

Abstract
Pig membrane cofactor protein (MCP; CD46) is a 50 000–60 000 MW glycoprotein that is expressed on a wide variety of cells, including erythrocytes. Pig MCP has cofactor activity for factor I-mediated cleavage of C3b and is an efficient regulator of the classical and alternative pathway of human and pig complement. A panel of 10 monoclonal antibodies (mAbs) was collected from two different laboratories; all of these mAbs were raised against pig leucocytes and all recognized the same complex banding pattern on sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) of erythrocyte membranes. All were shown to be reactive with pig MCP and were divided into four groups of mutually competitive antibodies based on competition studies for membrane-bound MCP and for soluble MCP, the latter by surface plasmon resonance (SPR) analysis. The antigenic properties of membrane-bound and soluble MCP were similar, although some interesting differences were revealed. None of the 10 mAbs were cross-reactive with human MCP and only one showed cross-reactivity with leucocytes from a panel of large mammals – a weak cross-reactivity with a subset of dog leucocytes. All antibodies in one of the epitope groups and some in a second epitope group were able to block the functional activity of pig MCP, as measured by inhibition of MCP-catalysed C3 degradation by factor I.
INTRODUCTION
Human membrane cofactor protein (MCP or CD46) is an important membrane-bound regulator of complement (C) activation. MCP serves as cofactor for the plasma serine protease factor I in the degradation of C3b and C4b deposited on self-tissues.1 MCP is expressed on a wide variety of cells but it is absent from erythrocytes. MCP is a glycoprotein consisting of four homologous short consensus repeats (SCR), a serine/threonine/proline (STP)-rich region, and transmembrane and cytoplasmic domains. The SCR are characteristic of the regulators of complement activation (RCA) family of C-regulators to which MCP belongs.2 In human MCP, SCR 3 and 4 are necessary for cofactor activity for the cleavage of C4b and C3b.3 Alternative splicing of the STP and cytoplasmic domains results in expression of multiple isoforms of MCP.4 On peripheral blood cells, individuals may express predominantly a 65 000 MW isoform, express predominantly a 45 000 MW isoform or express equal amounts of the two isoforms, and this characteristic is stable and inherited in an autosomal codominant fashion.5,6 In addition to its role in C regulation, human MCP is of interest in reproductive immunology because of its expression on sperm and at the maternal–fetal interface,7 to tumour immunology because of its high expression on malignant cells,8,9 and to microbiology because of its role as a measles virus receptor10 and as a receptor for the M protein of group A streptococci.11
We have recently purified and characterized the pig analogue of human MCP.12 Pig MCP is a 50 000–60 000 MW glycoprotein expressed on a wide variety of cells including, in contrast to human MCP, erythrocytes. Western blotting of pig leucocytes and erythrocytes revealed the presence of multiple isoforms – typically three distinct bands in the molecular weight range 45 000–65 000 are expressed.12 Molecular cloning of pig MCP revealed a 43% homology with human MCP and a very similar protein structure.13 We have shown previously that pig MCP is an efficient regulator of the classical and alternative pathway of pig and human C.12 The presence of a resident MCP on pig cells, which is capable of acting as a cofactor in the control of human C activation, has consequences for the use of pig organs in xenotransplantation.12
Following the original publication identifying pig MCP, it was realized that the uncharacterized pig leucocyte antigen recognized by three newly derived mAb (INIA6D8, INIA1C5 and INIA2C11; raised in CISA-INIA, Valdeolmos, Spain) resembled pig MCP in terms of behaviour on sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and tissue distribution.14 We here set out to confirm that these antibodies were reactive with pig MCP, to characterize the epitopes on MCP that were recognized by all available anti-pig MCP antibodies, and to examine whether any of the available antibodies blocked the cofactor activity of MCP. mAb that block the function of pig MCP might prove useful in analysing the role of endogenous pig MCP in the prevention of hyperacute rejection in xenotransplantation.
MATERIALS AND METHODS
Cell preparation
Fresh pig blood, obtained from the local abattoir, was collected into 0·38% sodium citrate as anticoagulant, and served as a source of pig leucocytes and erythrocytes (PgE). Blood from other species were obtained from the local animal facilities. Peripheral blood mononuclear cells (PBMC) were isolated by density-gradient centrifugation on Ficoll–Hypaque (Pharmacia, Uppsala, Sweden).
Proteins and antibodies
Purified pig MCP was obtained from PgE ghosts by immunoaffinity purification using the mAb, JM4C8, as previously described.12 Human and pig C3 were purified according to the method described for mouse C3.15 Human factor I was a kind gift of Dr T. Farries (Imutran Ltd, Cambridge, UK). mAbs JM2G11, JM3H6, JM4C8, JM5C3, JM6C5, JM6C11 and JM7A11 were produced by the fusion of splenocytes from a BALB/c mouse, previously immunized with porcine PBMC, and the Sp2/O-Ag14 mouse myeloma cell line, as previously described.12,16 mAbs INIA1C5, INIA2C11 and INIA6D8/8 were produced as previously described.17 mAb isotype was determined by sandwich enzyme-linked immunosorbent assay (ELISA), using rabbit antisera specific for mouse immunoglobulins and a peroxidase-conjugated goat anti-mouse immunoglobulin (Sigma, St Louis, MO). All mAb were of immunoglobulin G1 isotype except INIA1C5, which was immunoglobulin G2a.
mAb were purified from mouse ascitic fluid by affinity chromatography on a Sepharose protein A column (Pharmacia). Ascites was passed over a precolumn containing Sepharose 4B (Pharmacia) and then applied to the protein A column. After sequential washing with two to three column volumes of coupling buffer (15 mm glycine, 3 m NaCl, pH 8·9), the bound mAbs were eluted with 0·1 m citric acid, pH 3, in fractions of 1 ml. Fractions containing mAb were dialysed against 0·1 m sodium bicarbonate, pH 8·3. Antibodies (1 mg) were labelled with 0·125 mg of biotin-N-hydrosuccinimide (Sigma) at room temperature for 4 hr and finally dialysed against phosphate-buffered saline (PBS).
Flow cytometry
PBMC from several species (106/ml in PBS) were incubated on ice for 30 min with 50 μl of hybridoma supernatant (50 μg/ml), or an irrelevant mAb as a negative control. Cells were washed three times in flow cytometry buffer (FCB: PBS, 0·1% bovine serum albumin (BSA) and 0·1% NaN3). Cells were incubated at 4° for 30 min with 50 μl of fluorescein isothyocyanate (FITC)-conjugated goat anti-mouse immunoglobulin G (Sigma), diluted 1:500 in FCB. After three washes in FCB, cells were fixed with 0·5 ml of 1% paraformaldehyde in PBS. Fluorescence was analysed using a Becton-Dickinson FACScalibur (San Jose, CA) equipped with the CellQuest software. Cells were identified and gated by their characteristic forward and side scatter.
Competition assays
Cross-blocking experiments were performed as described previously.18 Briefly, biotin anti-MCP mAbs were titrated on 5×105 porcine PBMC to achieve a concentration that resulted in 70–90% maximal fluorescence staining. For inhibition studies, cells were first incubated with an excess of unlabelled mAb at 4° for 15 min. Thereafter, cells were incubated with biotin-labelled mAb, at the submaximal concentration identified by titration, at 4° for an additional 30 min. After washing, cells were incubated with streptavidin–phycoerythrin (Southern Biotechnology, Birmingham, AL) at 4° for 15 min and fluorescence was analysed by flow cytometry. An irrelevant mAb (immunoglobulin G1 isotype) was used as negative control for inhibition. Results were expressed as a ratio between fluorescence intensity (mean channel) of the cells preincubated with unlabelled mAb and the fluorescence intensity obtained for each biotinylated mAb alone. The percentage of inhibition (PI) was calculated by the following formula:
Immunoprecipitation analysis
Pig leucocytes (107) were resuspended in PBS and labelled by incubation with 0·4 mg/ml (final concentration) sulpho-NHS-biotin (Pierce, Chester, UK) at 4° for 15 min. After washing three times with PBS, cells were lysed with 0·5 ml of lysis buffer (10 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40 (NP-40), 1 mm phenylmethylsulphonyl fluoride (PMSF) and 1 mm EDTA). The lysate was precleared with 50 μl of a 25% (v/v) suspension of protein A–Sepharose (Pharmacia) in lysis buffer. Lysates (0·5 ml) were then incubated with 1 ml of hybridoma supernatant at 4° for 4 hr. Then, 50 μl of 25% (v/v) protein A–Sepharose was added and incubated at room temperature (RT) for 1 hr with gentle mixing. Beads were washed three times with lysis buffer, boiled in reducing electrophoresis sample buffer and the supernatant run on 5–15% SDS–PAGE19 and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA). Membrane-free binding sites were blocked with 5% non-fat powdered milk in PBS at RT for 1 hr. After three washes with PBS containing 0·1% Tween-20, the PVDF membrane was incubated with streptavidin horseradish peroxidase (Amersham, Bucks, UK) for 1 hr at room temperature. Finally, bands were visualized with the enhanced chemiluminiscense (ECLTM) reagents (Amersham), following the recommendations of the manufacturer.
Western blotting
Pig erythrocyte membranes (300 μl) were solubilized in 0·5 ml of lysis buffer (10 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1% NP-40 and 1 mm PMSF) at RT for 1 hr. After centrifugation at 12 000 gfor 15 min, the supernatant was mixed with sample buffer and run on 5–15% SDS–PAGE under reducing and non-reducing conditions. The separated proteins were then transferred to a nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). The nitrocellulose was blocked with PBS containing 5% non-fat powdered milk. Strips were cut and incubated with hybridoma supernatants at RT for 1 hr, followed by a 1-hr incubation with a peroxidase-labelled rabbit anti-mouse immunoglobulin (Sigma). Peroxidase activity was visualized using 1-chloro–4-naphthol (Sigma) as a substrate.
Inhibition of cofactor activity
Factor I cofactor activity in the affinity-purified pig MCP was assessed essentially as described previously.12 In brief, human C3 was inactivated by treatment with methylamine (100 mm, pH 8·0, at 37° for 1 hr) and dialysed against PBS. Pig MCP was preincubated with a threefold excess of mAb. Methylamine-treated C3 (200 ng) was then incubated with human factor I (40 ng) together with the purified pig MCP (200 ng; in the presence or absence of mAbs: 600 ng) or PBS in a total volume of 28 μl of PBS/0·05% NP-40 for 16 hr at 37°. Reducing buffer was added, and samples were run on 10% SDS–PAGE and Western blotted onto nitrocellulose. Blots were probed for C3 and C3-split products using a rabbit anti-C3c antibody. Blots were developed using GAR-HRPO (Bio-Rad, Hemel Hempstead, UK) and Supersignal chemiluminescent substrate (Pierce & Warriner, Chester, UK).
Surface plasmon resonance (SPR) analysis
SPR measurements were performed using a BIAcoreX system (Pharmacia Biosensor AB, Pharmacia). A rabbit anti-mouse immunoglobulin was covalently coupled to a CM5 sensor chip, using amine-coupling reagents, following the recommendations of the manufacturer. All analyses were performed at 25°, at a flow rate of 5 μl/min and using HEPES-buffered saline as the running buffer. The regeneration of the matrix was performed by injection of 20 μl of 50 mm glycine, pH 2·0. The capture antibody was added at 50 μg/ml for 4 min (typical accumulation between 500 and 1000 resonance units, RU). Unoccupied binding sites were blocked by injections of 20 μl of mouse immunoglobulin G (100 μg/ml) until no further response was observed (typical accumulation between 400 and 700 RU). Antigen was then injected at 25 μg/ml for 4 min (typical accumulation between 100 and 300 RU). Secondary mAbs, at 50 μg/ml, were finally introduced for 4 min and tested for binding to purified pig MCP. The binding of each secondary mAb (in RU) was compared with binding of the same antibody used for capture and binding of all other antibodies and expressed as a percentage of inhibition. All antibody combinations were tested in each direction so that all antibodies were, in turn, immobilized on the chip and tested with every antibody in the fluid phase.
RESULTS
Antibody specificities for pig MCP
mAbs JM2G11, JM3H6, JM4C8, JM5C3, JM6C5, JM6C11 and JM7A11 were characterized previously as specific for the pig analogue of MCP.12 In order to confirm the specificity of the new mAbs INIA6D8/8, INIA2C11 and INIA2C5 for pig MCP, immunoprecipitation experiments were performed. The immunoprecipitated molecules from biotinylated pig leucocytes were subjected to SDS–PAGE under reducing conditions. After blotting onto PVDF, the immunoprecipitated antigen was detected by streptavidin horseradish peroxidase. The antigen recognized by the mAbs consisted of several closely apposed bands of apparent Mr 50 000–60 000 Da, consistent with the Mr reported for pig MCP (Fig. 1). Western-blotting experiments performed with a porcine erythrocyte lysate showed that the three new antibodies and the seven previously characterized mAb all recognized precisely the same banding pattern. All antibodies recognized all bands from the erythrocyte lysate, indicating that all mAbs recognized epitopes common to all isoforms of pig MCP expressed on erythrocytes, under both reducing and non-reducing conditions (Fig. 2).
Figure 1.
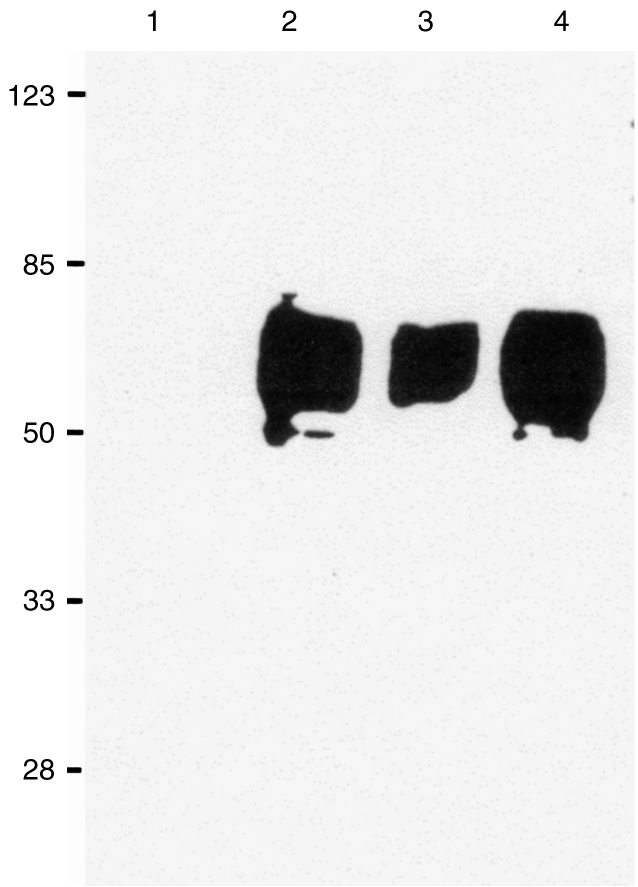
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) of the antigens immunoprecipitated from biotinylated pig peripheral blood mononuclear cells (PBMC) by anti-pig membrane cofactor protein (MCP) monoclonal antibodies (mAbs). The gel was blotted and developed with streptavidin horseradish peroxidase. Lanes: 1, negative control; 2: mAb INIA2C11; 3: mAb INIA1C5; 4: mAb INIA6D8. Molecular weight markers are shown on the left.
Figure 2.

Pig erythrocyte ghosts were solubilized, separated using sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and Western blotted. Strips of the blot were probed using different anti-pig membrane cofactor protein (MCP) monoclonal antibodies (mAbs). Lane 1, JM2G11; 2, JM3H6; 3, JM4C8; 4, JM5C3; 5, JM6C5; 6, JM6C11; 7, JM7 A11; 8, INIA1C5; 9, INIA2C11; 10, INIA6D8. 11, irrelevant mAb. (a) Reducing conditions; (b) non-reducing conditions. Molecular weight markers are shown on the left.
Cross-species reactivity
Leucocytes from other species were evaluated by flow cytometry to investigate if any of the 10 available mAb anti-pig MCP could recognize epitopes in MCP from different species (Table 1). Only mAb INIA1C5 showed evidence of cross-species reactivity, reacting with a subset of dog leucocytes at a relatively low intensity (Fig. 3). Given that this cross-reactivity was with only a small subpopulation of dog leucocytes, it seems unlikely that this represents dog MCP. No cross-reactivity of any of the antibodies was observed with sheep, guinea-pig, human, bovine or horse erythrocytes, or with the human promonocyte U937 cell line (data not shown).
Table 1.
Cross reactivity of the monoclonal antibodies (mAbs) to pig membrane cofactor protein with leucocytes from other species. Results are expressed as percentage positive cells by flow cytometry
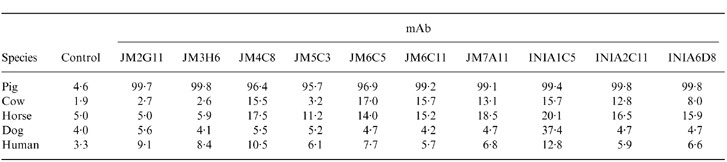
Figure 3.

Flow cytometric profiles of monoclonal antibody (mAb) INIA1C5 on pig, human, horse, dog and human peripheral blood mononuclear cells (PBMC). Unshaded profiles correspond to background staining with an irrelevant mAb.
Competition assays
Flow cytometric analyses were performed in order to determine the epitope specificity of the 10 mAbs. Unlabelled mAbs (hybridoma supernatants) were incubated with biotin-labelled purified mAb for competitive binding to pig leucocytes. PI was expressed as a ratio between fluorescence intensity (mean channel) of the cells preincubated with unlabelled mAbs and the fluorescence intensity obtained for each biotinylated mAb alone (Table 2). Inhibition was considered significant when the PI was 50% or higher. Taking this value as a cut-off, it was possible to place the mAbs into four groups of mutually competitive antibodies (Table 3). Group A comprised the mAbs INIA2C11, JM2G11, JM5C3 and INIA6D8. The second group (group B) was formed by mAbs JM4C8, JM6C11, JM7A11 and JM6C5. Groups C and D were represented by a single mAb in each: INIA1C5 and JM3H6, respectively.
Table 2.
Competition assays performed by flow cytometry
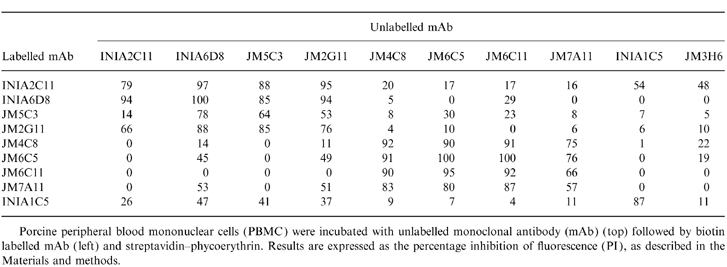
Porcine peripheral blood mononuclear cells (PBMC) were incubated with unlabelled monoclonal antibody (mAb) (top) followed by biotin labelled mAb ( left) and streptavidin–phycoerythrin. Results are expressed as the percentage inhibition of fluorescence (PI), as described in the Materials and methods.
Table 3.
Classification of monclonal antibodies (mAbs) to pig membrane cofactor protein (MCP) in four different groups, according to their percentage of inhibition (PI) values obtained in the competition assay
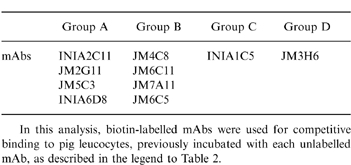
In this analysis, biotin-labelled mAbs were used for competitive binding to pig leucocytes, previously incubated with each unlabelled mAb, as described in the legend to Table 2.
Epitope mapping by SPR
In order to complement the competition assays utilizing pig MCP bound to cells, epitope analysis of the purified soluble molecule was performed using SPR technology, which measures real time interactions. In this system, binding events are monitored by observing the increase in RU and the signal is displayed in a sensorgram plot. The ability of each mAb to bind MCP captured on the chip was assessed using each of the mAb immobilized on the chip to capture. When the capturing and detecting mAbs can bind simultaneously to the purified antigen, they define separate and independent epitopes. If the detecting mAb fails to bind, then either the epitope defined by capturing and detecting mAbs are identical or overlapping, or binding of antigen causes conformational changes resulting in loss of epitopes. Table 4 shows the percentage of inhibition calculated as a ratio of the RU obtained from each detecting mAb, and the RU obtained when the capturing mAb and detecting mAb were the same. Inhibition values of 50% or higher were considered significant.
Table 4.
Epitope mapping by surface plasmon resonance (SPR) analysis
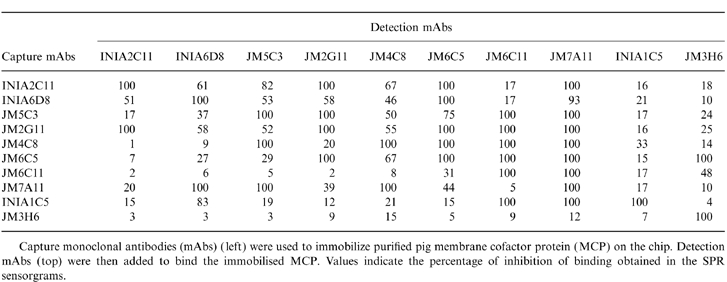
Capture monoclonal antibodies (mAbs) ( left) were used to immobilize purified pig membrane cofactor protein (MCP) on the chip. Detection mAbs (top) were then added to bind the immobilised MCP. Values indicate the percentage of inhibition of binding obtained in the SPR sensorgrams.
The antibodies placed in group A on the basis of binding to cell surface MCP (INIA2C11, INIA6D8, JM2G11 and JM5C3) were, in general, also competitive with one another in this system in both directions, the sole exception being JM5C3 which, when used as capture, competed strongly only with JM2G11 in this group and also competed with all mAbs of group B. Antibodies in group B were also in most instances competitive with one another, the few exceptions being loss of competition in one direction only. The antibody placed in group C (INIA1C5) competed with no other mAb when used as detection but when used as capture competed with several mAbs in groups A and B. Conversely, the antibody placed in group D (JM3H6) competed with no others when used as capture but strongly competed with JM6C11 (group B) when used as detection. The data make it clear that antibody capture can, in some cases, markedly alter the epitopes exposed on the antigen.
Functional assay
In order to establish whether any of the four epitopes recognized by the different mAbs were involved in the function of pig MCP, the effect of each mAb on MCP cofactor activity was examined. Human C3 and human factor I were used in this assay. Pig MCP was preincubated with mAb and human C3b and factor I were added subsequently. Cofactor activity was assessed by the cleavage of the α-chain of C3. Several mAbs blocked the function of MCP (Fig. 4). All mAbs in group B (JM4C8, JM6C5, JM6C11 and JM7 A11) completely inhibited cleavage of C3, indicating that the epitope recognized by these mAbs was involved in cofactor activity. Two of the mAbs in group A, JM5C3 and INIA6D8, did not inhibit cofactor activity; however, two mAbs from the same group (JM2G11 and INIA2C11) caused a partial inhibition of cofactor activity, as shown by a decrease in C3 cleavage product. The mAb from group C did not have any effect on cofactor activity. Owing to an insufficient amount of mAb, the activity of JMH36 (group D) could not be determined. Two mAbs kindly donated by Dr Taroh Kinoshita (Research Institute for Microbiological Diseases, Osaka University, Osaka, Japan), mAbs MCP6 and MCP7 raised against recombinant pig MCP,13 were also inactive in the inhibition assay (data not shown).
Figure 4.

Inhibition of cofactor activity of pig membrane cofactor protein (MCP) for cleavage of human C3 by human factor I. Pig MCP was incubated with an excess of individual anti-pig MCP monoclonal antibodies (mAbs), followed by addition of human C3 and factor I. The mixture was incubated for 16 hr at 37°, separated through 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) under reducing conditions, blotted onto nitrocellulose, and probed with anti-human C3. Cleavage is indicated by loss of the α-chain and the appearance of bands at 47 and 43 kDa. Lanes 1–3, group A mAbs (INIA6D8, JM5C3, JM2G11); lanes 4–7, group B mAbs (JM4C8, JM6C5, JM6C11, JM7A11); lane 8, mouse immunoglobulin G; lane 9, no antibody; lane 10, no pig MCP. The group A antibody, INIA2C11, not shown on this gel, caused partial inhibition, equivalent to that shown for JM2G11 (lane 3). The mAb INA1C5 (group C), also not shown on this gel, caused no inhibition of MCP.
DISCUSSION
Human CD46/MCP is an important regulator of C activation and is one of the three membrane-bound regulators of C that act at the stage of the C3/C5-convertase. We have recently identified the pig homologue of human MCP. Like human MCP, pig MCP is widely and abundantly expressed, but unlike human MCP it is also expressed on erythrocytes. Pig MCP was purified from pig erythrocyte ghosts by immunoaffinity chromatography and was shown to be a transmembrane glycoprotein of 50 000–60 000 MW. Pig MCP, like human MCP, consists of four SCRs followed by a carbohydrate-rich STP region, a transmembrane segment and a cytoplasmic tail.13 Like human MCP, pig MCP exhibits a range of different Mr on SDS–PAGE owing to the presence of multiple isoforms, probably generated by alternative splicing. MCP analogues have also been identified in primates,20 guinea-pigs21 and mice.22
Regulation of complement in the pig has become a subject of interest because of the planned use of pig organs for transplantation to humans. Pigs are currently being engineered to express human complement regulators in the expectation that organs from these pigs will be less susceptible to damage by human complement.23 We have undertaken to characterize membrane regulators of complement in the pig. Here we present further data on pig MCP. A panel of 10 mAbs was assembled, all of which recognize all isoforms of pig MCP, and these have been characterized in terms of the epitopes recognized on MCP, cross-species reactivity and function-blocking capacity. mAbs that block the cofactor activity of pig MCP will be particularly useful in analysing the likely contribution of pig MCP to protection in models of xenotransplantation.
None of the 10 mAbs cross-reacted to any significant degree with leucocytes from humans or from several other mammalian species. mAb INIA1C5 cross-reacted with a minor subset of dog leucocytes, but this has not been further investigated. Competition studies for binding to cell-surface MCP, using pairs of mAbs, identified four distinct epitope groups in pig MCP: mAbs in each group competing with one another but not with mAbs in other groups. Attempts to confirm this binding data using SPR technology and capture of soluble, purified MCP on individual mAbs, met with limited success. Although the epitope groups identified by competition on cell-surface MCP were apparent, some combinations of mAbs, not competitive in this first approach, appeared to compete, although often in one direction only. This confusing data suggested that capture of the antigen by some of the mAbs caused conformational changes in MCP such that epitopes remote from those utilized by the capture mAbs were altered. Conformational changes leading to the development of cryptic epitopes when antigen is bound to a solid support have been reported for several proteins, including the human complement regulator CR1.24,25
Support for the epitope groups identified from competitive binding assays was provided by the functional assay. mAbs of group B (JM4C8, JM6C5, JM6C11 and JM7A11) all strongly inhibited factor I cofactor activity of MCP, as shown by a complete inhibition of cleavage of C3 (Fig. 4). Group A mAbs JM5C3 and INIA6D8 did not inhibit MCP cofactor activity in this assay, but the two other mAbs from this group, JM2G11 and INIA2C11, both showed a partial inhibition of cofactor activity, as shown by the markedly reduced amount of C3 cleavage. This finding suggests that these two pairs of antibodies in group A bind different, but closely linked, epitopes, one of which is involved in MCP cofactor activity. Alternatively, mAbs INIA2C11 and JM2G11 may induce conformational changes in MCP such that the ‘active site’ epitope is altered. This latter interpretation is supported by the SPR data; using INIA2C11 as capture, subsequent binding of three of four group B mAbs was inhibited, and using JM2G11 as capture, binding of all four group B mAbs was inhibited. The single group C mAb (INA1C5) did not inhibit cofactor activity. Of interest, all the mAbs were also tested in a haemolysis assay to examine whether they enhanced lysis of pig erythrocytes by pig or human serum under classical pathway conditions. None of the mAbs caused a significant increase in pig erythrocytes (PgE) haemolysis in this assay (data not included). It has previously been reported that mAbs known to block human MCP function in a cofactor assay do not result in enhanced lysis when MCP is blocked on the cell surface.26 Enhancement of lysis by anti-MCP mAb was only observed when the two other major membrane-bound C regulators, CD59 and decay accelerating factor (DAF), were also blocked.26 Human MCP is considered to be mainly a regulator of the alternative pathway27 and it is possible that the classical pathway conditions used in our experiments militated against obtaining enhanced lysis. We were unable to obtain lysis of pig erythrocytes under alternative pathway conditions.
In conclusion, we describe the evaluation in terms of epitope specificity, species specificity and cofactor activity blocking of 10 mAbs against pig MCP. These mAbs will prove useful in characterizing the contribution of pig MCP to controlling complement in the pig and may facilitate investigations in models of xenotransplantation.
Acknowledgments
J.M.P.L. is a recipient of a ‘Marie Curie’ TMR fellowship, project ERB-FMBICT-972590. B.P.M. is a Wellcome Trust Senior Fellow. This work was supported by the Spanish Comision Interministerial de Ciencia y Tecnologia, project BIO97-0402. The authors acknowledge the collaboration with Dr E. Vinuela's group (Centro de Biologia Molecular, Universidad Autonoma de Madrid,Spain).
REFERENCES
- 1.Seya T, Turner JR, Atkinson JP. Purification and characterization of a membrane protein (gp45) that is a cofactor for cleavage of C3b and C4b. J Exp Med. 1986;163:837. doi: 10.1084/jem.163.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hourcade D, Holers VM, Atkinson JP. The regulators of complement activation (RCA) gene cluster. Adv Immunol. 1989;45:381. doi: 10.1016/s0065-2776(08)60697-5. [DOI] [PubMed] [Google Scholar]
- 3.Manchester M, Valsamakis A, Kaufman R, et al. Measles virus and C3 binding sites are distinct on membrane cofactor protein (CD46) Proc Natl Acad Sci USA. 1995;92:2303. doi: 10.1073/pnas.92.6.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liszewski MK, Post TW, Atkinson JP. Membrane cofactor protein (MCP or CD46) Annu Rev Immunol. 1991;9:431. doi: 10.1146/annurev.iy.09.040191.002243. [DOI] [PubMed] [Google Scholar]
- 5.Seya T, Ballard L, Bora N, Mcnearny T, Atkinson JP. Distribution of membrane cofactor protein (MCP) of complement on human peripheral blood cells. Eur j Immunol. 1988;18:1289. doi: 10.1002/eji.1830180821. [DOI] [PubMed] [Google Scholar]
- 6.Bora NS, Post TW, Atkinson JP. Membrane cofactor protein of the complement system. A Hin dIII restriction fragment length polymorphism that correlated with the expression polymorphism. Immunology. 1991;146:2821. [PubMed] [Google Scholar]
- 7.Petrányi G, Padányi Á, Szelényi J, et al. The polymorphic human TLX-B/CD46/MCP system and its implications in transplantation and reproduction. Eur J Immunogenet. 1995;22:147. doi: 10.1111/j.1744-313x.1995.tb00225.x. [DOI] [PubMed] [Google Scholar]
- 8.Seya T, Hara T, Matsumoto M, Akedo H. Quantitative analysis of membrane cofactor protein (MCP) of complement. J Immunol. 1990;145:238. [PubMed] [Google Scholar]
- 9.Hara T, Kojima A, Fukuda H, et al. Levels of complement regulatory proteins, CD35 (CR1), CD46 (MCP) and CD55 (DAF) in human haematological malignancies. Br J Haematol. 1992;82:368. doi: 10.1111/j.1365-2141.1992.tb06431.x. [DOI] [PubMed] [Google Scholar]
- 10.Naniche D, Varior-Krishnan G, Cervoni F, et al. Human membrane cofactor protein (CD46) acts a cellular receptor for measles virus. Virology. 1993;67:6025. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okada N, Liszewski MK, Atkinson JP, Caparon M. Membrane cofactor protein (MCP or CD46) is a keratinocyte receptor for the M protein of the group A streptococcus. Proc Natl Acad Sci USA. 1995;92:2489. doi: 10.1073/pnas.92.7.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Den Berg CW, Pérez De La Lastra JM, Llanes D, Morgan BP. Purification and characterization of the pig analogue of human membrane cofactor protein (CD46/MCP) J Immunol. 1997;158:1703. [PubMed] [Google Scholar]
- 13.Toyomura K, Fujimura T, Murakami H, et al. Molecular cloning of a pig homologue of membrane cofactor protein (CD46) Int Immunol. 1997;9:869. doi: 10.1093/intimm/9.6.869. [DOI] [PubMed] [Google Scholar]
- 14.Bullido R. Veterinary Faculty, University of Madrid; 1997. Obtención y caracterizacion de anticuerpos monoclonales frente a antigenos de membrana de leucocitos porcinos. PhD Thesis. [Google Scholar]
- 15.Van Den Berg CW, Van Dijk H, Capel PJA. Rapid isolation and characterization of native mouse complement components C3 and C5. J Immunol Methods. 1989;122:73. doi: 10.1016/0022-1759(89)90336-0. [DOI] [PubMed] [Google Scholar]
- 16.Perez De La Lastra JM, Moreno A, Perez J, Llanes D. Characterization of the porcine homologue to human platelet glycoprotein IIb-IIIa (CD41/CD61) by a monoclonal antibody. Tissue Antigens. 1997;49:588. doi: 10.1111/j.1399-0039.1997.tb02806.x. [DOI] [PubMed] [Google Scholar]
- 17.Bullido R, Gomez Del Moral M, Alonso F, et al. Monoclonal antibodies specific for porcine monocytes/macrophages: macrophage heterogeneity in the pig evidenced by the expression of surface antigens. Tissue Antigens. 1997;49:403. doi: 10.1111/j.1399-0039.1997.tb02769.x. [DOI] [PubMed] [Google Scholar]
- 18.Pescovitz MD, Aasted B, Canals A, et al. Analysis of monoclonal antibodies reactive with the porcine CD2 antigen. Vet Immunol Immunopathol. 1994;43:229. doi: 10.1016/0165-2427(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Nickells MM, Atkinson JP. Characterization of CR1 and membrane cofactor proteins of two primates. J Immunol. 1990;144:4262. [PubMed] [Google Scholar]
- 21.Hosokawa M, Nonaka M, Okada N, Nonaka M, Okada H. Molecular cloning of guinea-pig membrane cofactor protein, preferential expression in testis. J Immunol. 1997;157:4946. [PubMed] [Google Scholar]
- 22.Tsujimura A, Shida K, Kitamura M, et al. Molecular cloning of a murine homologue of membrane cofactor protein (CD46): preferential expression in testicular germ cells. Biochem J. 1998;330:163. doi: 10.1042/bj3300163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mccurry KR, Kooyman DL, Alvarado CG, et al. Human complement regulatory proteins protect swine-to-primate cardiac xenografts from humoral injury. Nature Med. 1995;1:423. doi: 10.1038/nm0595-423. [DOI] [PubMed] [Google Scholar]
- 24.Malmqvist M. Epitope mapping by label free biomolecular interaction analysis. Methods Enzymol. 1996;9:525. doi: 10.1006/meth.1996.0060. [DOI] [PubMed] [Google Scholar]
- 25.Nickells M, Hauhart R, Krych M, et al. Mapping epitopes for 20 monoclonal antibodies to CR1. Clin Exp Immunol. 1998;112:27. doi: 10.1046/j.1365-2249.1998.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris CL, Morgan BP. Characterization of a glycosyl-phosphatidylinositol anchor-deficient subline of Raji cells. An analysis of the functional importance of complement inhibitors on the Raji cell line. Immunology. 1995;86:311. [PMC free article] [PubMed] [Google Scholar]
- 27.Kojima A, Iwata K, Seya T, et al. Membrane cofactor protein (CD46) protects cells predominantly from alternative complement pathway-mediated C3-fragment deposition and cytolysis. J Immunology. 1993;151:1519. [PubMed] [Google Scholar]