

Abstract
Experimental autoimmune neuritis (EAN) is a CD4+ T-cell-mediated demyelinating disease of the peripheral nervous system (PNS) and serves as experimental model for human immune-demyelinating neurophathies, especially the Guillain–Barré syndrome. In this study, we examined the effect of recombinant rat interleukin-6 (rrIL-6) on chronic EAN in Lewis rats induced by immunization with P2 peptide 57-81 and Freund’s complete adjuvant (FCA). Nasal administration of rat rIL-6 (1 μg/rat/day) beginning in the initial phase of EAN as a therapeutic agent, decreased the severity and the duration of clinical EAN. Low-grade inflammation and suppression of regional demyelination within the sciatic nerves were seen in rrIL-6-treated rats. Hyporesponsiveness of lymph node T cells, down-regulation of serum tumour necrosis factor-α (TNF-α) and increased levels of P2-specific immunoglobulin G1 (IgG1) antibodies document that nasal administration of rrIL-6 was effective systemically. However, because of the non-specific nature of the treatment and multiple effects of IL-6, more experience and great caution are needed, before nasal administration of IL-6 can be considered as a treatment of human autoimmune demyelinating neurophathies.
INTRODUCTION
Several approaches have been proposed for immunotherapy of autoimmune diseases. It is possible to achieve immunotherapeutic effects by administration of recombinant cytokines which augment endogenous cytokines even when the site of cytokine administration is distant from that of target response.1 The inhibition of an ongoing immune response by nasal administration of cytokines is an important therapeutic issue. Nasal-associated lymphoid tissue (NALT) is a convenient drug delivery system that allows the use of lower doses of cytokines and provides efficacy via unique and potent immunoregulatory circuits without generating additional inflammatory cytokines.2,3
Interleukin 6 (IL-6) is a multifunctional cytokine with a broad range of activities, affecting haematopoiesis and neuroendocrine functions, as well as immune functions. It is one of the major mediators of the immune response.4–7 Various studies have indicated that IL-6 is directly or indirectly involved in the pathogenesis of immune-mediated inflammatory central nervous system (CNS) and peripheral nervous system (PNS) disorders and other autoimmune diseases and possesses pleiotropic activities.6–8 In the PNS, augmented IL-6 production is seen before the onset of clinical experimental autoimmune neuritis (EAN).9 The blood–nerve barrier (BNB) disturbance in EAN may also involve IL-6. High IL-6 concentrations were found in sera10 and cerebrospinal fluid (CSF)11 from patients with active Guillain–Barré syndrome (GBS) and correlated with clinical signs of the disease. Increased IL-6 release is associated with autoantibody production, considered to be involved in the pathogenesis of GBS. Although IL-6 was initially thought to be a proinflammatory cytokine,12–14 recent findings suggest that it has many anti-inflammatory and immunosuppressive effects,15–17 depending on the stage at which it is present and on the experimental system.18 It has varying effects on acute and chronic inflammatory processes by the direct suppression of IL-1 and tumour necrosis factor-α (TNF-α), the induction of glycocorticosteroid release, and the induction of natural antagonists of IL-1 and TNF-α.19 However, its specific role in the various aspects of inflammation and in the immune responses is not yet fully clarified. The function of this cytokine remains complex and controversial. Whether exogenous IL-6 plays a pro- or anti-inflammatory role in EAN has not yet been elucidated.
EAN is a CD4+ T-cell-mediated demyelinating inflammatory disease that can be actively induced in susceptible animals by immunization with PNS myelin,20 purified PNS proteins P221 or P0,22 or with P2 peptides23 emulsified with Freund’s complete adjuvant (FCA). EAN serves as an experimental model for the study of pathogenesis, immunoregulation and therapy of autoimmune demyelinating neurophathies24 and also as a model for CD4+ T-cell-mediated autoimmune diseases in general. Severity of clinical EAN and pathological changes correlate with the antigen dose used for immunization.24,25 Chronic EAN has been reported after administration of larger than usual doses of antigen.26,27
To evaluate the role of rrIL-6 in ongoing EAN, we administered rrIL-6 by the nasal route to Lewis rats with chronic EAN which more resembles the human GBS than acute EAN. Our data show that nasal rrIL-6 administration, in a dose-dependent manner, decreases the severity and the duration of clinical EAN. The beneficial clinical effects were associated with decreased lymphocyte proliferation and TNF-α levels as well as suppression of inflammation and demyelination within the sciatic nerves.
MATERIALS AND METHODS
Reagents
The neuritogenic P2 protein peptide corresponding to the aa 57-81 of rat PNS myelin P2 protein23 was synthesized by solid-phase stepwise elongation using a Tecan peptide synthesizer (Multisyntech, Bochum, Germany). Mass-spectrometry showed the expected masses as major components in the specta. RrIL-6 is a R & D systems product (Minneapolis, MN).
Induction of EAN and assessment of clinical signs
Male Lewis rats, 150–180 g, were purchased from Charles River Co. (Sulzfeld, Germany) and immunized by injection into both hind footpads with 200 μl of an inoculum containing 240 μg of P2 peptide 57-81 and 2 mg Mycobacterium turberculosis (strain H.37.RA; Difco, Detroit, MI) emulsified in 100 μl saline and 100 μl Freund’s incomplete adjuvant (FIA, Difco). Body weights and clinical signs of EAN were assessed before immunization (day 0) and thereafter every second day until day 120 post immunization (p.i.). Severity of paresis was graded as follows: 0=no illness; 1=flaccid tail; 2=moderate paraparesis; 3=severe paraparesis; 4=tetraparesis; 0·5=for intermediate clinical signs.28
In vivotreatment with rrIL-6
At the onset of the first apparent clinical signs of EAN on day 9 p.i., two groups of 16 rats received either high dose rrIL-6 (1 μg/rat/day) or low dose rrIL-6 (0·1 μg/rat/day) intranasally for 7 consecutive days (from days 9–15 p.i.). These doses are 1/30–1/3 compared to those which have been systemically applied in previous studies.15 rrIL-6 was administered daily in phosphate-buffered saline (PBS) by micropipette in a total volume of 60 μl (30 μl per nostril). A control group of eight rats received PBS only. Another control group of eight rats received bovine serum albumin (BSA), because BSA is the carrier protein of rrIL-6. At each administration, rats were gently anaesthetized with ether. Half of the animals were killed on day 18 p.i., 9 days after onset of treatment. The remaining animals were observed for clinical score up to day 120 p.i.
Histopathological assessment
Segments of sciatic nerves close to the lumbar spinal cord from killed animals were dissected, fixed in 4% formaldehyde and embedded in paraffin. Multiple longitudinal sections (5–6 μm slices) of sciatic nerves were stained with haematoxylin–eosin and replicate sections with luxol fast blue violet for evaluation of the extent of mononuclear cell (MNC) infiltration and of demyelination. Tissue areas were measured by image analysis and the numbers of inflammatory cells were calculated per mm2 at ×20 magnification. Degree of demyelination was expressed on a scale of five grades29 which corresponded to the extend of demyelination as follows: 5=100% of demyelinated nerve fibres; 4=80%; 3=60%; 2=40%; 1=10%-20%; 0<10% of demyelinated fibres. The cumulative percentage of demyelination in sciatic nerve sections, assigned to this scale was estimated for each animal of each experimental group.
Immunohistochemistry
Segments of sciatic nerves were dissected and snap-frozen in liquid nitrogen. Cryostat sections (10 μm) after fixation in acetone at −20°, were exposed to the mouse monoclonal antibodies ED1 (antirat, macrophages), W3/25 (antirat CD4, T helper cells), Ox8 (antirat CD8, T cytotoxic/suppressor cells) and Ox6 (antirat major histocompatibility complex (MHC) class II) (Serotec, Oxford, UK), respectively. Sections were stained according to the avidin–biotin technique (Vectastain Elite Kit; Vector Lab). After washing in PBS, the substrate aminoethyl carbazole was applied to the tissue. Omission of primary antibodies served as negative control. Specificity of the staining was also controlled on sections of peripheral lymphoid organs. Tissue areas were measured by image analysis and the numbers of stained cells were calculated per mm2 at ×20 magnification.
Preparation of mononuclear cells (MNC) from lymph nodes
The popliteal and inguinal lymph nodes from dead animals were removed under aseptic conditions on day 18 p.i. Lymph-node cell suspensions were prepared by grinding through a wire mesh. Single-cell suspensions of MNC from individual rats were prepared separately. The cells were washed three times in complete culture medium (CDME; Flow Lab, Irvine, UK), supplemented with 1% (v/v) non-essential amino acids, 50 IU/ml penicillin, 60 μg/ml streptomycin, 2 mm glutamine (Flow) and 3% (v/v) normal human AB+ serum without mercaptoethanol. Cells were adjusted to 2×106 lymph node MNC/ml.
Lymphocyte proliferation assay
For assessment of antigen-induced lymphocyte proliferation, a standard 3H-thymidine incorporation test was used. Lymph node MNC suspended in 200 μl aliquots, at cell density of 2×106 cells/ml, were cultured in round bottomed 96-well polystyrene microtiter plates (Nunc, Copenhagen, Denmark). For lymphocyte stimulation 10 μl aliquots of bovine peripheral myelin (BPM), P2 peptide 57-81 antigen or 10 μl of phytohraemagglutinin (PHA) (Difco) were added to cultures, respectively, to a final concentration of 10 μg/ml. The indicated concentrations had optimal stimulatory effects as assessed in preliminary experiments. Triplicate wells without antigen or mitogen served as background controls. After 60 hr of incubation at 37° in a humidified atmosphere with 7% CO2, cells were pulsed with 3H-methylthymidine (1 μCi/well; Amersham, Little Chalfont, UK) and cultured for additional 12 hr. Cells were harvested onto glass filters (Titertec, Skatron, Lierbyen, Norway).3H-thymidine incorporation was measured in a liquid β-scintillation counter. The results were expressed as counts per minute (c.p.m.) per culture.
Determination of P2 peptide 57-81-specific immunoglobulin G (IgG) antibodies of different isotypes
Serum was obtained from blood samples taken at days 18 and 36 p.i. Purified rat P2 peptide 57-81 was coated onto enzyme-linked immnuosorbent assay (ELISA) plates at 10 μg/ml in a volume of 100 μl/well. The plates were incubated overnight at 4° and washed three times with PBS plus 0·05% Tween-20. Non-specific binding was blocked with 1% normal horse serum for 2 hr. After three washings, serum samples diluted to 1:200 with PBS, were applied to wells and incubated for 2 hr at room temperature. After another three washings, either biotinylated mouse antirat IgG1 or IgG2b (1:2000, ams, Frankfurt, Germany) were added and incubated for 2 hr at room temperature. A further three washings were followed by incubation with avidin-biotin alkaline phosphatase complex (Vector, Burlingame, CA) for 1 hr at room temperature. The reaction was visualized with p-nitrophenyl phosphate substrate (Sigma) and read at 405 nm using an ELISA reader.
Measurements of TNF-α serum levels
Capture mouse antirat monoclonal antibody (mAb) and detecting polyclonal antibody reactive with rat TNF-α were produced at the Division of Neurology, Huddinge University Hospital, Karolinska Institute, Stockholm, Sweden. The specificity of antibody was examined and showed lack of cross-reactivities with various other cytokines. These antibodies were used by ELISA, which were adopted in this study for detection of serum TNF-α.30 Briefly, enzyme immunoassay (EIA)/ radioimmunoassay (RIA) flat-bottom, high-binding plates (Costar, Cambridge, MA) were coated with 100 μl anti-TNF-α mAb diluted to a concentration of 1 μg/ml in carbonate bicarbonate buffer (pH 9·6) and kept at 4° overnight. After four washes with 0·05 PBS–Tween-20, the wells were blocked with 100 μl per well of 5% BSA for 90 min at room temperature. After repeated washings with PBS, 100 μl sera diluted 1:20 in PBS, were added to each well. After 24 hr incubation at 4°, plates were washed repeatedly in PBS–Tween-20. To detect the bound TNF-α, anti-TNF-α polyclonal antibodies were incubated at concentrations of 10 μg/ml for 1 hr at 37°. After five washes, 100 μl of biotinylated rabbit antirat IgG (Vector) diluted 1:2000 in PBS were added. After another 1 hr of incubation at 37° and 4 washes, 100 μl of avidin–biotin alkaline phosphatase complex (ABC-AP; Vector Lab. Burlingame, CA) diluted 1:100 in PBS was added for 30 min. Unbound ABC-AP was removed by consecutive washings with PBS and 100 μl/well of freshly prepared enzyme substrate solution was added. Absorbance was measured after 15 min incubation in the dark in a 405 Multiscan photometer (mcc/340; Labsystem, Helsinki, Finland). In order to quantify serum TNF-α, standard TNF-α curves were obtained simultaneously by incubating different known concentrations (0, 0·009, 0·19, 0·39, 0·78, 1·56, 3·12, 6·25, 12·5, 25, 50, 100, 200 and 400 pg/ml) of TNF-α for 60 min at room temperature in wells precoated with anti-TNF-α mAb (obtained from TNO Primate Center, Rijswijk, The Netherlands). The procedure for developing the plates was essentially the same as described above and the absorbances measured from the standard concentration of TNF-α were used to plot TNF-α standard curves using computer software. Thereafter, the absorbances obtained from the specimens were automatically converted to pg/ml by the computer from the standard curve. In this assay, background absorbances (wells without coating mAb) were very low and they were subtracted from the absorbances of the specimens.
Statistical evaluation
Clinical scores of EAN between groups of animals at individual days, were compared by non-parametric Mann–Whitney test. Student’s t-test was used for comparison of inflammation and demyelination. Differences between the groups were tested by one-factor analysis of variance (anova). All tests were two-sided.
RESULTS
Nasal administration of high dose rr IL-6 reduces clinical signs of EAN
The time for initiating treatment of EAN rats with nasally administered rrIL-6 was chosen to simulate the human situation of starting therapy at the onset of the first apparent clinical deficit. Rats receiving 1 μg rrIL-6 intranasally daily, from 9 to 15 days post-immunization (p.i) showed a reduced severity of their ongoing EAN compared with low dose (0·1 μg/rat/day) rrIL-6-treated rats or with PBS- and BSA-treated control rats (Fig. 1). The difference was statistically significant after 7 days of treatment, i.e. from day 15 p.i. (P < 0·03 for all comparisons). High-dose rrIL-6-treated rats had a shorter duration of EAN (mean duration days 62·5±3·2) compared with low-dose rrIL-6-treated rats or with PBS- and BSA-treated control rats (mean duration days 118±9·53) (P < 0·05 for all comparisons).
Figure 1.

Effects of rrIL-6 treatment on clinical scores of EAN induced in Lewis rats by immunization on day 0 with P2 peptide 51-87 and Freund’s complete adjuvant (FCA). Rats in the treatment groups received intranasally either high dose rrIL-6 (1 μg/rat/day) or low dose rrIL-6 (0·1 μg/rat/day) for 7 consecutive days (from 9 to 15 days p.i.). The control groups received either PBS (a) or BSA (b). Values are given as mean±SD in groups of eight animals (P < 0·03 refers to comparisons between high dose rrIL-6-treated rats to low dose rrIL-6-treated and to BSA-or PBS-treated control EAN rats after 7 days of treatment, i.e. from day 1 p.i.).
Histopathology
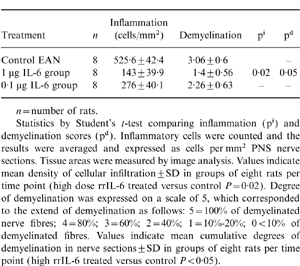
Histopathological evaluation revealed that high-dose rrIL-6-treatment resulted in less pronounced infiltration by macrophages, lymphocytes and granulocytes (Fig. 2a) as well as reduced regional demyelination (Fig. 2b) in the sciatic nerve sections compared with PBS-treated control rats (Fig. 2c,d) when examined on day 18 p.i. Focal areas of perivascular inflammation with only minimal demyelination were detected within PNS in high-dose rrIL-6-treated rats. The difference in inflammation and demyelination scores comparing high-dose rrIL-6-treated to PBS-treated control rats was statistically significant (P < 0·05 for inflammation and demyelination). There were no significant differences between low-dose rrIL-6-treated and PBS-treated rats (Table 1).
Figure 2.

Low-grade inflammation (a) the arrow indicates a inflammatory cell) and minimal demyelination (b) the arrow indicates an area of demyelination) within PNS were detected in rats treated nasally with high dose (1 μg/rat/day) of rrIL-6, and inflammatory infiltrates composed of macrophages and lymphocytes (c), as well as severe regional demyelination, (d) the arrows indicate areas of demyelination) in sciatic nerve sections from control EAN rats receiving PBS on day 18 p.i. (×2 00).
Table 1.
Sciatic nerve pathology scores (mean±SD) on day 18 p.i. in EAN Lewis rats treated nasally with rr IL-6
Immunohistochemical findings
Extensive CD4+, CD8+ T cells and macrophage infiltration as well as increased MHC class II expression were regularly detected in sciatic nerve sections from the PBS-treated control EAN rats on day 18 p.i. (Fig. 3). In contrast, only few macrophages, low levels of MHC class II expression as well as very few CD4+ and CD8+ T cells were seen in sciatic nerve sections from EAN rats treated with high doses of rrIL-6 at the same time point (high dose rrIL-6-treated versus PBS-treated control rats P < 0·001 for macrophages; P < 0·01 for MHC class II; P < 0·05 for CD4+ and CD8+ T cells). There were no significant differences between low-dose rrIL-6-treated and PBS-treated rats.
Figure 3.

Density of macrophages, MHC II positive cells, CD4+ and CD8+ T cells in sciatic nerve sections on day 18 p.i. from rats receiving rrIL-6 at different doses (0·1 or 1 μg/rat/day) or PBS by the nasal route. Tissue areas were measured by image analysis and the numbers of positive cells were counted in longitudinal serial sections at ×20 magnification. The results were averaged and expressed as cells per mm2 tissue sections. Values indicate mean density of cellular infiltration ±SD in groups of eight rats per time point. P-values refer to comparisons between high dose rrIL-6-treated rats with PBS-treated control EAN rats (***P < 0·001; **P < 0·01; *P < 0·05). There was no significant difference between low dose rrIL-6-treated and PBS-treated rats.
RrIL-6 suppresses P2 peptide 57-81-induced T-cell responses
Nasal administration of 1 μg rrIL-6 significantly suppressed the proliferative responses of T cells to the BPM as well as to the P2 peptide 57-81 in lymph node MNC cultures, when compared with low-dose rrIL-6-treated rats or PBS-treated control rats (P < 0·05 for both comparisons). Low-dose rrIL-6 does not induce a significant suppression of autoantigen reactivity of T cells (Fig. 4). These data document that nasal administration of rrIL-6 induces suppression of autoantigen reactivity of T cells in a dose-dependent manner.
Figure 4.

Proliferation of lymph node MNC on day 18 p.i. from rats receiving rrIL-6 at different doses (0·1 or 1 μg/rat/day) or PBS by the nasal route. MNC were cultured in the presence of P2 peptide 51-87 and BPM as antigens or PHA as mitogen as well as without antigen or mitogen. The left y-axis refers to the proliferation of T cells in response to the BPM and P2 peptide 57-81, as well as without antigen in lymph node MNC cultures. The right-hand y-axis refers to the proliferation of T cells in response to PHA. Mean values and SD are indicated. P-values refer to comparisons between rrIL-6-treated rats with PBS-treated control EAN rats. *P < 0·05.
P2 peptide 57-81-specific antibodies of IgG isotype are altered in rats treated with rrIL-6
We analysed the levels of specific antibodies to the P2 peptide 57-81 in the rat sera on days 18 and 36 p.i. Elevated serum levels of IgG1 and decreased levels of IgG2b isotype antibodies were observed at day 18 p.i. (Fig. 5) but not at day 36 p.i. in high dose rrIL-6-treated EAN rats compared to PBS-treated control rats (P < 0·05 for both comparisons). Levels of IgG1 and IgG2b antibodies in low-dose rrIL-6 treated rats were not significantly altered compared to control rats.
Figure 5.

Serum levels of IgG1 and IgG2b isotype antibodies to the P2 peptide 57-81 observed on day 18 p.i. from EAN rats receiving different doses of rrIL-6 nasally. Mean values and SD are indicated. P-values refer to comparisons between rrIL-6-treated rats with PBS-treated control EAN rats. *P < 0·05.
Serum levels of TNF-α
Although cytokines act mainly in an autocrine or paracrine way, with rather short half-lifes and high affinity to nearby receptors, measurable levels of TNF-α were detected in sera from rats in this study (Fig. 6). High-dose rrIL-6-treated rats showed significantly lower serum levels of TNF-α at days 18 and 36 p.i (P < 0·05 for both comparisons) compared to PBS-treated rats. Levels of TNF-α in low-dose rrIL-6-treated rats were not significantly altered, compared to control rats in any time point.
Figure 6.

Serum levels of TNF-α observed on days 18 and 36 p.i. from EAN rats receiving different doses of rrIL-6 or PBS by the nasal route. Mean values and SD are indicated. P-values refer to comparisons between high dose rrIL-6-treated rats to PBS-treated control EAN rats. *P < 0·05.
DISCUSSION
This study demonstrates that nasal administration of rrIL-6 at the initial phase of EAN decreases the severity of EAN in Lewis rats with a chronic course of the disease. The usage of the chronic EAN model may have two advantages over the conventional ones: (i) the disease course more resembles human GBS, which is often characterized by neurological deficits over a larger period of time than the acute EAN; and (ii) it may be more suitable for demonstrating discrete cytokine effects than the short-lasting acute EAN. The duration of the disease in PBS-treated control rats (about 120 days) is considerably prolonged compared to the ‘normal’ acute EAN induced by active immunization with whole peripheral myelin. The reason for the chronic course of the EAN in our study may be the application of larger dose of the P2 peptide 57-81 used for immunization, i.e. 240 μg.26
Among the cytokines that orchestrate cellular interactions during immune responses, IL-6 is considered one of the major mediators.6 In experimental autoimmune encephalomyelitis (EAE), neutralizing antibodies to IL-6 increased the bioactivity of the cytokine in the circulation and CNS and decreased the severity of the disease31 suggesting a disease-downregulatory role of IL-6. However, the effect of IL-6 on ongoing demyelination is not known so far. Furthermore, we were interested in the therapeutic effect of IL-6 when treatment was usally applied in the initial phase of ongoing EAN (day 9 p.i.). Previous studies indicated that the protective effects of IL-6 are related to the time of administration.15,18 In our study, administration of a high dose of rrIL-6 (1 μg rrIL-6/rat/day) reduced the EAN when given at the onset of the first clinical signs of the disease. The dose is small compared to systemically applied doses15 and is not associated with side effects from other systems and organs32 (data not shown). Intranasal administration of rrIL-6 is an easy route of administration, safe and effective. The lower tested dose of 0·1 μg rrIL-6/rat/day was not effective on the clinical course of EAN suggesting a ‘threshold dose’ for therapeutic effects between 0·1 μg and 1 μg rrIL-6/rat/day.
Nasal treatment of chronic EAN with high-dose rrIL-6 was accompanied by reduced T lymphocyte proliferative responses to P2 peptide 57-81 as well as reduced levels of TNF-α in serum. These results, being in agreement with other studies7,16,18 indicate that rrIL-6 exerts its protective effects through the suppression of autoantigen reactivity of T cells and the reduction of TNF-α production, thereby supporting an anti-inflammatory role for IL-6. IL-6 negatively regulates the production of TNF-α, whereas TNF-α functions conversely as a positive modulator of IL-6 production.19,33 Additionally, IL-6 is the major inducer of the acute phase response, whose reactants include TIMP (tissue inhibitor of metalloproteinase) and other proteins with anti-inflammatory potential.34 Furthermore, IL-6 induces adrenocorticotropic hormone, which induces glucocorticosteroid synthesis and synergizes with adrenocorticotropic hormone to enhance glucocorticosteroid release.35 Corticosteroids exert in turn an inhibitory effect on the production of inflammatory cytokines, thus activating a negative feedback loop.36 That the effects of nasal rrIL-6 were systemic, is further indicated by the result that serum levels of IgG1 antibodies to P2 peptide were increased in rats treated with high dose of rrIL-6. These effects were obvious at day 18 p.i. but not at day 36 p.i., most likely caused by the relatively short duration of exposure to rrIL-6. The changes in serum levels of specific IgG1 antibodies could be caused by direct effects of IL-6 on B cells. It is well known that IL-6 enhances immunoglobulin secretion by activated B cells and causes their differentiation into plasma cells.37
The systemic immune alterations in peripheral lymphocytes were reflected at the pathological level in target organs of EAN, i.e. in sciatic nerves. High-dose rrIL-6 diminished the extent of demyelination and reduced inflammation characterized by infiltration of macrophages, CD4+ and CD8+ T cells as well as MHC class II expression. This suggests that factors that influence the development of demyelination and inflammation in PNS were reduced. The pathological hallmark of EAN and GBS is a demyelination of spinal nerve roots and sciatic nerves, with a predominant infiltration of T lymphocytes and macrophages.38,39 T cells open the BNB and allow circulating autoantibodies to have access to myelin antigens which are then responsible for the nerve damage.40,41 Furthermore, in PNS the endoneural macrophages are most likely the antigen-presenting cells.39 These cells exert most of their effects and immunoregulatory functions through the release of cytokines. TNF-α, a primary mediator of inflammation and autoimmune tissue injury42 and IL-1, another potent proinflammatory cytokine43 represent triggering mechanisms for EAN and GBS including selective damage to peripheral myelin. As mentioned above the protective effect of IL-6 may be mediated by various mechanisms including its function as a negative modulator of TNF-α and IL-1 production.18,19,33 There is the possibility that reduction of TNF-α levels may be verified at the tissue level by determining the production of TNF-α in the target organs, as was shown previously. IL-6 inhibits lipopolysaccharide (LPS)-induced synthesis of TNF-α protein in glial cells and suppresses demyelination in a viral model of multiple sclerosis15 as well as inhibiting the development of adjuvant arthritis.44 Suppression of TNF-α activity in the early stages of EAN and GBS provides an avenue to a more effective therapy.45
In conclusion, intranasal administration of rrIL-6 ameliorates chronic EAN, modulates systemic immune response by reducing autoantigen T-cell reactivity and TNF-α activity and suppresses the inflammation and the demyelination in sciatic nerves of treated rats, even when it is given at the onset of ongoing EAN. The reduction immune response to PHA indicates that this treatment is not specific. Therefore, more experience with long-term application of the cytokine and great caution are needed, before IL-6 is administered nasally to humans with autoimmune diseases such as immune-demyelinating mediated neuropathies.
Acknowledgments
The study was supported by grants from the Swedish Medical Research Council, Swedish Doctor Society and funds from Karolinska Institute.
REFERENCES
- 1.Fearon ER, Pardoll DM, Itala T, et al. Interleukin 2 production by tumor cells bypasses T helpers function in the generation of an anti-tumor response. Cell. 1990;60:397. doi: 10.1016/0092-8674(90)90591-2. [DOI] [PubMed] [Google Scholar]
- 2.Mentzel B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5:1159. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 3.Xiao BG, Bai XF, Zhang GX, Link H. Suppression of acute and protracted-relapsing experimental allergic encephalomyelitis by nasal administration of low-dose IL-10 in rats. J Neuroimmunol. 1998;84:230. doi: 10.1016/s0165-5728(97)00264-6. [DOI] [PubMed] [Google Scholar]
- 4.Hirano T, Yasukawa K, Harada H, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 5.Hirano T. Interleukin 6. In: Thomson AW, editor. The Cytokine Handbook. 2. London: Academic Press; 1994. p. 145. [Google Scholar]
- 6.Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16:249. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 7.Barton B. Molecule of the month. IL-6: Insights into the novel biological activities. Clin Immunol Immunophathol. 1997;85:16. doi: 10.1006/clin.1997.4420. [DOI] [PubMed] [Google Scholar]
- 8.Wong GW, Clark SC. Multiple actions of interleukin 6 within a cytokine network. Immunol Today. 1988;9:137. doi: 10.1016/0167-5699(88)91200-5. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Mix E, Link H. Cytokine production and the pathogenesis of experimental autoimmune neuritis and Guillain–Barré syndrome. J Neuroimmunol. 1998;84:40. doi: 10.1016/s0165-5728(97)00238-5. [DOI] [PubMed] [Google Scholar]
- 10.Weller M, Stevens A, Sommer N, Melms A, Dichans J, Witholter H. Comparative analysis of cytokine patterns in immunological disorders. J Neurol Sci. 1991;104:215. doi: 10.1016/0022-510x(91)90313-v. [DOI] [PubMed] [Google Scholar]
- 11.Sivieri S, Ferrerini AM, Lolli F, et al. Cytokine pattern in the cerebrospinal fluid from patients with GBS and CIDP. J Neurol Sci. 1997;147:93. doi: 10.1016/s0022-510x(96)00319-x. [DOI] [PubMed] [Google Scholar]
- 12.Strassmann G, Fong M, Windsor S, Neta R. The role of interleukin-6 in lipopolysaccharide-induced weight loss, hypoglycemia and fibrinogen production, in vivo. Cytokine. 1993;5:285. doi: 10.1016/1043-4666(93)90058-d. [DOI] [PubMed] [Google Scholar]
- 13.Baumann H, Gauldie J. The acute response. Immunol Today. 1994;15:74. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 14.Fattori E, Cappelletti M, Costa P, et al. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med. 1994;180:1243. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez M, Pavelco KD, Mckinney CW, Leibowitz JL. Recombinant human IL-6 suppresses demyelination in a viral model of multiple sclerosis. J Immunol. 1994;153:3811. [PubMed] [Google Scholar]
- 16.Tilg H, Dinarello CA, Mier J. IL-6 and APPs: anti-inflammatory and immunosuppressive mediators. Immunol Today. 1997;18:428. doi: 10.1016/s0167-5699(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 17.Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell RA. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med. 1997;185:461. doi: 10.1084/jem.185.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizuhara H, O’neil E, Seki N, et al. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin-6. J Exp Med. 1994;179:1529. doi: 10.1084/jem.179.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aderka D, Le J, Vilcek J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor-α production in cultured human monocytes, U937 cells and in mice. J Immunol. 1989;143:3517. [PubMed] [Google Scholar]
- 20.Waksman NH, Adams RD. Allergic neuritis: an experimental disease of rabbits induced by the injection of peripheral nervous tissue and adjuvant. J Exp Med. 1955;102:213. doi: 10.1084/jem.102.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaldubowski M, Hughes RA. Identification of the neuritogen for experimental allergic neuritis. Nature. 1979;277:140. doi: 10.1038/277140a0. [DOI] [PubMed] [Google Scholar]
- 22.Milner PA, Lovelidge CA, Taylor WA, Hughes RA. P0 myelin protein produces experimental allergic neuritis. J Neurol Sci. 1987;79:275. doi: 10.1016/0022-510x(87)90235-8. [DOI] [PubMed] [Google Scholar]
- 23.Olee T, Powell HC, Brostoff SW. New minimum length requirement for a T cell epitope for experimental allergic neuritis. J Neuroimmunol. 1990;27:187. doi: 10.1016/0165-5728(90)90068-x. [DOI] [PubMed] [Google Scholar]
- 24.Hanh AF. Experimental allergic neuritis as a model for the immune-mediated demyelinating neurophathies. Rev Neurol. 1996;152:328. [PubMed] [Google Scholar]
- 25.Hanh AF, Feasby TE, Steele A, Lovgren DS, Berry J. Demyelination and axonal degeneration in Lewis rat experimental allergic neuritis depend on the myelin dosage. Lab Invest. 1988;59:115. [PubMed] [Google Scholar]
- 26.Harvey GK, Polland JD, Schindhelm K, Antony J. Experimental allergic neuritis. An electrophysiological and histological study in the rabbit. J Neurol Sci. 1987;81:215. doi: 10.1016/0022-510x(87)90097-9. [DOI] [PubMed] [Google Scholar]
- 27.Adams AM, Atkinson PF, Hall SM, Hughes RA, Taylor WA. Chronic experimental allergic neuritis in Lewis rats. Neuropathol Appl Neurobiol. 1989;15:249. doi: 10.1111/j.1365-2990.1989.tb01226.x. [DOI] [PubMed] [Google Scholar]
- 28.Rostami A, Gregorian SK, Brown MJ, Pleasure DE. Induction of severe experimental allergic neuritis with a synthetic peptide corresponding to the 53 amino acid sequence of the myelin P2 protein. J Neuroimmunol. 1990;30:145. doi: 10.1016/0165-5728(90)90098-8. [DOI] [PubMed] [Google Scholar]
- 29.Said G, Hontebeyrie-Jockowicz M. Nerve lesions induced by macrophage activation. Res Immunol. 1992;143:589. doi: 10.1016/0923-2494(92)80040-r. [DOI] [PubMed] [Google Scholar]
- 30.Bakhiet M, Diab A, Mustafa M, Zhu J, Lindqvist L, Link H. Potential role of autoantibodies in the regulation of cytokine responses during bacterial infections. Infect Immun. 1997;65:3300. doi: 10.1128/iai.65.8.3300-3303.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gijbels K, Brocke S, Abrams J, Steiman L. Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated level of IL-6 bioactivity in central nervous system and circulation. Mol Med. 1995;1:795. [PMC free article] [PubMed] [Google Scholar]
- 32.Kammuller M, Ryffel B. Extrapolation of experimental safety data to humans: the interleukin-6 case. Clin Immunol Immunopathol. 1997;83:15. doi: 10.1006/clin.1996.4305. [DOI] [PubMed] [Google Scholar]
- 33.Schindler R, Mancilla J, Enders S, Ghorbani R, Clark SC, Dinarello CA. Correlations and interactions in the production of IL-6, tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood. 1990;75:40. [PubMed] [Google Scholar]
- 34.Baumann H, Strassmann G. Suramin inhibits the stimulation of acute phase protein genes by IL-6-type cytokines in rat hepatoma cells. J Immunol. 1993;151:1456. [PubMed] [Google Scholar]
- 35.Salas MA, Evans SW, Levell MJ, Whicher JT. Interleukin-6 and ACTH act synergistically to stimulate the release of corticosterone from adrenal gland cells. Clin Exp Immunol. 1990;79:470. doi: 10.1111/j.1365-2249.1990.tb08114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Besedovsky H, Del Rey A, Sorkin E, Dinarello CA. Immunoregulatory feedback between interleukin-1 and glucocorticosteroid hormones. Science. 1986;233:652. doi: 10.1126/science.3014662. [DOI] [PubMed] [Google Scholar]
- 37.Muraguchi A, Hirano T, Tang B, et al. The essential role of B cell stimulatory factor 2 (BSF-2/IL-6) for the terminal differentiation of B cells. J Exp Med. 1988;167:332. doi: 10.1084/jem.167.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartung HP, Toyka KV. T cell and macrophage activation in experimental autoimmune neuritis and Guillain–Barré syndrome. Ann Neurol. 1990;27(Suppl.):S 57. doi: 10.1002/ana.410270716. [DOI] [PubMed] [Google Scholar]
- 39.Hartung HP, Kiefer R, Cold R, Toyka K. Autoimmunity in the peripheral nervous system. Baillière’s Clin Neurol. 1996;5:1. [PubMed] [Google Scholar]
- 40.Spies JM, Pollard JD, Westald KW, Bonner JG, McLeod J. Synergy between antibody and P2-reactive T cells in experimental autoimmune neuritis. J Neurol. 1995;57:77. doi: 10.1016/0165-5728(94)00164-j. [DOI] [PubMed] [Google Scholar]
- 41.Spies JM, Westald KW, Bonner JG, Pollard JD. Intraneural activated T cells cause focal breakdown of the blood–nerve barrier. Brain. 1995;118:857. doi: 10.1093/brain/118.4.857. [DOI] [PubMed] [Google Scholar]
- 42.Redford EJ, Hall SM, Smith KJ. Vascular changes and demyelination induced by the intraneural injection of tumor necrosis factor. Brain. 1995;118:869. doi: 10.1093/brain/118.4.869. [DOI] [PubMed] [Google Scholar]
- 43.Dinarello C, Wolff S. The role of interleukin-1 in disease. N Engl J Med. 1993;118:869. doi: 10.1056/NEJM199301143280207. [DOI] [PubMed] [Google Scholar]
- 44.Mihara M, Ikuta M, Koishihara Y, Ohsugi Y. Interleukin 6 inhibits delayed-type hypersensitivity and the development of adjuvant arthritis. Eur J Immunol. 1991;21:2327. doi: 10.1002/eji.1830211006. [DOI] [PubMed] [Google Scholar]
- 45.Sharief MK, Ingmar DA, Swash M. Circulating tumor necrosis factor-α correlates with electrodiagnostic abnormalities in Guillain–Barré syndrome. Ann Neurol. 1997;42:68. doi: 10.1002/ana.410420112. [DOI] [PubMed] [Google Scholar]