

Abstract
A rat monoclonal antibody (mAb) that neutralizes mouse interleukin-12 (IL-12) was administered to female non-obese diabetic (NOD) mice of different ages to dismantle the role of endogenous IL-12 in murine autoimmune diabetogenesis. This mAb was effective in preventing clinical, but not histological signs of spontaneous diabetes when treatment was started early in life at the age of 4 weeks and consecutively continued for 10 weeks. Delaying commencement of anti-IL-12 mAb prophylaxis until the age of 18 weeks, when NOD mice suffer from advanced insulitis, was ineffective. Anti-IL-12 mAb did not influence the course of the accelerated model of diabetes induced by cyclophosphamide. These data prove that the pathogenetic role of endogenous IL-12 in NOD mouse diabetes is restricted to the very early diabetogenic events presumably occurring prior to insulitis development.
A delayed-type hypersensitivity (DTH) reaction mounted from autoreactive macrophages and T lymphocytes against relevant β-cell autoantigens, may be involved in the pathogenesis of type 1 diabetes both in humans and animal models [non-obese diabetic (NOD) mouse, diabetes-prone biobreeding (DP-BB) rat].1 Studies in the NOD mouse have also indicated that the anti-β-cell response is driven by the T helper 1 (Th1) cell subset of T lymphocytes that produce type 1 cytokines such as interleukin-2 (IL-2), tumour necrosis factor-α/β (TNF-α/β) and interferon-γ (IFN-γ) that stimulate DTH reactions.2–4
Interleukin-12, a heterodimeric cytokine composed of the p30 and p45 subunits, is produced by antigen-presenting cells (APC) such as macrophages and dendritic cells. Because IL-12 powerfully up-regulates IFN-γ production from T cells and natural killer (NK) cells and polarizes Th0 cells into a Th1 secretory penotype, a disregulated production of IL-12 from APC has been hypothesized to be an important pathogenetic movens in eliciting the type 1 cytokine-mediated anti-β-cell response.5 Studies in NOD mice and BB rats have demonstrated an association between IL-12 gene expression and disease development both in cyclophosphamide-(CY) induced (NOD mouse) and spontaneous type-1 diabetes.6–8 Moreover, exogenous IL-12 accelerates the course and augments the incidence of type 1 diabetes in female NOD mice, apparently by inducing Th1 cells,9 and treatment with IL-12p(40)2, the homodimer of IL-12 p40 that antagonizes IL-12p75 bioactivities, may inhibit CY-induced type-1 diabetes in NOD mice.10 Local expression of the IL-12p40 gene also prolongs syngeneic islet graft survival in diabetic NOD mice by dampening immuno-inflammatory destruction of the islet cells.11 Further support for a pathogenetic role of IL-12 in type-1 diabetes comes from in vitro studies showing that high-risk type-1 diabetes first-degree relatives produce more IL-12 than controls12 and that IL-12 exerts a weak suppressive effects on normal rodent islets.13
Other reports have however, questioned or limited the pathogenicity of IL-12 in type-1 diabetes. When administered under a different treatment regimen than that of Trembleau et al., IL-12 inhibits NOD mouse diabetes,14 and IL-12p(40)2 is only capable of preventing disease development in NOD mice if treatment is started early during the pre-diabetic period when the mice are devoid of insulitis.15 Moreover, unlike Rothe et al.,10 CY-induced diabetes could not be suppressed by exogenous IL-12p(40)2 in this study.15
These conflicting data prompted us to undertake this study where we used a neutralizing rat anti-mouse IL-12 monoclonal antibody (mAb) to treat NOD mice at different stages during development of spontaneous and CY-induced type-1 diabetes. Our data clearly demonstrated that blockage of endogenous IL-12 only significantly reduced type-1 diabetes development in these animals if the anti-IL-12 mAb is given early in life within the age of 4 weeks. Starting anti-IL-12 mAb prohylaxis in NOD mice at 18-weeks old was ineffective, as it was in CY-induced type-1 diabetes.
Euglycaemic female NOD mice purchased from Charles River Laboratories (Calco, Italy) were kept under standard laboratory conditions (non-specific pathogen free) and provided with food and water ad libitum.
The mice were screened for diabetes development by twice weekly examination of glycosuria. Diabetes was diagnosed in the presence of two consecutive days of glycosuria followed by fasting glycaemia above 11·8 mmol/l.
The rat IgG2a anti-mouse IL-12 mAb, C17.8 neutralizes mouse IL-12 in vivo and exerts biological effects in a mouse model of experimental colitis and experimental allergic encephalomyelitis. The C17.8 mAb is widely used for neutralization of IL-12, and an isotype-matched control is no longer considered necessary.17 Therefore, irrelevant rat immunoglobulin G (IgG; Sigma, St Louis, MO) were used as irrelevant control.
The degree of mononuclear cell infiltration was graded in a blind fashion as described elsewhere:18 0, no infiltrate; 1, peri-ductular infiltrate; 2, peri-islet infiltrate; 3, intraislet infiltrate; 4, intraislet infiltrate associated with beta-cell destruction. At least 15 islets were counted for each mouse. A mean score for each pancreas was calculated by dividing the total score by the numbers of islets examined.
Four-week old, euglycaemic female NOD mice were randomly allocated into six groups (A-F) that received anti-IL-12 mAb, irrelevant rat IgG or no treatment, for 10 consecutive weeks. This period of age was chosen to evaluate the effects of anti-IL-12 mAbs on the early diabetogenic pathways of the NOD mice because virtually all mice are free from insulitis until 4–5 weeks of age.1 At the age of 14 weeks euglycaemic mice from groups A–C were killed for histological examination of insulitis, while the mice from the other groups D–F were left untreated and followed for diabetes development until the age of 30 weeks.
The extent of insulitis (mean values±SD) was comparable between anti-IL-12 mAb-treated (1·9±0·7, n = 8) and control mice either untreated (2·1±0·7, n = 8) or treated with irrelevant rat IgG (2·4±0·5, n = 6) (P > 0·05 versus both control groups by anova). However, in spite of this, the number of mice becoming overtly diabetic was significantly reduced by anti-IL-12 mAb, the so-treated mice having a reduced incidence of disease that appeared at an older age as compared with the controls (P < 0·02 versus both control groups by Logrank (Cox-mantel, see Fig. 1). However, continuous treatment with anti-IL-12 mAb seemed necessary for the anti-diabetogenic effects to be maintained as type-1 diabetes occurred in the majority of the mice after 2–3 months from treatment withdrawal (Fig. 1).
Figure 1.

Early prophylaxis with anti-IL-12 mAb reduces the incidence of type-1 diabetes in NOD mice. Euglycaemic 4-week-old female NOD mice, received for 10 consecutive weeks 1 mg/mouse of either anti-IL-12 mAb or irrelevant rat IgG. An additional control group received no treatment. At the age of 14-weeks old, treatments were withdrawn and the mice were followed for diabetes development until the age of 30 weeks. Each group consisted of 12 mice. Data from two independent experiments are shown.
To ascertain whether blockage of endogenous IL-12 later during advanced phases of pre-diabetes was also effective in diminishing type-1 diabetes development, NOD mice were treated with either anti-IL-12 mAb or irrelevant rat IgG for eight consecutive weeks from the age of 18-weeks old, a period of life when these animals have developed severe insulitis.1 An additional control group of mice received no treatment. These mice were followed for type-1 diabetes development until the age of 30-weeks old. As shown in Fig. 2, anti-IL-12 mAb was ineffective under this late prophylactic regimen, most of the so-treated mice developing type-1 diabetes with a similar kinetic and incidence as the control mice.
Figure 2.

Late prophylaxis with anti-IL-12 mAb does not reduce the incidence of type-1 diabetes in NOD mice. From the age of 18-weeks old, euglycaemic NOD mice were continuously treated for 8 weeks with 1 mg/mouse weekly of either anti-IL-12 mAb (n = 12) or irrelevant rat IgG (n = 12). An additional control group of mice received no treatment (n = 11). These mice were followed for type-1 diabetes development until the age of 30 weeks. Data from two independent experiments are shown.
Finally, we examined the effects of anti-IL-12 mAb in the accelerated model of diabetes that can be induced in NOD mice by one or two large injections with CY.1 Euglycaemic 105-day-old female NOD mice were randomly divided into three groups that received anti-IL-12 mAb, irrelevant rat IgG or no treatment. The mice were challenged i.p. with 250 mg/kg CY (Endoxan Asta, Schering Plough, Milan, Italy) on day 0. As shown in Table 1, 14 days after CY injection the majority of the control mice were either treated with irrelevant rat IgG or untreated developed type-1 diabetes. Anti-IL-12 mAb treatment had no effect on the course of CY-induced type-1 diabetes as shown by the comparable rate of diabetes incidence that was observed in these mice as compared with the controls (Table 1). Significant differences could not be found in the severity of insulitis between the three groups of mice killed on day +14 (Table 1).
Table 1.
Failure of anti-IL-12 mAb to modify CY-induced diabetes and insulitis in NOD mice.
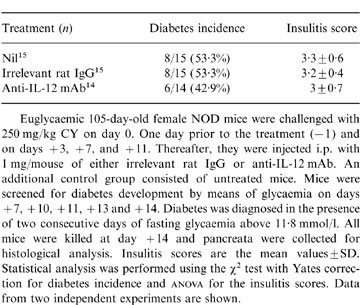
Euglycaemic 105-day-old female NOD mice were challenged with 250 mg/kg CY on day 0. One day prior to the treatment(−1) and on days +3, +7, and +11. Thereafter, they were injected i.p. with 1 mg/mouse of either irrelevant rat IgG or anti-IL-12mAb. An additional control group consisted of untreated mice. Mice were screened for diabetes development by means of glycaemia on days +7, +10, +11, +13 and +14. Diabetes were diagnosed in the presence of two consecutive days of fasting glycaemia above 11·8 mmol/1. All mice were killed at day +14 and pancreata were collected for histological analysis. Insulitis scores are the mean values±SD. Statistical analysis was performed using the x2 test with Yates correction for diabetes incidence and anova for the insulitis scores. Data from two independent experiments are shown.
We have demonstrated that type-1 diabetes development can be temporarily suppressed in NOD mice if the action of endogenous IL-12 is negated early in life when the insulitis process is likely not to have started or it is still in its very early stages. Blockage of IL-12 in older NOD mice that are expected to suffer from actively ongoing insulitis or in the accelerated model of diabetes induced by CY was ineffective. These data are consistent with those of Trembleau et al.15 showing that exogenously administered IL-12(p40)2 only reduces in a reversible fashion the clinical development of spontaneous type-1 diabetes in NOD mice if treatment started in 3-week-old mice is continued until the age of 12 weeks. Besides reducing spontaneous type-1 diabetes, this early and prolonged treatment also renders the mice resistant to the diabetogenic action of two injections of CY given at 8 and 10 weeks of age.15 However, in agreement with our study, when IL-12(p40)2 prophylaxis is first started in 9-week-old NOD mice, only a partial and not significant protection against type-1 diabetes development is achieved even if treatment is prolonged up to 30 weeks of age. Moreover, the so-treated mice are fully susceptible to the diabetogenic action of CY.15 This concurs with our observed failure to prevent CY-induced type-1 diabetes with anti-IL-12 mAb. The reasons for the discrepancies of our and Trembleau et al.’s data15 with another study,10 which claims protection against clinical and histological signs of CY-induced diabetes by exogenous IL-12 (p40)2, are not known. Note however, that the diabetogenic response to CY can be highly variable in individual NOD mice and that environmental factors may influence the course of the disease.19
In agreement with the study by Trembleau et al.,15 was also our observation that the clinical protection against spontaneous type-1 diabetes afforded by early prophylaxis with anti-IL-12 mAb was not accompanied by a milder insulitis. Dissociation between clinical efficacy and lack of histological effects has already been reported in NOD mice with anti-CD4 mAb,20 soluble (s) IL-1 receptor (R)21 and anti-IL-1β antibody.22 Trembleau et al.15 report that exogenous IL-12(p40)2 deviates pancreas-infiltrating CD4+, but not CD8+, T cells from a Th1 into a Th2/Th0 phenotype, diminishing the percentages of IFN-γ-positive cells and augmenting those of IL-4 and IL-10. Because anti-IFN-γ mAb or the sIFN-γR18,23–25 and exogenously administered IL-426 and IL-1023 prevent NOD mouse diabetes, the above immunomodulatory effects may be related to the anti-diabetogenic action of early IL-12(p40)2 prophylaxis. In fact, IL-12(p40)2-treated normoglycaemic mice have lower numbers of Th1 pancreas-infiltrating lymphocytes than those NOD mice eventually developing type-1 diabetes in spite of IL-12(p40)2 treatment. In as much as late prophylaxis with either IFN-γ inhibitors18 or IL-426 also prevent NOD mouse type-1 diabetes, the inability of exogenous IL-12 (p40)2 (and anti-IL-12 mAb here) to significantly do so under late treatment regimen may be secondary to the lower capacity of late versus early IL-12(p40)2 prophylaxis to skew pancreas-infiltrating lymphocytes towards Th2/Th0 phenotype. The reason for the reduced immunomodulatory and clinical efficacy of IL-12 antagonists in NOD mice with advanced pre-diabetes is not known One possibility would be that other cytokines that appear late during the insulitis process can compensate for the deficit in IL-12-mediated IFN-γ production. A likely candidate may be IL-18, an IL-1-related cytokine that stimulates IFN-γ production and whose intra-insular expression coincides with and perhaps promotes the onset of Th1 destructive insulitis lesions.27
In conclusion, this clinical and histological study proves that the pathogenetic role of endogenous IL-12 in NOD mouse diabetes is restricted to the very early diabetogenic events presumably occurring prior to insulitis development. IL-12 has, if any, a replaceable role in the advanced efferent phases of β-cell destruction.
Acknowledgments
The Authors thank Professor G. Teti, Institute of Microbiology, University of Messina, Itlay, for providing them with the hybridoma producing the C17.8 antimouse IL-12 mAb. This hybridoma was kindly provided to Prof. Teti by Dr G. Trinchieri, the Wistar Institute, PA.
REFERENCES
- 1.Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev. 1994;15:516. doi: 10.1210/edrv-15-4-516. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM. Therapeutic intervention by immunostimulation? Diabetes. 1994;43:613. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- 3.Mandrup-Poulsen T, Nerup J, Reimers JJ, et al. Cytokines and the endocrine system. II. Roles in substrate metabolism, modulation of thyroidal and pancreatic endocrine cell functions and autoimmune endocrine diseases. Eur J Endocrinol. 1996;134:21. doi: 10.1530/eje.0.1340021. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovitch A. An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabets Metab Rev. 1998;14:129. doi: 10.1002/(sici)1099-0895(199806)14:2<129::aid-dmr208>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 5.Rothe H, Kolb H. The APC1 concept of type 1 diabetes. Autoimmunity. 1998;27:179. doi: 10.3109/08916939809003865. [DOI] [PubMed] [Google Scholar]
- 6.Rabinovitch A, Suarez PW, Sorensen O. Interleukin-12 mRNA expression in islets correlates with beta-cell destruction in NOD mice. J Autoimmun. 1996;9:645. doi: 10.1006/jaut.1996.0084. [DOI] [PubMed] [Google Scholar]
- 7.Rothe H, Burkart V, Faust A, Kolb H. Interleukin-12 gene expression is associated with rapid development of diabetes mellitus in non-obese diabetic mice. Diabetologia. 1996;39:119. doi: 10.1007/BF00400422. [DOI] [PubMed] [Google Scholar]
- 8.Zipris D, Greiner DL, Malkani S, Whalen B, Mordes JP, Rossini AA. Cytokine gene expression in islets and thyroids of BB rats. IFN-gamma and IL-12p40 mRNA increase with age in both diabetic and insulin-treated nondiabetic rats. J Immunol. 1996;156:1315. [PubMed] [Google Scholar]
- 9.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin. 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothe H, O’hara RM, Martin S, Kolb H. Suppression of cyclophosphamide induced diabetes development and pancreatic Th1 reactivity in NOD mice treated with the interleukin (IL) -12 antagonist IL-12 (p40)2. Diabetologia. 1996;39:119. doi: 10.1007/s001250050728. [DOI] [PubMed] [Google Scholar]
- 11.Yasuda H, Nagata M, Arisawa K, et al. Local expression of immunoregulatory IL-12p40 gene prolonged syngeneic islet graft survival in diabetic NOD mice. J Clin Invest. 1998;102:1807. doi: 10.1172/JCI2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szelachowska M, Kretowski A, Kinalska I. Increased in vitro interleukin-12 production from by peripheral blood in high-risk IDDM first degree relatives. Horm Metab Res. 1997;29:168. doi: 10.1055/s-2007-979014. [DOI] [PubMed] [Google Scholar]
- 13.Sternesjo J, Sandler S. Effects of interleukin-12 in vitro on pancreatic islets isolated from normal rodents and from non-obese diabetic mice. J Endocrinol. 1998;158:69. doi: 10.1677/joe.0.1580069. [DOI] [PubMed] [Google Scholar]
- 14.O’hara RJ, Henderson SL, Nagelin A. Prevention of a Th1 disease by a Th1 cytokine: IL-12 and diabetes in NOD mice. Ann N Y Acad Sci. 1996;759:241. doi: 10.1111/j.1749-6632.1996.tb52673.x. [DOI] [PubMed] [Google Scholar]
- 15.Trembleau S, Penna G, Gregori S, Gately MC, Adorini L. Deviation of pancreas-infiltrating cells to Th2 by interleukin-12 antagonist administration inhibits autoimmune diabetes. Eur J Immunol. 1997;27:2330. doi: 10.1002/eji.1830270930. [DOI] [PubMed] [Google Scholar]
- 16.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Constantinescu CS, Wysocka M, Hilliard B, et al. Antibodies against IL-12 prevent superantigen-induced and spontaneous relapses of experimental autoimmune encephalomyelitis. J Immunol. 1998;161:5097. [PubMed] [Google Scholar]
- 18.Nicoletti F, Zaccone P, Di Marco R, et al. The effects of a nonimmunogenic form of murine soluble interferon-gamma receptor on the development of autoimmune diabetes in NOD mouse. Endocrinology. 1996;137:5567. doi: 10.1210/endo.137.12.8940385. [DOI] [PubMed] [Google Scholar]
- 19.Parish NM, Hutchings PR, O’reilly L, et al. Tolerance induction as a therapeutic strategy for the control of autoimmune endocrine disease in mouse models. Immunol Rev. 1995;144:269. doi: 10.1111/j.1600-065x.1995.tb00073.x. [DOI] [PubMed] [Google Scholar]
- 20.Kurasawa K, Sakamoto A, Maeda T, et al. Short-term administration of anti-L3T4 MoAb prevents diabetes in NOD mice. Clin Exp Immunol. 1993;91:376. doi: 10.1111/j.1365-2249.1993.tb05912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicoletti F, Di Marco R, Barcellini W, et al. Protection from experimental autoimmune diabetes in the nonobese diabetic mouse by soluble interleukin-1 receptor. Eur J Immunol. 1994;24:1843. doi: 10.1002/eji.1830240818. [DOI] [PubMed] [Google Scholar]
- 22.Cailleau C, Diu-Hercend A, Ruuth E, Westwood R, Carnaud C. Treatment with neutralizing antibodies specific for IL-1 beta prevents cyclophosphamide-induced diabetes in nonobese diabetic mice. Diabetes. 1997;46:937. doi: 10.2337/diab.46.6.937. [DOI] [PubMed] [Google Scholar]
- 23.Pennline KJ, Roque-Gaffney E, Monaham R. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin Immunol Immunopathol. 1994;71:169. doi: 10.1006/clin.1994.1068. [DOI] [PubMed] [Google Scholar]
- 24.Campbell IL, Kay TW H, Oxbrow, Harrison LC. Essential role for interferon-γ and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Whei mice. J Clin Invest. 1991;87:739. doi: 10.1172/JCI115055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debray-Sachs M, Carnaud C, Boitard C, et al. Prevention of diabetes in NOD mice treated with antibody to murine IFN-Γ. J Autoimmun. 1991;4:237. doi: 10.1016/0896-8411(91)90021-4. [DOI] [PubMed] [Google Scholar]
- 26.Cameron MJ, Arreaza GA, Zucker P, et al. IL-4 prevents insulitis and insulin-dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper-2 cell function. J Immunol. 1997;159:4686. [PubMed] [Google Scholar]
- 27.Rothe H, Jenkins NA, Copeand NG, Kolb H. Active stage of autoimmune diabetes is associated with the expression of a novel cytokine, IGIF which is located near Idd2. J Clin Invest. 1997;99:469. doi: 10.1172/JCI119181. [DOI] [PMC free article] [PubMed] [Google Scholar]