 37°. The cells were harvested and washed, and then total RNA was prepared, and HDC mRNA was analysed by Northern hybridization as described in the Materials and Methods. The β-actin probe was used to verify that an equal amount of total RNA (20 μg) was loaded in each lane.
Effect of NaSal on anti-DNP IgE-induced TNF-α secretion from RPMC
We next examined whether NaSal could also regulate TNF-α secretion by a RPMC. NaSal inhibited anti-DNP IgE-induced TNF-α secretion from RPMC at concentrations ranging from 10−3 to 10−1 mm (Table 4). Only concentrations ranging between 10−2 and 10−1 inhibited TNF-α secretion significantly.
Table 4.
Effect of NaSal on anti-DNP IgE-induced TNF-α secretion in RPMCqc

RPMC (1×106 cells) were sensitized with anti-DNP IgE (2 μg/ml)for 16 hr and incubated for 10 min with various concentrations of NaSal before the challenge with DPN-HSA (0·2 μg/ml) for 4 hr. The data represent the mean±SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
Effect of NaSal on cyclic AMP level of RPMC
Finally, we investigated the cyclic AMP content to clarify the mechanism by which NaSal inhibits histamine release from RPMC. When RPMC were incubated with 1 mm NaSal, the cyclic AMP content significantly increased. It peaked 1 min after the NaSal was added, then decreased to basal value about 3 min later (Fig. 4).
Figure 4.

Time–course of increase in the cAMP level of RPMC caused by NaSal. RPMC (105 cells/ml) were pretreated with NaSal (○) or not (•) at 37° (n = 3 rats/group). The data represent the mean±SEM of three independent experiments. *P < 0·05; significantly different from the saline value.
DISCUSSION
The present study showed that NaSal treatment profoundly affected compound 48/80-induced systemic anaphylaxis and anti-DNP IgE-induced PCA. There is absolutely no doubt that stimulation of mast cells with compound 48/80 or anti-DNP IgE initiates the activation of a signal-transduction pathway, which leads to histamine release. Some recent studies have shown that compound 48/80 and other polybasic compounds are able, apparently directly, to activate G-proteins.29,30 The evidence indicates that the protein is G inhibitory-like and that the activation is inhibited by benzalkonium chloride.31 Tasaka et al. reported that compound 48/80 increased the permeability of the lipid bilayer membrane by causing a perturbation of the membrane.32 This result indicates that the permeability increase of the cell membrane may be an essential trigger for the release of the mediator from mast cells. In this sense, anti-allergic agents having a membrane-stabilizing action may be desirable. NaSal might act on the lipid bilayer membrane affecting the prevention of the perturbation being induced by compound 48/80. The NaSal-administered rats are protected from IgE-mediated allergic reaction. It is conceivable that NaSal inhibits the initial phase of immediate-type allergic reactions, probably through interference with the mast cell/histamine system.
In this study, the compound 48/80-induced or anti-DNP IgE-induced histamine release from RPMC was significantly inhibited by NaSal at the concentrations of 10−1 and 1 mm. We have demonstrated that NaSal suppressed substance P-induced accumulation of HDC mRNA. The mechanism by which the NaSal suppression results in increased HDC gene transcription is of interest. In previous studies, it was reported that substance P could specifically bind to murine mast cells to trigger release of histamine.33,34 NaSal may work as an antagonist in receptor interaction. This is supported by previous reports that benzalkonium chloride and other selective antagonists inhibit the histamine release incurred by compound 48/80 and basic neuropeptides such as substance P.28,35 TNF-α is a multifunctional cytokine that has a pro-inflammatory role. Our data showed that NaSal inhibited anti-DNP IgE-induced TNF-α secretion from mast cells. NaSal inhibited TNF-α secretion at lower concentrations than those needed for the inhibition of degranulation, suggesting the differential regulation of the degranulation process and of TNF-α secretion in mast cells. The effect of NaSal on mast cell cytokine secretion in vivo and the relative importance of mast cells as a source of TNF-α during inflammatory and immune responses are important areas for future studies. The release of histamine is known to be depressed by an increase in the intracellular cyclic AMP content due to the activation of adenylate cyclase or inhibition of cyclic AMP phosphodiesterase.36 The intracellular cyclic AMP content of the mast cells, when incubated with NaSal (1 mm), increased about sixfold in comparison with that of basal cells. Our results, however, must be confirmed by further studies because cyclic AMP levels returned to basal values after 3 min of incubation, even though inhibition of histamine release appeared after 10 min of incubation. In addition, inhibition of phospholipase A2 has been shown to suppress histamine release from activated mast cells,37 suggesting that NaSal may reduce histamine release by inhibiting phospholipase A2. It is tempting to speculate that phospholipase A2 may also modulate anaphylaxis, but more study is needed to confirm this hypothesis.
Our results demonstrated that NaSal inhibited the mast cell-dependent anaphylaxis in vivo and in an in vitro murine model. To our knowledge, this is the first report of such selective inhibition of immediate-type allergic reactions by NaSal. However, it may be necessary to study further the relevance of these findings with respect to the previous report on the clinical responses and mediator-release profiles of an aspirin-sensitive man with systemic mast cell disease during desensitization.38
Acknowledgments
This paper is supported by the Oriental Medicine Found of Ministry of Health and Welfare, Republic of Korea.
REFERENCES
- 1.Grilli M, Pizzi M, Memo M, Spano PF. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 2.Farivar RS, Chobanian AV, Brecher P. Salicylate or aspirin inhibits the induction of the inducible nitric oxide synthase in rat cardiac fibroblasts. Circ Res. 1996;78:759. doi: 10.1161/01.res.78.5.759. [DOI] [PubMed] [Google Scholar]
- 3.Jurivich D, Sistonen L, Kroes R, Morimoto R. Effect of sodium salicylate on the human heat shock response. Science. 1992;255:1243. doi: 10.1126/science.1546322. [DOI] [PubMed] [Google Scholar]
- 4.Malamy J, Carr JP, Klessig DF, Raskin I. Salicylic acid: a likely endogenous signal in the resistance response of tobacco to viral infection. Science. 1990;250:1002. doi: 10.1126/science.250.4983.1002. [DOI] [PubMed] [Google Scholar]
- 5.Métraux J, Signer H, Ryals J, et al. Increase in salicylic acid at the onset of systemic acquired resistance in cucumber. Science. 1990;250:1004. doi: 10.1126/science.250.4983.1004. [DOI] [PubMed] [Google Scholar]
- 6.Kim HM, Kim KS, Lee EH. Specific inhibition of Immunoglobulin E-mediated allergic reaction using antisense FcεRIα oligodeoxynucleotides. Immunology. 1998;93:589. doi: 10.1046/j.1365-2567.1998.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen LJ, Mosbech H, Skov P. Allergen-induced histamine release in intact human skin in vivo assessed by skin microdialysis technique: Characterization of factors influencing histamine releasability. J Allergy Clin Immunol. 1996;97:672. doi: 10.1016/s0091-6749(96)70313-5. [DOI] [PubMed] [Google Scholar]
- 8.Lagunoff D, Martin TW, Read G. Agents that release histamine from mast cells. Annu Rev Pharmacol Toxicol. 1983;23:331. doi: 10.1146/annurev.pa.23.040183.001555. [DOI] [PubMed] [Google Scholar]
- 9.Ennis M, Pearce FL, Weston PM. Some studies on the release of histamine from mast cells stimulated with polylysine. Br J Phamacol. 1980;70:329. doi: 10.1111/j.1476-5381.1980.tb07940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allansmith MR, Baird RS, Ross RN, Barney NP, Bloch KJ. Ocular anaphylaxis induced in the rat by topical application of compound 48/80. Dose response and time course study. Acta Ophthalmol. 1989;192(suppl.):145. doi: 10.1111/j.1755-3768.1989.tb07106.x. [DOI] [PubMed] [Google Scholar]
- 11.Segal DM, Taurog J, Metzger H. Dimeric immunoglobulin E serves as a unit signal for mast cell degranulation. Proc Natl Acad Sci USA. 1977;74:2993. doi: 10.1073/pnas.74.7.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metzger H, Alcaraz G, Hohman R, Kinet JP, Pribluda V, Quarto R. The receptor with high affinity for immunoglobulin E. Annu Rev Immunol. 1986;4:419. doi: 10.1146/annurev.iy.04.040186.002223. [DOI] [PubMed] [Google Scholar]
- 13.Alber G, Miller L, Jelsema C, Varin-Blank N, Metzger H. Structure/function relationships in the mast cell high-affinity receptor for IgE (FcεRI): Role of cytoplasmic domains. J Biol Chem. 1991;266:22613. [PubMed] [Google Scholar]
- 14.Saito H, Nomura Y, et al. Screening methods for durg evaluation 3. In: Suzuki L, Tanaka H, Yajima H, editors. Pharmaceutical Research and Development. Tokyo: Hirokawa; 1989. p. 22. [Google Scholar]
- 15.Plaut M, Pierce JH, Watson CJ, Hanley-Hyde J, Nordon RP, Paul WE. Mast cell lines produce limphokines in response to cross-linkage of FcεRI or to calcium ionophores. Nature. 1989;339:64. doi: 10.1038/339064a0. [DOI] [PubMed] [Google Scholar]
- 16.Wodnar-filipowicz A, Heusser CH, Moroni C. Production of the haemopoietic growth factors GM-CSF and interleukin-3 by mast cells in response to IgE receptor-mediated activation. Nature. 1989;339:150. doi: 10.1038/339150a0. [DOI] [PubMed] [Google Scholar]
- 17.Burd PR, Rogers HW, Gordon JR, et al. Interleukin 3-dependent and -independent mast cells stimulated with IgE and antigen express multiple cytokines. J Exp Med. 1989;170:245. doi: 10.1084/jem.170.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurish MF, Ghildyal N, Arm J, et al. Cytokine mRNA are preferentially increased relative to secretory granule protein mRNA in mouse bone marrow-derived mast cells that have undergone IgE-mediated activation and degranulation. J Immunol. 1991;146:1527. [PubMed] [Google Scholar]
- 19.Galli SJ, Gordon N, Wershil BK. Cytokine production by mast cells and basophils. Curr Opin Immunol. 1991;3:865. doi: 10.1016/s0952-7915(05)80005-6. [DOI] [PubMed] [Google Scholar]
- 20.Katayama S, Shionoya H, Ohtake S. A new method for extraction of extravasated dye in the skin and the influence of fasting stress on passive cutaneous anaphylaxis in guinea pigs and rats. Microbiol Immunol. 1978;22:89. doi: 10.1111/j.1348-0421.1978.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 21.Shore PA, Burkhalter A, Cohn VH. A method for fluorometric assay of histamine in tissues. J Pharmacol Exp Ther. 1959;127:182. [PubMed] [Google Scholar]
- 22.Shin BK, Lee EH, Kim HM. Suppression of l-histidine decarboxylase mRNA expression by methyleugenl. Biochem Biophysics Res Commun. 1997;232:188. doi: 10.1006/bbrc.1997.6260. [DOI] [PubMed] [Google Scholar]
- 23.Yurt RW, Leid RW, Austen KF. Native heparin from rat peritoneal mast cells. J Biol Chem. 1977;252:518. [PubMed] [Google Scholar]
- 24.Yamamoto J, Yatsunami K, Ohmori E, et al. cDNA-derived amino acid sequence of l-histidine decarboxylase from mouse mastocytoma P-815 cells. FEBS Lett. 1990;276:214. doi: 10.1016/0014-5793(90)80545-t. [DOI] [PubMed] [Google Scholar]
- 25.Kim HM, Hirota S, Chung HT, et al. Differential expression of protein kinase C genes in cultured mast cells derived from normal and mast cell-deficient mice and mast cell lines. Int Arch Allergy Immunol. 1994;105:258. doi: 10.1159/000236766. [DOI] [PubMed] [Google Scholar]
- 26.Scuderi P, Sterling RE, Lam KS, et al. Raised serum levels of tumor necrosis factor in parasitic infections. Lancet. 1986;2:1364. doi: 10.1016/s0140-6736(86)92007-6. [DOI] [PubMed] [Google Scholar]
- 27.Peachell PT, Macglashan DW, Lichtenstein LM, Schleimer RP. Regulation of human basophil and lung mast cell function by cyclic adenosine monophosphate. J Immunol. 1988;140:571. [PubMed] [Google Scholar]
- 28.Piotrowski W, Foreman JC. On the actions of substance P, somatostatin, and vasoactive intestinal polypeptide on rat peritoneal mast cells and in human skin. Naunyn Schmiedeberg's Arch Pharmacol. 1985;331:364. doi: 10.1007/BF00500821. [DOI] [PubMed] [Google Scholar]
- 29.Mousli MC, Bronner C, Bockaert J, Rouot B, Landry Y. Interaction of substance P, compound 48/80 and mastoparan with α-subunit C-terminal of G protein. Immunol Lett. 1990;25:355. doi: 10.1016/0165-2478(90)90207-7. [DOI] [PubMed] [Google Scholar]
- 30.Mousli MC, Bronner C, Landry Y, Bockaert J, Rouot B. Direct activation of GTP-binding regulatory proteins (G proteins) by substance P and compound 48/80. FEBS Lett. 1990;259:260. doi: 10.1016/0014-5793(90)80023-c. [DOI] [PubMed] [Google Scholar]
- 31.Bueb J-L, Mousli MC, Bronner C, Rouot B, Landry Y. Activation of Gi-like proteins, a receptor-independent effect of kinins in mast cells. Mol Pharmacol. 1990;38:816. [PubMed] [Google Scholar]
- 32.Tasaka K, Mio M, Okamoto M. Intracellular calcium release induced by histamine releasers and its inhibition by some antiallergic drugs. Ann Allergy. 1986;56:464. [PubMed] [Google Scholar]
- 33.Shibata H, Mio M, Tasaka K. Analysis of the mechanism of histamine release induced by substance P. Biochim Biophys Acta. 1985;846:1. doi: 10.1016/0167-4889(85)90102-8. [DOI] [PubMed] [Google Scholar]
- 34.Krumins SA, Broomfield CA. C-terminal substance P fragments elicit histamine release from a murine mast cell line. Neuropeptides. 1993;24:5. doi: 10.1016/0143-4179(93)90035-9. [DOI] [PubMed] [Google Scholar]
- 35.Piotrowski W, Devoy MAB, Jordan CC, Foreman JC. The substance P receptor on rat mast cells and in human skin. Agents Actions. 1984;14:420. doi: 10.1007/BF01973842. [DOI] [PubMed] [Google Scholar]
- 36.Makino H, Saijo T, Ashida Y, Kuriki HH, Maki Y. Mechanism of action of an antiallergic agent, Amlexanox (AA-673), in inhibiting histamine release from mast cells. Int Arch Allergy Immunol. 1987;82:66. doi: 10.1159/000234292. [DOI] [PubMed] [Google Scholar]
- 37.Murakami M, Kudo I, Suwa Y, Inoue K. Release of 14-kDa group II phospholipase A2 from activated mast cells and its possible involvement in the regulation of the degranulation process. Eur J Biochem. 1992;209:257. doi: 10.1111/j.1432-1033.1992.tb17284.x. [DOI] [PubMed] [Google Scholar]
- 38.Butterfield JH, Kao PC, Klee GC, Yocum MW. Aspirin idiosyncrasy in systemic mast cell disease: a new look at mediator release during aspirin desensitization. Mayo Clin Proc. 1995;70:481. doi: 10.4065/70.5.481. [DOI] [PubMed] [Google Scholar]