

Abstract
Langerhans' cells (LC) are found in high numbers infiltrating skin tumours, the functional significance of which remains unknown. To study the mechanism by which tumours increase the number of LC we developed a procedure whereby supernatant from cultured T7 tumour cells applied topically increases the number of LC. Tumour factors increased the number of resident epidermal LC and did not attract LC precursors into parental murine skin grafted onto F1 hybrids. There was no evidence for increased LC division in response to the tumour-derived factors. LC migration from the epidermis to local lymph nodes, induced by topical fluorescein isothiocyanate (FITC), was inhibited by the tumour supernatant. To examine the functional significance of this, FITC-induced migration of LC from the epidermis overlying progressor tumours, which evade immunological destruction, and regressor tumours, which are immunologically destroyed, was examined. The progressor tumour T7 growing subcutaneously in syngeneic mice inhibited FITC-induced migration of LC from overlying epidermis. Furthermore, two progressor, but not two regressor murine skin tumour lines growing in BALB/c nu/nu mice inhibited LC migration from the epidermis. Our results demonstrate that progressor skin tumours produce factor(s) which inhibit LC migration from the epidermis to lymph nodes, leading to LC accumulation. Inhibition of LC migration by tumour-derived factors may enable tumours to evade the activation of protective immunity as regressor tumours do not interfere with the normal trafficking of LC.
INTRODUCTION
Primary immune responses to antigens encountered in the skin are initiated when naive T cells are activated by antigen presented to them in the draining lymph nodes by Langerhans' cells (LC). LC are dendritic cells (DC) that are located within the epidermis in a plane just above basal keratinocytes. The number of epidermal LC is tightly maintained under normal circumstances, even though there is a steady migration of LC from the epidermis to the draining local lymph nodes.1 LC can be stimulated to migrate from the epidermis to the local lymph nodes in response to various cutaneous stimuli including the application of fluorescein isothiocyanate (FITC).2 The maintenance of LC numbers in the epidermis is thought to be by replacement of LC predominantly from precursors which migrate into the epidermis from the blood,3 with a minority of repopulation caused by in situ cell division.4
DC have been shown to induce protective immunity against human cancer.5 It has previously been reported that actively growing human skin tumours become infiltrated with LC,6 although in mice higher LC numbers were shown not to be related to antitumour immunity.7,8 Effective immunity achieved with the number of LC in normal skin is dependent on their ability to migrate to local lymph nodes and activate T cells, as well as being present at a critical density. Hence, increased numbers of these cells may not always aid tumour rejection. The mechanism by which tumours increase the number of DC is unknown, and clarification of this would aid our understanding of the role of these cells in tumour biology.
We have previously shown that topical application of supernatants taken from a cultured skin tumour cell line, T7, were able to mimic the effect of an underlying tumour and increase the local LC density.9 This indicated that tumours produce a factor(s) which leads to an accumulation of LC. It also provided a biological model suitable to determine the cellular mechanisms by which tumours increase the number of LC. This could occur via: (a) increased migration of LC precursors into the epidermis; (b) increased epidermal LC mitosis; or (c) reduced migration of epidermal LC to the local lymph nodes. In this study we have examined the mechanism by which tumours increase LC numbers, and, by comparing progressor tumours that evade immunological destruction with regressor tumours that the immune system is able to eradicate, the functional significance of high LC numbers in tumours has been addressed.
MATERIALS AND METHODS
Animals
Six to 12-week-old female BALB/c or BALB/c×C3H HeJ F1 hybrid mice were obtained from the University of Sydney, HRA:Skh-1 mice from the Animal Resources Centre, Perth, and BALB/c nu/nu mice from the Combined University Laboratory Animal Supply, Little Bay, Sydney. Mice were supplied with food and water ad libitum and were used with approval of the animal ethics committee.
Tumour cell lines
The T7 and T79 cell lines8 (kind gifts from Dr V. Reeve, Department of Veterinary Pathology, University of Sydney) were derived from squamous cell carcinomas which arose in HRA:Skh-1 mice exposed to chronic ultraviolet radiation (UVR). The 13.1 and LK2 cell lines were derived from squamous cell carcinomas which arose in C3H/HeN mice exposed to chronic UVR. The 13.1 line was a kind gift from Dr M. Kripke (University of Texas), and LK2 was derived in this department.10 Tumour cell lines were maintained in culture in 75 cm3 cell culture flasks (Costar, Cambridge, MA) at 37° in 5% CO2-in-air. The media used throughout was Dulbecco's minimum essential medium (DMEM; Commonwealth Serum Laboratories (CSL), Melbourne, Australia), supplemented with 10% newborn calf serum (NCS; CSL), 23 mm NaHCO3, and 2 mm l-glutamine (Sigma Chemical Company, St. Louis, MO).
T7 tumour cell supernatant
Tumour supernatant was prepared as described previously.9 Cream containing tumour supernatant (TC) was prepared by mixing T7 supernatant with cetomacrogol cream (Davis Chemicals, Perth, Australia) 1:1 (v/w). A control cream (CC) was prepared by mixing DMEM with cetomacrogol cream. The creams were stored at 4° for the duration of an experiment.
Mice were treated topically for 5 consecutive days with TC or CC. Either shaved and depilated (Veet, Reckitt and Colman Pharmaceutical's, Auckland, New Zealand) dorsal skin or ear surfaces were treated with creams. Cellophane tape (Tesa, BDF Australia, Sydney, Australia) was gently applied and peeled from the skin once to remove the stratum corneum. The creams were applied topically and rubbed into the skin surface until saturated with cream, typically 1 min per site.
Subcutaneous growth of tumours
Tumour cell lines grown in culture were resuspended at a viable cell concentration of between 2 and 10×105 viable cells/50 μl phosphate-buffered saline (PBS), depending on cell line. Fifty microlitres of tumour cells were inoculated subcutaneously into each side of the dorsal flank of groups of mice and the tumours were allowed to grow to a maximum diameter of 10 mm, typically taking 3 weeks.
Primary antibodies
Major histocompatibility complex class II (MHC class II)-positive cells were detected using TIB 120 (American Type Culture Collection (ATCC), Rockville, MD), which is a rat immunoglobulin G2b (IgG2b), anti I-Ab+d+q, I-Ed+k+, or TIB 93 (ATCC), which is a mouse IgG2b anti I-Ak antibody, or MKD6 (ATCC), which is a mouse IgG2a anti I-Ad antibody. NLDC-145,11 a rat antibody was also used to detect LC. Antibodies were used as supernatants collected from cultured hybridomas.
Preparation of epidermal sheets and LC staining
Epidermal sheets were prepared and immunohistochemically stained, as described previously.9 Immunostained cells were quantitated using a true color fully automated image analysis system (Chromatic Color Image Analysis System, L. R. Jarvis, Wild-Leitz, Sydney, Australia). For each mouse, randomly selected fields were counted until the total area evaluated approached 1 mm2.
Grafting studies
To differentiate LC precursors recently migrated into the epidermis from LC of epidermal derivation, parental skin (BALB/c) was grafted onto F1 hybrid BALB/c [Iad]×C3H HeJ [Iak] mice where it was permanently accepted. To detect host-derived LC we used an Iak+ specific antibody, TIB 93. To detect all of the LC present in the graft we used the broadly MHC class II reactive antibody TIB 120 (I-Ab+d+q, I-Ed+k+). The numbers of graft-derived Iad+ LC were calculated by subtraction of the number of Iak+ LC from the numbers of Iad+k+ LC. BALB/c parental full thickness skin ≈3×4 cm was surgically excised from the dorsum of killed donor mice. This skin was not shaved or depilated and was placed into a Petri dish containing 1 part Betadine® (Faulding Pharmaceuticals, Adelaide, Australia) to 3 parts sterile PBS, until grafted. Recipient host F1 mice were anaesthetised using a Metofane® (Pitman-Moores, Washington Crossing, NY)-soaked mask. The dorsal surface of the recipient was close shaved with clippers, and washed with 70% ethanol. Approximately 4×5 cm areas of skin were surgically removed and the grafted skin stitched into place using nylon sutures (B. Braun, Melsungen AG, Germany). The grafted area was then painted with Betadine/PBS and bandaged using a layer of sterile gauze, covered by Elastoplast® adhesive bandage (Smith-Nephew, Auckland, New Zealand), and a thin layer of plaster of Paris. The grafts were allowed to become established, and then starting at week 2 post grafting the grafted skin was depilated (Veet, Reckitt and Coleman Pharmaceuticals, Auckland) and treated topically with creams for 5 days as described above. After a further 2 days the skin grafts were removed and epidermal sheets prepared for LC determination, as described above.
Measurement of LC mitosis
BALB/c mice were supplied with drinking water containing 1 mm of the thymidine analogue 5′ bromodeoxyuridine (BrdU; Sigma) and 4 g of sucrose/100 ml ad libitum from day 0. From day 0 they were treated on shaved and depilated dorsal skin with creams as described above. On day 6, the mice were sacrificed, the treated skin was removed, epidermal cell suspensions were prepared as described previously10 and fixed with 0·2% paraformaldehyde in PBS for 5 min, then immunolabelled, as described by Holt et al.,12 with rat anti-MHC class II antibody (TIB 120) and murine anti-BrdU (Becton Dickinson, San Jose, CA).
Single or double positive cells were determined by microscopic examination. At least 180 MHC class II+ cells were examined per animal with respect to double positivity. Total MHC class II+ ells were stained red while MHC class II+/BrdU+ cells were a deep purple and, hence, could be easily differentiated. Appropriate specificity controls, including the omission of primary antibodies were included with each experiment.
Contact sensitizer induced migration
LC remaining in the epidermis
BALB/c mice were treated topically with TC or CC on all ear surfaces for 5 consecutive days, as described above. The following day the ear surfaces were treated with 10 μl of either 0·1 or 0·5% FITC (Sigma) dissolved in acetone:dibutylphthalate (1:1 (v/v); BDH, Poole, UK) or 3% 2, 4, 6-trinitrochlorobenzene (TNCB; Kasei, Tokyo, Japan) dissolved in acetone. 18 hr later the animals were killed and epidermal sheets prepared from the ear surfaces as described above. LC were stained with TIB 120 primary antibody and LC densities were determined as described above. The percentage of LC which had migrated from the epidermis in response to FITC or TNCB was calculated from the following formula:
LC migrated to the draining auricular lymph node
BALB/c mice were treated topically with TC or CC on all ear surfaces for 5 consecutive days, followed by 10 μl of 0·5% FITC dissolved in acetone:dibutyl phthalate, as described above. 18 hr later the animals were killed and the draining auricular lymph nodes removed. Lymph nodes were teased into PBS and total cell numbers determined using a haemocytometer. The cells were washed in 2 ml of PBS, and pelleted by centrifugation at 200 g for 5 min. They were then fixed using 0·1% paraformaldehyde (BDH) in PBS and washed as described above. MHC class II+ cells were then labelled by incubation in TIB 120 antibody for 60 min at 37° followed by goat antirat IgG conjugated to phycoerythrin (Caltag Laboratories, San Francisco, CA) in PBS containing 10% NCS. The cells were washed, resuspended at 106 per ml in PBS containing 10% NCS, and filtered through 80 μm nylon mesh (Swiss Screens, Sydney, Australia). The percentage of MHC class II+/FITC+ lymph node cells was determined using fluorescence-activated cell sorting (FACScan flow cytometer and Cellquest™ analysis; Becton Dickinson) for each individual mouse and this was used to calculate the number of LC which had migrated to the lymph node (total number of MHC class II+/FITC+ cells per lymph node).
Statistics
Statistical comparisons between two groups were performed using unpaired two-tailed Student's t-tests, P-values of <0·05 were considered statistically significant.
RESULTS
LC which accumulate in tumour-supernatant-treated epidermis are derived from the epidermis, not a bone marrow precursor
The initial approach used to determine the mechanism by which LC accumulate in TC-treated skin was to establish whether they originated from within the skin or from a bone-marrow-derived LC precursor. To determine this we grafted parental skin onto F1 hybrid host mice. The grafts became established and were permanently accepted by the host mice. During this period epidermal LC migrate from the graft to local lymph nodes and are replaced by host bone-marrow-derived precursors. The different MHC class II molecules present on the graft-derived (Iad) and host-derived (Iad,k) LC enabled their origin to be determined.
Graft-derived LC were reduced from 1089±27 mm−2 (mean±SEM) to a density of 308±86 mm−2 in the CC treated group by 3 weeks after grafting (Fig. 1). The TC-treated epidermis contained a significantly higher number (P < 0·05) of graft-derived LC (686±101 mm−2). In contrast, the CC- and TC-treated grafts had been infiltrated with similar numbers of host-derived LC precursors (means; 692±52 and 527±67 mm−2, respectively). The total number of LC significantly increased from a mean of 1001±52 mm−2 in CC-treated skin to 1214±62 mm−2 in TC-treated skin. Thus, the significantly increased number of total LC within the epidermis of skin grafts treated with TC all arose from within the epidermis and were not derived from increased migration of bone-marrow precursors into the epidermis.
Figure 1.

LC which accumulate in tumour supernatant treated skin are derived from the epidermis, not a bone marrow precursor. BALB/c parental skin was grafted onto BALB/c X C3H HeJ F1 recipients. The grafts were allowed to heal for 2 weeks and were then treated with control cream or tumour-supernatant-containing cream for 5 consecutive days. Graft- and host-derived LC were determined after a further 2 days by differential expression of I-Ad (graft-derived) and I-Adk (total) LC. *denotes a statistically significant difference between control (N = 7) and tumour (N = 8) groups (P < 0·05), NS, not significant (unpaired two-tailed Student's t-test). Mean±SEM shown for each group.
LC division is similar in epidermis treated with control (CC) and tumour supernatant (TC) creams
We determined the percentage of LC that had divided in the epidermis over the course of CC and TC treatments. The drinking water of the mice was supplemented with BrdU during the course of treatment. During this period, TC increased the number of epidermal LC and BrdU was incorporated into dividing cells. Dividing cells were detected by staining with anti-BrdU antibodies, LC were identified by anti-MHC class II antibody staining. Thus, double positive cells indicated LC which had divided during the experiment. Treatment with TC had no significant effect on the percentage of LC that had divided, 3·2±0·6% compared to 4·3±0·2% in the CC-treated group (Fig. 2).
Figure 2.
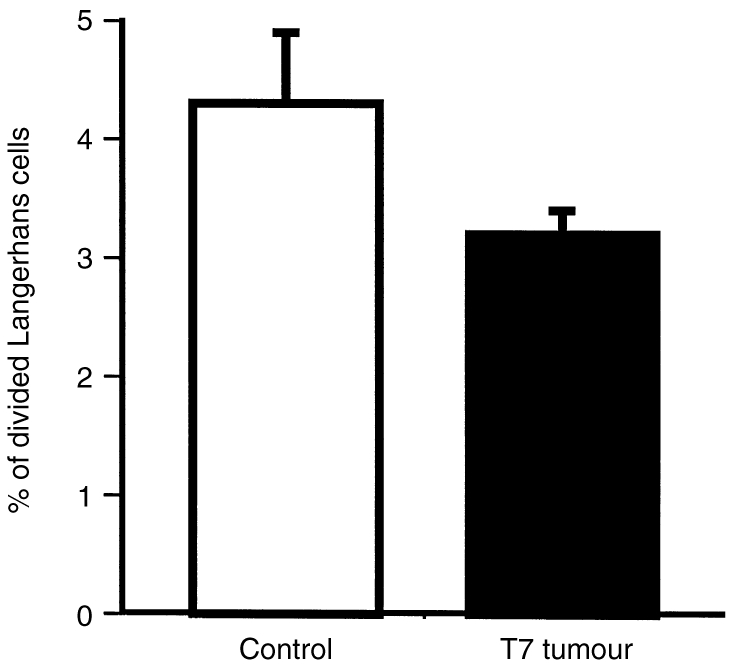
LC division is similar in epidermis treated with control (CC) or tumour supernatant (TC) creams. Mice were treated by applying CC or TC topically for 5 consecutive days. They were simultaneously supplied with 5′ bromodeoxyuridine (BrdU) in their drinking water throughout the 5 days of treatment. Epidermal cell suspensions prepared from treated skin were immunohistochemically labelled for MHC class II and incorporated BrdU. The percentage of the LC which had incorporated BrdU into their nucleus was determined by counting total LC, and double positive LC. Mean±SEM shown for each group, N = 6 for each group. There was no significant difference in the percentage of divided LC within CC and TC samples (unpaired two-tailed Student's t-test).
LC migration from the epidermis to local lymph nodes is inhibited by treatment with tumour supernatant
To study LC migration from the epidermis to local lymph nodes, groups of 12 mice were treated topically on their ears with CC or TC for 5 consecutive days. On the following day half of each group (six mice) had FITC or TNCB dissolved in solvent applied to their treated ears, the other mice received the solvent alone. The number of LC which were retained in the epidermis 18 hr after treatment with FITC or TNCB was determined. The groups that received solvent alone determined the baseline LC levels in the absence of the migration-inducing stimulus, and were statistically compared to the mice that received FITC or TNCB to determine whether significant LC migration had occurred. Results are presented as the percentage of LC that migrated from the epidermis (Fig. 3).
Figure 3.

LC migration from the epidermis is inhibited by treatment with tumour supernatant. Mice were treated with control or tumour-supernatant-containing cream for 5 consecutive days. 24 hr later half of the treated mice (6 mice) were painted with either 0·1, 0·5% FITC or 3% TNCB, the other half received solvent alone. 18 hr later epidermal sheets were prepared and stained with anti-MHC class II antibodies to detect LC. LC numbers were determined using video image analysis. For each group, the percentage of LC which had migrated from the epidermis was calculated as described in Materials and Methods, mean±SEM shown for each group. To determine the statistical significance of FITC or TNCB -induced migration, each of these groups was compared to its respective group which received solvent alone (unpaired two-tailed Students t-test). *P < 0·01, **P < 0·001, NS not significant for FITC or TNCB compared to solvent-treated groups.
FITC (0·1%) significantly reduced the LC density from 858±33 to 681±33 LC mm−2 (P < 0·001), and 0·5% FITC significantly reduced the LC density from 760±55 to 557±23 LC mm−2 (P < 0·01) in CC-treated epidermis. In contrast, neither concentration of FITC significantly decreased the number of epidermal LC in TC-treated mice (Fig. 3).
Application of 3% TNCB significantly reduced the LC density from 990±26 to 304±32 LC mm−2 (P < 0·0001) in CC-treated mice. A similar reduction of LC was detected in TC-treated epidermis, from 1132±67 to 282±46 LC mm−2 (P < 0·0001, Fig. 3).
To confirm that the cells being studied were LC they were labelled with NLDC-145 after mice received 0·5% FITC to induce LC migration. This FITC concentration induced 30% of the NLDC-145+ LC to migrate from the epidermis of CC-treated mice (Fig. 4) which is similar to the level of MHC class II+ LC-induced migration (Fig. 3). FITC did not significantly decrease the number of LC in TC-treated mice, confirming results obtained using MHC class II to identify LC.
Figure 4.
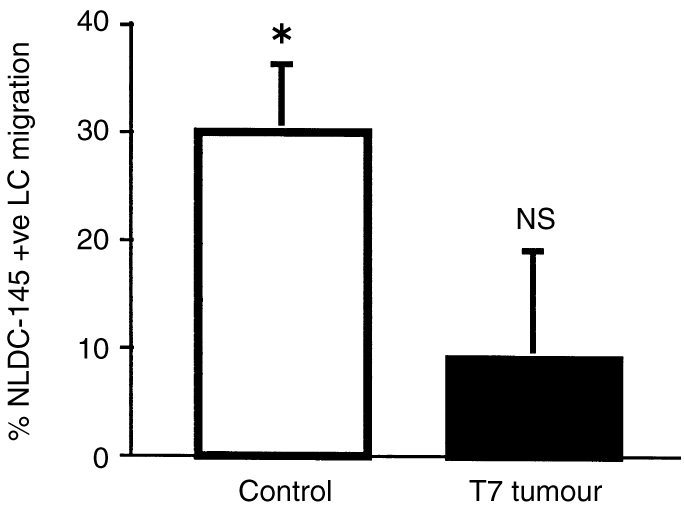
NLDC-145+ LC migration from the epidermis is inhibited by treatment with tumour supernatant. Legend the same as for Fig. 3, except that the mice were only treated with 0·5% FITC, and LC were stained with NLDC-145 antibody. To determine the statistical significance of FITC-induced migration, this group was compared to the solvent-treated group. *P < 0·01, NS not significant for FITC compared to solvent-treated groups (unpaired two-tailed Students t-test).
To study LC at the end-point of their migration, mice were treated on their ear surfaces with CC or TC, followed by FITC, and draining lymph nodes were collected 18 hr later. LC, which were labelled with FITC whilst in the epidermis and had migrated to the lymph node, could be observed as green fluorescent cells. To confirm specificity the lymph node cells were also stained with anti-MHC class II antibodies. Lymph nodes draining the epidermis of mice treated with CC and FITC contained significantly larger numbers of migrated LC than did the lymph nodes of mice treated with TC and FITC (means±SEM of 7668±1181, compared to 3911±742, respectively; P < 0·05). Thus, the tumour supernatant inhibited LC migration from the epidermis to local lymph nodes (Fig. 5).
Figure 5.
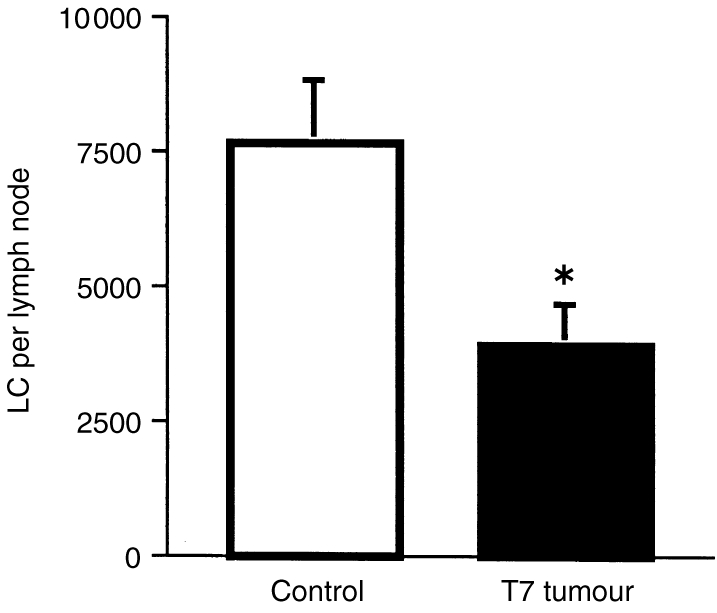
Tumour supernatant inhibits LC migration to local lymph nodes in response to 0·5% FITC. Groups of six mice were treated topically for 5 consecutive days with either control cream (CC) or tumour-supernatant-containing cream (TC). Twenty-four hours later 0·5% FITC was painted onto the treated skin. The number of LC that had migrated to draining auricular lymph nodes 18 hr following FITC was determined. Paraformaldehyde (0·1%)-fixed lymph node cells were labelled with anti-MHC class II–phycoerythrin (PE) and examined by flow cytometry for MHC class II and FITC double positivity. *denotes a statistically significant difference between control and tumour supernatant treated groups (P < 0·05, unpaired Student's t-test). Mean±SEM shown for each group.
Skin tumour inhibition of LC migration from overlying epidermis
We next examined whether growth of the T7 tumour in vivo also inhibited epidermal LC migration from the overlying epidermis. Groups of 16 syngeneic mice were transplanted subcutaneously with T7 cells. The tumours were allowed to grow for 3 weeks, and then the overlying epidermis was treated topically with either FITC or a mixture of FITC and TNCB, or solvent alone. Control skin on non-tumour-bearing mice was also treated in the same manner. The number of MHC class II+ LC which were retained in the epidermis 18 hr after treatment was determined and used to calculate the percentage of LC which migrated from the epidermis (Fig. 6).
Figure 6.
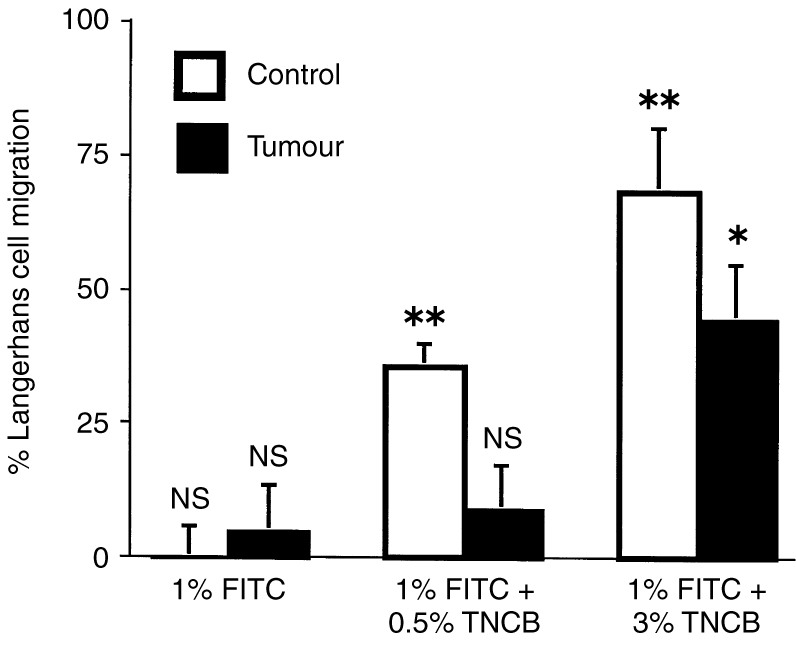
LC migration from the epidermis overlying the T7 progressor tumour is inhibited. Groups of 16 HRA:Skh-1 mice were transplanted with 2×105 T7 tumour cells into each side of the dorsal flank, and the tumours were allowed to grow to a diameter of 10 mm. Control groups did not receive tumour cells. Half of the mice in each group (eight mice) were painted with either 1% FITC, 1% FITC and 0·5% TNCB, or 1% FITC and 3% TNCB. The other half of each group received solvent alone. Eighteen hours later epidermal sheets were prepared and stained for LC with anti-MHC class II antibodies. LC numbers were determined using video image analysis. For each group, the percentage of LC which had migrated from the epidermis was calculated as described in Materials and Methods, mean±SEM shown for each group. To determine the statistical significance of FITC- or FITC+TNCB-induced migration, each of these groups was compared to its respective group which received solvent alone (unpaired two-tailed Students t-test). *P < 0·01, **P < 0·001, NS not significant for FITC or FITC+TNCB compared to solvent-treated groups.
A significant level of LC migration was detected in control epidermis 18 hr following application of 1% FITC and 0·5% TNCB (35%) or 1% FITC and 3% TNCB (69%). FITC (1%) alone was unable to induce LC migration in this mouse strain (Fig. 6).
LC within the epidermis overlying the T7 tumour were unable to migrate in response to 1% FITC and 0·5% TNCB. Significant migration (44%) from the epidermis overlying the T7 tumour did occur in response to the stronger stimulus of 1% FITC and 3% TNCB.
Progressor, but not regressor, murine tumours inhibit LC migration
To determine whether other tumours inhibit LC migration from the epidermis, a range of progressor and regressor murine tumours were examined. The use of athymic immunodeficient mice enabled the effect of similar-sized progressor and regressor tumours to be examined. In each experiment, groups of 16 female BALB/c nu/nu mice were transplanted with tumour cells into both sides of their dorsal flank, and the tumours allowed to grow to a diameter of 10 mm. Other groups of 16 mice did not receive tumour inoculation and served as non-tumour-bearing controls. The epidermis overlying the tumours was then treated topically with 1% FITC, or solvent alone. Control skin on non-tumour-bearing mice was also treated in the same manner. The number of MHC class II+ LC which were retained in the epidermis 18 hr after treatment was determined and used to calculate the percentage of LC that had migrated from the epidermis (Fig. 7) in each experiment. Topical application of 1% FITC to control mice induced 53–68% of LC to migrate from the epidermis. These levels of migration were statistically significant in each experiment.
Figure 7.
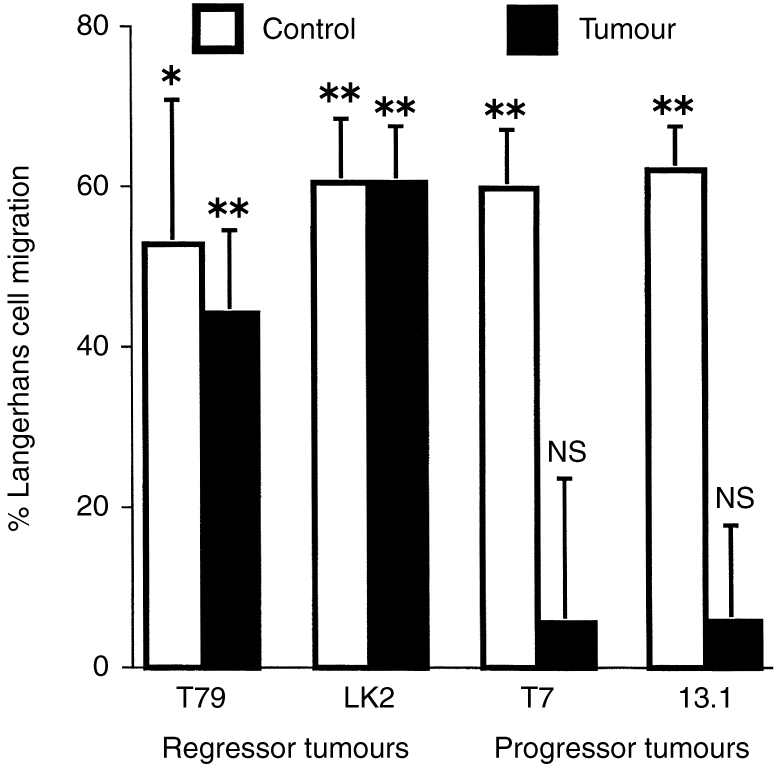
LC migration is inhibited from the epidermis overlying progressor but not regressor tumours. Groups of 16 BALB/c nu/nu mice were transplanted with progressor or regressor tumour cell lines, and the tumours were allowed to grow to a diameter of 10 mm. Other groups did not receive tumour cells and served as non-tumour-bearing controls. Half of the mice were painted with 1% FITC, the other half received solvent alone. Eighteen hours later epidermal sheets were prepared and stained for LC with anti-MHC class II antibodies. LC numbers were determined using video image analysis. For each group, the percentage of LC which had migrated from the epidermis was calculated as described in Materials and Methods, mean±SEM are shown for each group. To determine the statistical significance of FITC-induced migration, each of these groups was compared to its respective group which received solvent alone (unpaired two-tailed Students t-test). *P < 0·05, **P < 0·01, NS not significant for FITC-compared to solvent-treated groups.
The response of LC in the epidermis overlying the various tumours, however, contrasted markedly (Fig. 7). There was a significant decrease in LC density in the epidermis overlying the regressor tumours T79 and LK2. The magnitude of FITC-induced migration of LC from the epidermis overlying these tumours (44–66%) was similar to that which occurred from control epidermis. In contrast, no significant migration of LC from the epidermis overlying the progressor tumours T7 or 13·1 was observed.
DISCUSSION
Our initial approach in establishing the source of the increased LC in skin tumours was to determine whether they were derived from bone marrow precursors or resident epidermal LC. To determine this we grafted parental skin onto F1 hybrid mice which enabled the discrimination of newly immigrated bone-marrow-derived host LC from graft-derived LC because of their different MHC class II phenotypes. This approach was previously used to determine the bone-marrow origin of LC precursors.13 The grafts treated with TC from 2–3 weeks had a significantly higher LC density than those treated with CC. The LC population being affected was the graft-derived LC, with no difference being observed in LC migration into the epidermis. Thus, the tumour supernatant slowed the fall in number of graft-derived LC.
To evaluate the effects of tumour-derived factors on LC migration, we measured the response of LC to topically applied contact sensitizing chemicals. Two different contact sensitizers were used in this study: FITC and TNCB. Topical application of FITC labels the LC with fluorescein and induces them to migrate to local lymph nodes where they can be identified by their fluorescein label. Fluorescently conjugated LC have been detected in the dermis after 6 hr14 and in the draining lymph nodes 24 hr following skin painting.15 Furthermore, skin grafting studies have confirmed that the fluorescent cells in the draining lymph nodes of FITC-treated mice are derived from the skin.2 Topical application of another contact sensitizing chemical, TNCB, also caused LC to migrate from the epidermis and the degree of migration stimulated by this treatment is greater than FITC. The magnitude of the TNCB response is similar to that described for dinitrofluorobenzene.16
We found that application of 0·1% or 0·5% FITC and 3% TNCB onto murine ear skin pretreated with our control cream could significantly reduce MHC class II+ LC numbers in the epidermis after 18 hr. FITC stimulated approximately a 20–30% reduction in MHC class II+ LC, whilst TNCB induced a 70% reduction. In contrast, in ear skin pretreated with tumour-derived-factor-containing creams there was no significant change in LC numbers in response to either concentration of topically applied FITC; however, there was a significant and similar reduction in LC in response to TNCB.
A number of studies have reported that the reduction in immunohistochemically labelled LC detectable in the epidermis following corticosteroid treatment17,18 or exposure to UVR19 is caused by a rapid and reversible downregulation of MHC class II from the cell surface. To confirm that the reduction in LC numbers in the epidermis following contact sensitizer application was caused by active migration from the epidermis, we examined the local lymph nodes draining this treated skin, following the application of 0·5% FITC, for MHC class II and FITC double positive LC, using FACS analysis.
Examination of lymph nodes draining skin pretreated with CC and FITC showed a population of double positive cells that averaged 6×103 per node. This compares with the levels found by other groups at 24 hr following FITC skin painting of 10×103 per node20 and confirmed that FITC was inducing MHC class II+ cell migration from the epidermis to the draining lymph nodes. In contrast, the number of double positive cells that were detected in nodes draining TC- pretreated skin, was significantly less: 3×103 per node. When taken together, these experiments indicate that tumour-derived factors inhibited LC migration from treated epidermis, and this could explain in part the accumulation of LC in topically treated skin.
These experiments could not exclude LC mitosis from contributing, at least in part, to the increased number of LC found in TC-treated skin. A low level of LC mitosis has been demonstrated by electron microscopy,21,22 incorporation of 3H-thymidine into normal or tape-stripped epidermis23,24 and DNA densitometric analysis of FACS-sorted human LC.25
To study LC mitosis we fed mice with the thymidine analogue BrdU over the course of topical treatment with TC. Incorporation of BrdU into the DNA of dividing cells has become a common tool for the measurement of cell division in vivo because of the availability of antibodies that bind to BrdU groups incorporated in the DNA. In our system, 4% of LC showed evidence of division after 1 week of treatment. This compares with the instantaneous labelling index of LC in normal murine skin determined using 3H-thymidine incorporation of 0·01%,26 and a level of 4·9% of LC in human skin grafted onto nude mice showing BrdU incorporation over a 6-hr pulse.25 In our system TC treatment did not significantly affect the rate of LC mitosis.
When these three separate investigations are taken together, alterations in LC numbers by tumour derived factors applied topically in a cream was not caused by increased attraction of precursors or stimulation of in situ LC division, but there was evidence for a reduced responsiveness of epidermal LC to migrate both from the epidermis and reduced numbers of migrated LC detectable in draining local nodes. This is strongly suggestive that some tumour-derived factors can inhibit LC migration from the epidermis.
LC migration was also inhibited from the epidermis overlying the T7 tumour cells grown subcutaneously in syngeneic mice. Similar to the response following topical application of tumour derived factors, inhibition of LC migration could be demonstrated in response to some doses of contact sensitizer, which could be overcome using a higher dose. A stronger stimulus, 1% FITC combined with 0·5% TNCB, was required to cause significant LC migration from HRA:Skh-1 mice. LC in epidermis overlying the T7 tumour did not migrate in response to this stimulus. Increasing the dose to 1% FITC combined with 3% TNCB caused higher levels of LC migration from the control epidermis, and significant migration from epidermis overlying the T7 tumour. Thus, LC from above the T7 tumour were able to migrate, but only in response to a stronger stimulus.
To study the functional significance of skin tumour inhibition of LC migration, progressor and regressor tumours were compared. Regression of human skin tumours is immunologically mediated.27 Similarly, an effective immune response destroys the two regressor tumours used in this study when they are transplanted into syngeneic, immunocompetent, host mice.10,28 The progressor tumours used in this study are antigenic, as they can induce immunity in syngeneic mice. Thus, a study of the progressor and regressor tumours enabled the effects of tumours which evade immunological destruction (progressors) and tumours which activate effective immunity (regressors) to be compared. The regressor tumours could not be studied in immunocompetent mice, because the immune response mediating tumour destruction would be likely to interfere with the accurate identification of LC, and also because insufficient tumour material remains for study. Therefore, these tumours were grown in immunodeficient mice, where both tumour groups had similar growth characteristics. We have previously shown that LC accumulate in the epidermis overlying progressor tumours growing in athymic mice.8 The LC overlying both progressor tumours did not respond to FITC, whereas the LC overlying both the regressor tumours migrated from the epidermis in response to the same stimulus, as did LC in control epidermis. Thus, progressor tumours, which evade immunological destruction, inhibit migration of LC from the epidermis, whereas regressor tumours, which do not evade the immune response, do not interfere with the mobilization of dendritic cells.
It is unknown how the progressor tumours inhibit LC migration. Our demonstration that supernatant from cultured progressor tumour cells has the same effect as the growing tumour indicates that the tumours produce soluble factors which interfere with LC migration. The tumours may produce inhibitors of the signals which stimulate LC migration from normal epidermis, or may downregulate LC receptors, so that they are less responsive to migration-inducing stimuli. Rauscher leukaemia virus infection of murine epidermis inhibits LC migration from the epidermis to the draining lymph nodes.29 Thus, inhibition of LC migration may be a mechanism used by both viruses and tumours to evade activation of immune responses.
There has been considerable speculation regarding the role of LC in immune responses against tumours. Our data suggests that if they are present in large numbers, this may be because their ability to migrate from the tumour has been inhibited. This could prevent the activation of an immune response, and therefore be detrimental to the host. The development of procedures that stimulate LC migration from tumours may enable dendritic cells to be used more effectively for immunotherapy of cancer.
Acknowledgments
We would like to thank Linda Johnston for kind assistance with the flow cytometry and the National Health and Medical Research Council of Australia for financial support.
Abbreviations
- LC
Langerhans' cells
- DC
dendritic cells
- EC
epidermal cells
- TC
tumour supernatant cream
- CC
control supernatant cream
- FITC
fluorescein isothiocyanate
- TNCB
2,4,6-trinitrochlorobenzene
- BrdU
5′ bromodeoxyuridine
REFERENCES
- 1.Larsen CP, Steinman RM, Witmer PM, Hankins DF, Morris PJ, Austyn JM. Migration and maturation of Langerhans' cells in skin transplants and explants. J Exp Med. 1990;172:1483. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kripke ML, Munn CG, Jeevan A, Tang JM, Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol. 1990;145:2833. [PubMed] [Google Scholar]
- 3.Katz SI, Tamaki K, Sachs DH. Epidermal Langerhans' cells are derived from cells originating in bone marrow. Nature. 1979;282:324. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 4.Czernielewski J, Vaigot P, Prunieras M. Epidermal Langerhans' cells – a cycling cell population. J Invest Dermatol. 1985;84:424. doi: 10.1111/1523-1747.ep12265523. [DOI] [PubMed] [Google Scholar]
- 5.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nature Med. 1998;4:328. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 6.McArdle JP, Knight BA, Halliday GM, Muller HK, Rowden G. Quantitative assessment of Langerhans' cells in actinic keratosis, Bowen's disease, keratoacanthoma, squamous cell carcinoma and basal cell carcinoma. Pathology. 1986;18:212. doi: 10.3109/00313028609059461. [DOI] [PubMed] [Google Scholar]
- 7.Bergfelt L, Bucana C, Kripke ML. Alterations in Langerhans' cells during growth of transplantable murine tumors. J Invest Dermatol. 1988;91:129. doi: 10.1111/1523-1747.ep12464151. [DOI] [PubMed] [Google Scholar]
- 8.Halliday GM, Reeve VE, Barnetson RS. Langerhans' cell migration into ultraviolet light-induced squamous skin tumors is unrelated to anti-tumor immunity. J Invest Dermatol. 1991;97:830. doi: 10.1111/1523-1747.ep12491503. [DOI] [PubMed] [Google Scholar]
- 9.Halliday GM, Lucas AD, Barnetson RS. Control of Langerhans' cell density by a skin tumour-derived cytokine. Immunology. 1992;77:13. [PMC free article] [PubMed] [Google Scholar]
- 10.Cavanagh LL, Halliday GM. Dendritic epidermal T cells in ultraviolet-irradiated skin enhance skin tumor growth by inhibiting CD4+ T-cell-mediated immunity. Cancer Res. 1996;56:2607. [PubMed] [Google Scholar]
- 11.Kraal G, Breel M, Janse M, Bruin G. Langerhans' cells, veiled cells, and interdigitating cells in the mouse recognized by a monoclonal antibody. J Exp Med. 1986;163:981. doi: 10.1084/jem.163.4.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holt PG, Haining S, Nelson DJ, Sedgwick JD. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J Immunol. 1994;153:256. [PubMed] [Google Scholar]
- 13.Stingl G, Tamaki K, Katz SI. Origin and function of epidermal Langerhans' cells. Immunol Rev. 1980;53:149. doi: 10.1111/j.1600-065x.1980.tb01043.x. [DOI] [PubMed] [Google Scholar]
- 14.Carr MM, Botham PA, Gawkrodger DJ, et al. Early cellular reactions induced by dinitrochlorobenzene in sensitized human skin. Br J Dermatol. 1984;110:637. doi: 10.1111/j.1365-2133.1984.tb04697.x. [DOI] [PubMed] [Google Scholar]
- 15.Macatonia SE, Edwards AJ, Knight SC. Dendritic cells and the initiation of contact sensitivity to fluorescein isothiocyanate. Immunology. 1986;59:509. [PMC free article] [PubMed] [Google Scholar]
- 16.Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans' cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124:445. [PubMed] [Google Scholar]
- 17.Berman B, France DS, Martinelli GP, Hass A. Modulation of epidermal Langerhans' cell properties following in situ exposure to glucocorticosteroids. J Invest Dermatol. 1983;80:168. doi: 10.1111/1523-1747.ep12533397. [DOI] [PubMed] [Google Scholar]
- 18.Pakes WL, Muller HK, Schwarz MA, Marks R. Langerhans' cells – a reduction in numbers and their reappearance following steroid and cytotoxic therapy in humans. Clin Exp Dermatol. 1986;11:450. doi: 10.1111/j.1365-2230.1986.tb00492.x. [DOI] [PubMed] [Google Scholar]
- 19.Aberer W, Schuler G, Stingl G, Honigsmann H, Wolff K. Ultraviolet light depletes surface markers of Langerhans' cells. J Invest Dermatol. 1981;76:202. doi: 10.1111/1523-1747.ep12525745. [DOI] [PubMed] [Google Scholar]
- 20.Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med. 1987;166:1654. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyauchi S, Hashimoto K. Epidermal Langerhans' cells undergo mitosis during the early recovery phase after ultraviolet-B irradiation. J Invest Dermatol. 1987;88:703. doi: 10.1111/1523-1747.ep12470379. [DOI] [PubMed] [Google Scholar]
- 22.Breathnach AS. Prosser White Oration 1976. Electron micrographs from a collection. Clin Exp Dermatol. 1977;2:1. doi: 10.1111/j.1365-2230.1977.tb01531.x. [DOI] [PubMed] [Google Scholar]
- 23.Giacometti L, Montagna W. Langerhans' cells: uptake of tritiated thymidine. Science. 1967;157:439. doi: 10.1126/science.157.3787.439. [DOI] [PubMed] [Google Scholar]
- 24.Schellander F, Wolff K. On the autoradiographic labelling of Langerhans' cells with H3-thymidine. Archiv Fur Klinische und Exp Dermatologie. 1967;230:140. [PubMed] [Google Scholar]
- 25.Czernielewski J, Demarchez M. Further evidence for the self-reproducing capacity of Langerhans' cells in human skin. J Invest Dermatol. 1987;88:17. doi: 10.1111/1523-1747.ep12464659. [DOI] [PubMed] [Google Scholar]
- 26.Mackenzie IC. Labelling of murine epidermal Langerhans' cells with H3-thymidine. Am J Anat. 1975;144:127. doi: 10.1002/aja.1001440202. [DOI] [PubMed] [Google Scholar]
- 27.Halliday GM, Patel A, Hunt MJ, Tefany FJ, Barnetson RS. Spontaneous regression of human melanoma/nonmelanoma skin cancer: association with infiltrating CD4+ T cells. World J Surg. 1995;19:352. doi: 10.1007/BF00299157. [DOI] [PubMed] [Google Scholar]
- 28.Patel A, Halliday GM, Barnetson RS. CD4+ T lymphocyte infiltration correlates with regression of a UV-induced squamous cell carcinoma. J Dermatol Sci. 1995;9:12. doi: 10.1016/0923-1811(94)00344-e. [DOI] [PubMed] [Google Scholar]
- 29.Gabrilovich DI, Woods GM, Patterson S, Harvey JJ, Knight SC. Retrovirus-induced immunosuppression via blocking of dendritic cell migration and down-regulation of adhesion molecules. Immunology. 1994;82:82. [PMC free article] [PubMed] [Google Scholar]