

Abstract
Dendritic cells (DCs) are the most powerful of all antigen-presenting cells and play a critical role in the induction of primary immune responses. DC-based vaccination represents a potentially powerful strategy for cancer immunotherapy. In this study, a new approach for a DC-based melanoma vaccine was described. Splenic DCs from C57BL/6 mice were fused with B16 melanoma cells, and the resultant B16/DC hybrid cells expressed major histocompatibility complex (MHC) molecules – B7 as well as the B16 tumour marker M562 – which were enriched by Ia-mediated positive selection with a MiniMACS column. The fusion rates were 12·7–26·8%. To generate hybrid tumour vaccines with potentially greater potent therapeutic efficacy, we genetically engineered DCs with granulocyte–macrophage colony-stimulating factor (GM-CSF) prior to cell fusion. Recombinant adenovirus vector was used to mediate gene transfer into DCs with high efficiency and DCs expressed GM-CSF at 96–138 ng/105cells/ml 24 hr after GM-CSF gene transfer. GM-CSF gene-modified DCs (DC.GM) exhibited higher expression of B7 and co-stimulatory capacity in mixed lymphocyte reaction (MLR). Fusion of DC.GM with B16 cells generated B16/DC.GM hybrid cells secreting GM-CSF at 59–63 ng/105 cells/ml. Immunization of C57BL/6 mice with the B16/DC hybrid vaccine elicited a specific cytotoxic T-lymphocyte (CTL) response and protected the immunized mice from B16 tumour challenge, reduced pulmonary metastases and extended the survival of B16 tumour-bearing mice. The B16/DC.GM hybrid vaccine was able to induce a CTL response and protective immunity more potently and tended to be therapeutically more efficacious than the B16/DC vaccine. In vivo depletion of T-cell subsets demonstrated that both CD8+ and CD4+ T cells were essential for the therapeutic effects of B16/DC and B16/DC.GM hybrid vaccines. Additionally, other non-specific effector cells may also contribute to tumour rejection induced by the B16/DC.GM hybrid vaccine. These data indicate that a DC-based hybrid tumour vaccine may be an attractive strategy for cancer immunotherapy, and that GM-CSF gene-modified DCs may lead to the generation of hybrid vaccines with potentially increased therapeutic efficacy.
INTRODUCTION
Malignant transformed cells express tumour antigens that may act as targets for immune rejection mediated by the host immune system.1–3 In the last few decades, great efforts have been directed towards cancer immunotherapy with tumour vaccines. Tumour cells are usually poor antigen-presenting cells (APC), which express low levels of major histocompatibility complex (MHC) antigens and co-stimulatory molecules, and often have some alteration in antigen processing or transport, which results in an inability to present tumour antigens to host T cells.4–6 The gene transfer technique brings new strategies and promise to tumour vaccine-mediated cancer immunotherapy. Some immunoregulatory molecules, including MHC antigens, the co-stimulatory molecule (B7), and cytokines such as granulocyte–macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-2, IL-4, or interferon-γ (IFN-γ), have been used to genetically engineer tumour cells to improve their immunogenicity and potential antigen-presenting capability and to intensify their induced antitumour immunity.4,7–9 The precise therapeutic effects of gene-modified tumour vaccines are under clinical evaluation. However, without knowing the specific signalling defect of tumour cells derived from individual patients, a single-gene modified tumour vaccine is unlikely to be effective.
Dendritic cells (DCs) are regarded as ideal natural adjuvants and represent a potentially powerful strategy for cancer immunotherapy,10,11 the potency of which can be further augmented by genetic modification with cytokines (GM-CSF, IL-12 or IFN-γ) and T-cell attracting chemokines.12–14 To develop DC-based tumour vaccines, DCs could be charged with tumour antigenic information in the form of antigen proteins, peptides, tumour antigen-encoding cDNA or tumour-cell derived mRNA.15–21 Fusion of two different kinds of cells can generate hybrid cells that presumably have phenotypic characteristics of both of the progenitor cells. This technique has been successfully applied in the area of monoclonal antibody (mAb) production, and was recently demonstrated to have potential in the generation of novel tumour vaccines.22–24 Hence, fusion of DCs with tumour cells may be an alternative approach for developing DC-based tumour vaccines. Malignant melanoma is poorly immunogenic and putatively an ideal model for cancer immunotherapy. In this study, we tested the feasibility of generating DC-based tumour vaccines by fusion of DCs with mouse melanoma cells, subsequently evaluated their therapeutic efficacy and attempted to further improve their therapeutic efficacy by genetically engineering DCs prior to cell fusion.
The exceptional capability of DCs to induce T-cell mediated immune responses is attributed to their high expression of antigen-presenting molecules (MHC class I and II antigens) as well as accessory molecules that mediate T-cell binding and co-stimulation.25 The maturation and functional status of DCs are highly regulated by cytokines such as GM-CSF, tumour necrosis factor-α (TNF-α), IL-1, transforming growth factor-β (TGF-β), IL-10, etc., and membrane-anchored ligands (such as CD40L), which consequently affect the vaccination efficacy of DC-based vaccines.26,27 GM-CSF is one of the most crucial cytokines involved in DC maturation and functional regulation, being capable of promoting their proliferation and differentiation, maintaining cell viability, regulating their in vivo migration and distribution, and enhancing their co-stimulation and antigen presentation.28–32 GM-CSF gene-modified tumour vaccines have been demonstrated to be capable of inducing antitumour immunity, presumably by augmenting the antigen presentation of APC, e.g. DCs.8 In our previous study, GM-CSF gene-modified DCs pulsed with B16 tumour-cell extracts were able to induce a potent specific tumour rejection against B16 tumour cells.12 In this study, the therapeutic effects and related immunological mechanisms of DC-based hybrid vaccines, generated by fusion of tumour cells with GM-CSF gene-modified DCs, were also evaluated.
MATERIALS AND METHODS
Animals
C57BL/6 (denoted B6) and BALB/c mice (female; 5–6 weeks old) were purchased from Shanghai Experimental Animal Centre and the Charles River Laboratories (Wilmington, MA), and were used at 6–8 weeks of age.
Tumour cell lines
B16.F10, a melanoma cell line of B6 (H-2b) mouse origin, was a gift from Dr S. A. Rosenberg’s laboratory (National Cancer Institute, Bethesda, MD). B16 tumour cells express the specific membrane protein of murine leukaemia virus (MuLV) protein AKV-Env gp85 on their cell surfaces, which can be detected by mAb M562.33 GM-CSF gene-transfected B16 cells (B16.GM) were generated in our laboratory by transfecting B16.F10 tumour cells with recombinant adenovirus harbouring the mouse GM-CSF gene, which secreted GM-CSF at 84 ng/105 cells/24 hr. EL-4 is a dimethylbenzanthrene-induced lymphoma of B6 origin. The B16, B16.GM and EL-4 tumour-cell lines were maintained in culture in complete medium (CM) consisting of RPMI-1640 (Gibco BRL, Gaithersburg, MD) supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mm glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin. The transformed human embryonic kidney line 293 was from the American Type Culture Collection (Rockville, MD), and was grown in Dulbecco’s modified Eagle’s minimal essential medium (DMEM; Gibco BRL) supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin and 100 mg/ml streptomycin.
Recombinant adenoviruses
The recombinant Ad5 adenoviruses harbouring mouse GM-CSF or the LacZ gene were constructed as previously described.34 The recombinant viruses were propagated with 293 cells, the titres of which were determined by plaque assay on 293 cells. The adenovirus solution was stored at −80°. The recombinant adenoviruses of LacZ or mouse GM-CSF, with titres of 3·6×109 plaque-forming units (PFU)/ml and 5×109 PFU/ml, respectively, were used to mediate gene transfer in this study.
Preparation and gene modification of spleen dendritic cells
DCs were prepared and enriched from splenocytes of normal B6 mice. In brief, a single cell suspension of splenocytes from B6 mice was depleted of red blood cells (RBC) using NH4Cl (ACK) lysis buffer, washed twice, and resuspended at 1×108 cells/ml in RPMI-1640 for gradient centrifugation. Each 4 ml of cell suspension was overlaid onto 4-ml 14·5% metrizamide gradients (Sigma Chemical Co., St. Louis, MO; 14·5 g in 100 ml of phosphate-buffered saline [PBS], pH 7·0). After centrifugation for 15 min at 600 g, the DC-enriched fraction at the interface was collected, washed twice, resuspended at 5×106 cells/ml in CM, and placed in six-well plates at 2×107 cells/well. After 2 hr of incubation at 37°, the plates were washed twice with prewarmed Hanks’ balanced salt solution (HBSS) to remove non-adherent cell populations. The remaining adherent cells in the plates were incubated overnight (16–18 hr) in CM. Non-adherent cells from the second incubation were harvested and used as purified DCs. Phenotype staining with N418 mAb showed that the enriched cell population contained ≈80% DCs. For gene modification of DCs, 500 μl of recombinant adenovirus containing GM-CSF or LacZ was added (at a multiplicity of infection [MOI] of 100) into each well of six-well plates containing enriched DCs. After 1 hr of infection, the adenovirus solution was removed and replaced with 4 ml/well of fresh CM. Twenty-four hr after subsequent incubation at 37°, the culture supernatants from DCs were collected for detection of GM-CSF with capture enzyme-linked immunosorbent assay (ELISA; Endogen Inc., Cambridge, MD). The non-adherent cells from overnight cultures were collected and used as GM-CSF gene-modified DCs (DC.GM) or LacZ gene-modified DCs (DC.LacZ). The transfer efficiencies of DCs with adenovirus–LacZ were confirmed to be higher than 90% at a MOI of 100.
Cell fusion and selection
Purified DCs from B6 mice were labelled with rat antimouse Ia mAb (M5/114, Boehringer Mannheim, Mannheim, Germany) in 0·01 m PBS (pH 7·4) at 4° for 30 min, washed twice with PBS and subsequently labelled with minimagnetic bead-conjugated goat antirat IgG (Militenyi Biotec GmbH, Bergisch Galdbach, Germany) in PBS at 10° for 15 min. The labelled DCs were fused with B16 cells at a ratio of 10:1 (DC:tumour) using polyethylene glycol/dimethylsulphoxide solution (50% PEG/10% DMSO in PBS; Sigma) by stirring. The cells were centrifuged at 400 g for 5 min. The magnetically positive cells were enriched by using a MiniMACS separation column (Militenyi Biotec GmbH). The enriched Ia+ cells were washed, resuspended in CM and seeded in 12-well plates at 106 cells/well. The cells were cultured overnight and the non-adherent cells of unfused DCs were removed by pipette. The remaining adherent cells were regarded as fused cells. B16 fused with DCs, or GM-CSF gene-modified DCs, were termed as B16/DC and B16/DC.GM, respectively.
Phenotype staining
DCs, DC.GM, B16 and B16/DC.GM were analysed by staining with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated mAbs (Pharmingen, San Diego, CA) against MHC antigens (H-2 Db, Kb, IAb), adhesion and co-stimulatory molecules (B7.1, B7.2, intracellular adhesion molecule-1 [ICAM-1]) and other antigens (Thy-1.2, SmIg), at 4° for 45 min. Dendritic cells were identified by labelling with N418 35 for 45 min at 4°, washed twice, then stained with secondary FITC-conjugated mouse antihamster immunoglobulin G (IgG) antibody for 30 min at 4°. B16 and B16/DC.GM fused cells were stained with mAb against the AKV-Env gp85 protein (M562, kindly provided by Dr Masaru Taniguchi, Ciba University, Tokyo, Japan) as a B16-specific tumour marker. All cells were washed twice with HBSS and fixed with 0·2% paraformaldehyde. Fluorescence intensity and positive cell percentage were measured on a fluorescence-activated cell sorter (FACStar Plus; Becton-Dickinson, San Jose, CA).
Mixed leucocyte reaction (MLR)
Splenocytes from normal BALB/c mice were depleted of RBC using ACK lysis buffer, incubated for 60 min on nylon wool at 37°, and subsequently depleted of Ia+ cells by labelling with antimouse Ia mAb (M5/114) and then purified by using a MiniMACS separation column. Graded doses (103–105/well) of irradiated (3000 rad) DCs (with or without overnight incubation with 20 ng/ml recombinant GM-CSF [rGM-CSF]), DC.LacZ, or DC.GM were placed in 96-well round-bottom culture plates as stimulators and co-incubated with purified allogeneic BALB/c T cells as responders (2×105/well). Diluted rabbit anti-mGM-CSF mAb (1:500, Gibco BRL) was added to culture wells containing DC.GM to block the endogenous expression of GM-CSF. The plates were incubated in a humidified atmosphere, under 5% CO2, at 37° for 96 hr, and then pulsed for 16 hr with 1 μCi [3H]thymidine/well before harvesting. All determinations were carried out in triplicate, and thymidine incorporation was determined with a β-counter (Wallac 1409; Wallac Oy, Turku, Finland). The basal proliferation levels of the irradiated stimulators were less than 350 counts per minute (c.p.m.) in all experiments.
Cytotoxicity assay
Groups of B6 mice were immunized twice, at weekly intervals, by intraperitoneal (i.p.) injection of 5×106 irradiated (5000 rad) B16, B16.GM, DC.GM, B16/DC.GM, or B16/DC cells. Four mice from each vaccination group were killed on day 7 after the second immunization and their spleens were harvested. Splenic lymphocytes from each group were pooled and stimulated in vitro (4×106/well) with irradiated (15 000 rad) 50 U/ml IFN-γ-pretreated B16 tumour cells (2×105/well) for 5 days in 24-well culture plates containing CM. After 5 days of mixed tumour–lymphocyte culture, lymphocytes were harvested and depleted of dead cells by Ficoll gradient centrifugation. The viable lymphocytes were used as effector cells for testing cytotoxicity against B16. Target cells (B16, EL-4) were incubated at 37° with 200 μCi 51Cr (Amersham, Little Chalfont, Bucks, UK) in 1 ml of RPMI-1640, 20% FCS, for 1 hr. The labelled targets were washed three times with HBSS and resuspended in RPMI-1640, 20% FCS, at 105 cells/ml. The 51Cr-labelled target cells (104 cells in 100 μl) were placed in each well of 96-well plates and 100 μl/well of each dilution of immune T cells, as effectors, was added. To confirm CTL-mediated cytotoxity, anti-CD8 mAb (2.43, Lyt2.2) was added to some culture wells. Plates were incubated at 37° for 4 hr. The supernatant from each well was harvested and the amount of 51Cr released was counted in a gamma counter. The spontaneous release value was obtained from target cells cultured with CM alone, and the maximum release value was obtained from target cells cultured with 0·1% Nonidet P-40 (NP-40). The percentage of specific lysis was calculated. All determinations of cytotoxicity were carried out in triplicate.
Tumour challenge
Groups of B6 mice were immunized twice, at weekly intervals, by i.p. injection with 5×106 irradiated (5000 rad) B16, B16.GM, DC.GM, B16/DC.GM, or B16/DC cells. One week following the final immunization, the mice were challenged subcutaneously (s.c.) with 106 viable B16 tumour cells. Tumour growth and the survival time of each group of mice were recorded on a regular basis. The size of the tumour on each mouse was measured, at regular intervals after tumour challenge, in two perpendicular dimensions with a vernier caliper. Tumour incidence was considered positive when the average diameter of the tumour exceeded 3 mm.
Therapy of pulmonary metastases with fusion vaccines
B6 mice were administered 2×105 B16 tumour cells by tail vein injection on day 0. Three days after tumour inoculation, the tumour-bearing mice were randomly divided into several groups to receive treatment, by s.c. injection, with 2×106 irradiated B16, B16.GM, DC.GM, B16/DC.GM, or B16/DC cells. For evaluation of lung metastases, the tumour-bearing mice received two vaccinations on days 3 and 10 after tumour inoculation. On day 15, mice were killed and the number of metastatic nodules on the pulmonary surface of each mouse were counted. For observation of the survival time, tumour-bearing mice were treated with three vaccinations on days 3, 10 and 17. The survival time of each group of mice was recorded.
In vivo depletion of T-cell subsets
Tumour-bearing mice were injected i.p. with rat IgG2b mAb (1 mg/mouse/injection) against murine CD4 (GK1.5, L3T4) and/or CD8 (2.43, Lyt2.2) 2 days before each of the three vaccinations. Normal rat IgG (Sigma) was given as a negative control. This procedure has previously been shown to be effective in depleting specific T cells in splenocytes and produces long-term T-cell subset depletion.
Statistical analysis
The significant differences in the numbers of pulmonary metastases and the survival time between groups were determined by the Wilcoxon rank-sum test. Two-sided P-values below 0·05 were considered statistically significantly different.
RESULTS
Genetic modification of DCs with GM-CSF enhances their cell viability, B7 expression and co-stimulatory capacity
Splenic DCs isolated by metrizamide gradients exhibited typical morphologies of DCs, expressed high levels of MHC-I, MHC-II, B7 and ICAM-1 (Fig. 1a), and were capable of stimulating the proliferation of T cells in an allogenic MLR (data not shown). N418 antibody staining showed that the isolated splenic DCs were greater than 80% pure. The gene transfer efficiency of splenic DCs was evaluated using the LacZ reporter gene, and X-gal staining showed that greater than 90% of DCs could be efficiently transfected by the LacZ recombinant adenovirus at a MOI of 100. Twenty-four hr after infection of DCs with GM-CSF recombinant adenovirus (at a MOI of 100), GM-CSF could be detected in the culture supernatants at 96–138 ng/105cells/ml, whereas there was no detectable GM-CSF (<5 pg/ml) in the counterparts of untransfected DCs or LacZ gene-transfected DCs (DC.LacZ). Compared with untransfected DCs, DC.LacZ had the same expression patterns of MHC-I, MHC-II, B7 and ICAM-1, and the same stimulatory capability in MLR (data not shown). The indicated adenovirus-mediated gene transfer did not itself alter the phenotypic characteristics of splenic DCs.
Figure 1.

Phenotypic analysis of B16, granulocyte–macrophage colony-stimulating factor gene-modified dendritic cells (DC.GM) and their fusion hybrid. (a) B16, DCs, DC.GM and purified B16/DC.GM fused cells were analysed by flow cytometry for expression of major histocompatibility complex (MHC) class I (Db, Kb) and class II (IAb) antigens, co-stimulatory molecule (B7.1), DC marker CD11c (N418) and B16 tumour marker (M562), as detailed in the Materials and methods. (b) DC.GM were fused with B16 at a fusion ratio of 10:1 by polyethylene glycol (PEG). A bulk B16/DC.GM fusion preparation was cultured overnight and the non-adherent cells (unfused DCs and dead cells) were removed. The retained adherent cells were harvested and double stained with M562 monoclonal antibody (mAb), or with M562 plus mAbs against IAb, or Db or B7.1. Numbers in the quadrants of dual-parameter histograms represent the percentage of cells contained within that quadrant. FITC, fluorescein isothiocyanate; PE, phycoerythrin.
After transfection with the GM-CSF gene, DCs expressed comparable levels of MHC class I and II and ICAM-1, but their expression of B7 was highly augmented (Fig. 1a). To confirm that the augmented B7 expression of DC.GM was attributable to its GM-CSF expression after gene transfer, we added recombinant GM-CSF (20 ng/ml) during DC overnight culture, which also led to enhanced expression of B7 in the untransfected DCs (data not shown). Furthermore, the DC.GM co-stimulatory capacity of DC.GM was studied by MLR, where rabbit antimouse GM-CSF mAb was added to block the endogenous GM-CSF expressed by DC.GM. It was found that DC.GM were more potent stimulators of T-cell proliferation than untransfected DCs or DC.LacZ, but comparable to DCs pretreated with recombinant GM-CSF (data not shown). Moreover, DC.GM maintained higher cell viability in short-term culture than their counterparts, but without an obvious increase in cell number. These results suggest that modification of DCs with GM-CSF gene could promote the maturation of DCs and their T-cell stimulatory capacity. We hypothesized that the GM-CSF production of B16/DC hybrid cells could facilitate the in vivo induction of immune responses, so DC.GM, rather than GM-CSF pretreated DCs, were used to fuse with tumour cells to generate a DC-based hybrid vaccine.
Fusion of DCs with B16 tumour cells produces hybrid cells that acquire the phenotypes of both DCs and tumour cells
Consistent with their poor immunogenicity, B16 tumour cells expressed very low amounts of MHC class I, and were negative for MHC class II and the B7 co-stimulatory molecule (Fig. 1a). Their cell surface protein MuLV AKV-Env gp85 was detectable with the M562 mAb, which served as a B16 tumour marker (Fig. 1a). B16 cells were fused with DCs, DC.LacZ or DC.GM, by PEG, and the resultant hybrid cells were termed B16/DC, B16/DC.LacZ or B16/DC.GM, respectively. According to our preliminary experiments, the optimal fusion rate was achieved at a ratio of 10:1 (DC:tumour cells). In our eight independent experiments, the average fusion rate was 18·9% (ranging from 12·7 to 26·8%). The fusion hybrids of B16 and DCs were detected by double staining with the M562 mAb and mAbs against IAb, Db, CD11c or B7.1, which were demonstrated by fusion of B16 cells with DC.GM (Fig. 1b). The enriched hybrid cells, B16/DC.GM, were positive for the B16 tumour marker, M562, as well as for Kb, IAb, B7.1 and CD11c (Fig. 1a). X-gal staining demonstrated that B16/DC.LacZ fused cells were positive for β-galactosidase. The B16/DC.GM fused cells were shown to secrete GM-CSF at 59–63 ng/105 cells/ml in the 24-hr culture supernatants. These observations indicate that the fused cells acquired the phenotypes of both B16 and dendritic cells. Injection of hybrid cells into B6 mice showed that the in vivo tumorigenicity of B16/DC, B16/DC.LacZ and B16/DC.GM hybrid cells decreased markedly (data not shown).
Vaccination of B6 mice with hybrid cells elicits tumour-reactive CTL and resistance to B16 challenge
All of the mice vaccinated with irradiated B16, B16.GM or DC.GM cells were tumour positive (tumour diameter >3 mm) within 2 weeks of B16 challenge. In contrast, B16/DC, B16/DC.LacZ and B16/DC.GM hybrid vaccines rendered the immunized mice resistant to B16 tumour challenge, exhibiting delayed tumour development and inhibited tumour growth (Fig. 2). The B16/DC and B16/DC.LacZ hybrid vaccines protected the immunized mice almost to the same extent (P < 0·01, compared with the B16 control vaccine), with nearly 50% of immunized mice completely resistant to B16 tumour challenge. This suggests that adenovirus-mediated gene transfer into DCs, prior to cell fusion, does not affect the antitumour immunity induced by DC-based hybrid vaccines, despite the high immunogenicity of adenoviruses. The B16/DC.GM vaccine was able to protect the immunized mice more potently (P < 0·05, compared with the B16/DC vaccine), with 90% of mice free of tumour 25 days after B16 tumour challenge (Fig. 2), and 70–80% of mice completely resistant to long-term B16 tumour challenge (data not shown). Neither B16/DC, B16/DC.LacZ or B16/DC.GM hybrid vaccines induced protection from subsequent EL4 tumour challenge (data not shown).
Figure 2.

Immunization of B6 mice with hybrid tumour vaccine protects the mice from B16 tumour challenge. Normal B6 mice were immunized twice, intraperitoneally (i.p.), at a 1-week interval, with 5×106 irradiated B16, B16.GM, DC.GM, B16/DC.GM or B16/DC. One week after the last immunization, the mice were challenged by subcutaneous (s.c.) injection with 1×106 B16 cells. The tumour size was monitored at regular intervals and is represented by the mean of the maximal perpendicular diameters. The tumour sizes presented were measured 25 days after tumour challenge. Dots represent individual tumour diameters (10 mice/group). Data are representative of three experiments performed. Mice were killed when the challenge tumours reached 2·5 cm in diameter or if severe ulceration developed.
CTL results (Fig. 3) showed that vaccination with B16, B16.GM or DC.GM cells induced a poor CTL response against B16 cells. By contrast, vaccination with B16/DC or B16/DC.LacZ hybrid cells induced a marked and comparable CTL cytotoxicity specifically against B16 tumour cells but not against syngeneic EL-4 tumour cells. The B16/DC.GM hybrid vaccine was able to induce higher CTL activity, specifically against B16 cells, than either the B16/DC or B16/DC.LacZ vaccines. The CTL cytotoxicity could be neutralized by anti-CD8 mAb (data not shown), which was indicative of CD8 T-cell mediated cytotoxicity.
Figure 3.

Vaccination of B6 mice with B16/DC.GM fusion vaccine elicits a tumour-specific cytotoxic T-lymphocyte (CTL) response against B16. Groups of naive B6 mice were immunized twice, intraperitoneally (i.p.) with 5×106 irradiated B16, B16.GM, DC.GM, B16/DC.GM or B16/DC cells. One week after the last vaccination, four mice from each vaccination group were killed. The splenocytes from each group of mice were harvested, pooled, restimulated with irradiated B16 tumour cells for 5 days in vitro and subsequently measured for cytotoxicity against B16 (a) or EL-4 (b) cells as targets in a standard 4-hr 51Cr-release assay.
Therapy with the B16/DC.GM vaccine can effectively reduce established pulmonary metastases
As the B16/DC.LacZ and B16/DC hybrid vaccines induced protective antitumour immunity against B16 tumours almost to the same extent, we eliminated the B16/DC.LacZ control vaccine in a subsequent therapeutic model. The results showed that vaccination with irradiated B16, B16.GM or DC.GM had no therapeutic effect on established tumours. All tumour-bearing mice in these treatment groups had developed more than 180 lung metastases by day 15 (Fig. 4) and died within 35 days after tumour inoculation (Fig. 5). By contrast, treatment with B16/DC or B16/DC.GM vaccines reduced pulmonary metastases and significantly extended the survival time of tumour-bearing mice (P < 0·001, compared with B16-, B16.GM- or DC.GM-treated groups). In the B16/DC vaccine-treated group, the lung metastatic nodules averaged 31·8±34·5 (Fig. 4), and the mean survival time was 54·4±8·0 days, with 50% of mice free of lung metastatic nodules on day 15 and 37·5% surviving long term (Fig. 5). In the B16/DC.GM-treated group, the lung metastatic nodules averaged 14·6±22·7 (Fig. 4), and the mean survival time was greatly extended to 61·0±5·6 days, with 70% of mice free of lung metastatic nodules on day 15 and 62·5% surviving long term (Fig. 5). These results demonstrated that treatment with either B16/DC.GM or B16/DC hybrid vaccines could induce effective tumour rejection in tumour-bearing mice. Although there were no significant differences in lung metastases and animal survival between B16/DC and B16/DC.GM vaccines (P > 0·05), the B16/DC.GM hybrid vaccine tended to be more efficacious.
Figure 4.

Therapy with B16/DC.GM fusion vaccine greatly reduces the established pulmonary metastasis of tumour-bearing mice. B6 mice were given an intravenous (i.v.) injection of 2×105 B16 tumour cells to establish pulmonary metastases. Three days after tumour inoculation, mice were treated by two vaccinations, at a 1-week interval, with 2×106 irradiated B16, B16.GM, DC.GM, B16/DC.GM or B16/DC cells. On day 15 after tumour inoculation, mice were killed and pulmonary metastases nodules of each mouse were counted. The data presented were pooled from two separate experiments.
Figure 5.

Therapy with the B16/DC.GM fusion vaccine prolongs the survival time of tumour-bearing mice. B6 mice were given an intravenous (i.v.) injection of 2×105 B16 tumour cells to establish tumours. Three days after tumour inoculation, mice were divided into five groups and were treated by three vaccinations, at weekly intervals, with 2×106 irradiated B16, B16.GM, DC.GM, B16/DC.GM or B16/DC cells. The survival times of each group of mice were monitored. The data presented were pooled from two separate experiments.
Involvement of T cells in the tumour rejection induced by vaccination with B16/DC.GM
Mice were depleted of CD4+ and/or CD8+ T cells, before each immunization, by mAb against murine CD4 and/or CD8. The results demonstrated that depletion of either CD4+ or CD8+ T cells could significantly diminish the therapeutic effects of B16/DC.GM and B16/DC (Table 1), suggesting that both CD4+ and CD8+ T lymphocytes are essential for the therapeutic antitumour immunity induced by B16/DC and B16/DC.GM hybrid vaccines. In vivo depletion of both CD4+ and CD8+ T cells completely blocked the therapeutic effects induced by vaccination with B16/DC. However, depletion of both CD4+ and CD8+ T cells only partially blocked the therapeutic effects of the B16/DC.GM vaccine, suggesting that other non-specific effector cells (i.e. macrophages, granulocytes or natural killer cells), besides CD4+ and CD8+ T cells, were involved in B16/DC.GM vaccine-induced therapeutic antitumour responses.
Table 1.
In vivo depletion of T-lymphocyte subsets inhibits the therapeutic effects induced by B16/DC and B16/DC.GM fusion vaccines
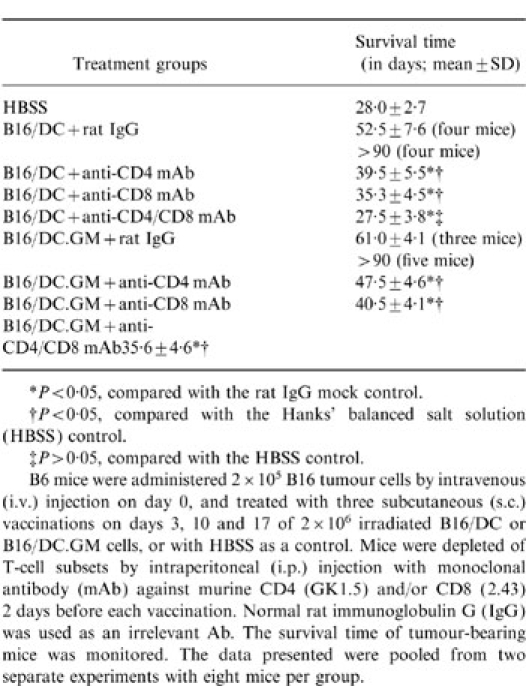
*P < 0·05, compared with the rat IgG mock control.
†P < 0·05, compared with the Harnks' balanced salt solution (HBSS) control.
‡P > 0·05, compared with the HBSS control.
B6 mice were administered 2 × 105 B16 tumour cells by intravenous (i.v) injection on day 0, and treated with three subcutaneous (s.c.) vaccinations on days 3, 10 and 17 of 2 × 106 irradiated B16/DC or B16/DC.GM cells, or with HBSS as a control. Mice were depleted of T-cell subsets by intraperitoneal (i.p.) injection with monoclonal antibody (mAb) against murine CD4 (GK 1.5) and/or CD8 (2.43) 2 days before each vaccination. Normal rat immunoglobulin G (IgG) was used as an irrelevant Ab. The survival time of turnour-bearing mice was monitored. The data presented were pooled from two separate experiments with eight mice per group.
DISCUSSION
Several approaches have been used to develop DC-based vaccines. With identified tumour antigens, DCs can be pulsed with tumour antigens in the form of proteins or peptides,15,16,18 or even transfected with tumour antigen-encoding cDNA.21,22,36 However, these approaches are greatly limited in the clinical situation as most tumour antigens are currently unidentified. In this situation, it is more appropriate to pulse DCs with unfractionated tumour cell 19,37 or tumour cell-derived RNA or mRNA,17,38 which abrogates the need to identify tumour antigens. Fusion of tumour cells with DCs may be a potentially attractive strategy for developing DC-based tumour vaccines. The resultant hybrid cells have been hypothesized to be able to acquire both tumour antigen expression and immunostimulatory capability. Compared with tumour cell extract-pulsed DCs, tumour/DC hybrid cells facilitate longer term antigen processing and presentation, and CTL antigenic epitopes could be produced endogenously and presented to the cell surface via the MHC-I pathway, which may facilitate the induction of antitumour immunity. In the recent report of Gong et al., immunogenic MC38 murine adenocarcinoma cells were fused with syngeneic DCs to generate a tumour vaccine with the ability to elicit tumour rejection.23 We tested this approach to DC-based tumour vaccine in the tumour model of malignant melanoma, which is putatively an ideal model for cancer immunotherapy.
In this study we used mature splenic DCs for fusion, which requires the use of a large number of mice for preparation of DCs, and a higher DC:tumour cell ratio (10:1) to achieve a 12·7–26·8% fusion rate. Recent work has shown that the limited number of naturally resident DCs can be overcome by treating mice with Flt3L, a haematopoietic growth factor.39 In mice treated s.c. with Flt3L, an increase in the number of DCs, of more than 17-fold (from 0·5 to 1·0×106 cells/mouse to 16×106 cells/mouse) was observed in spleen. Generation of DCs from bone marrow (BM) cultures supplemented with GM-CSF represents another effective way to acquire large numbers of DCs.40 Our fusion experiments using BM-derived DCs showed that large number of MHC-I+, MHC-II+, B7.1+, and CD11c+ DCs (25–30×106 cells/mouse), with greater than 95% purity, could be obtained by culturing BM cells in CM containing GM-CSF plus IL-4 for 7 days. The BM-derived DCs are relatively large cells and easier to fuse with tumour cells. After fusion, the B16/DC hybrid cells acquired the phenotypes of both DCs and B16 tumour cells, expressing MHC and B7 molecules as well as the B16 tumour marker, M562.
The method was established to enrich B16/DC hybrid cells from unfused tumour cells, DCs, or the hybrid tumour cells generated by fusion between tumour cells. As most human malignant tumour cells are poorly immunogenic and express low levels of MHC molecules, while DCs express high levels of MHC molecules, we chose the MHC-II molecule as a marker to enrich tumour/DC hybrid cells by MiniMACS positive selection, and DCs could be easily removed after subsequent overnight culture. In the report of Gong et al., DC/MC38 fused cells were selected in hypoxanthine aminopterm thymidine (HAT) medium, as the parental MC38/MUC1 and MC38 tumour cells were sensitive to HAT selection medium, whereas the fused cells were not.23 This method of selection may not be applicable to most tumour cell lines. Our experience indicated that MHC-II-mediated positive selection by the MiniMACS column is a simple and reproducible method for obtaining purified B16/DC hybrid cells.
In our previous study, GM-CSF gene-modified DCs pulsed with B16 tumour-cell extracts could induce potent and specific tumour rejection against B16 cells.12 To further improve the therapeutic effects of the B16/DC hybrid tumour vaccine, we genetically engineered DCs with GM-CSF prior to cell fusion. The genetic modification of DCs has been well evaluated in recent years. Adenovirus vectors have been demonstrated to be capable of mediating gene transfer into DCs with high efficiency.41,42 In our study, the β-galactosidase gene was used as a marker to evaluate gene transfer efficiency, and X-gal staining indicated that more than 95% of DCs could be efficiently transfected by replication-deficient recombinant adenoviruses. DC.LacZ cells have the same MHC and B7 expression patterns as untransfected DCs, indicating that adenovirus infection itself does not alter the physiological and phenotypic characteristics of DCs. After transfection with GM-CSF gene, DCs expressed a higher level of B7 co-stimulatory molecules and acquired more potent stimulatory capacity in MLR than the mock control. The gene-modified DCs were fused with B16 tumour cells, and it was found that both B16/DC.LacZ and B16/DC.GM hybrid cells retained the expression of exogenous genes introduced into DCs by their adenovirus vectors. The biological and immunological features of B16/DC.GM hybrid cells are currently under further investigation.
In vivo data demonstrated that the B16/DC hybrid vaccine, rather than the B16 vaccine, could induce a CTL response, and protective and therapeutic antitumour immunity, specifically against B16 tumours. Vaccination with inactivated B16/DC hybrid cells could protect the immunized mice against B16 tumour challenge at lethal dosage, and significantly reduce the pre-established pulmonary metastases and extend the survival time of tumour-bearing mice. The B16/DC.LacZ hybrid vaccine induced protective immunity as potently as the B16/DC vaccine, indicating that adenovirus-mediated transfection did not alter the induction of antigen-specific immune responses, despite their high immunogenicity. The B16/DC.GM hybrid vaccine could induce a more potent CTL response and tended to be more therapeutically effective than the B16/DC vaccine, although there were no statistically significant differences between them relative to the survival time and reduced pulmonary metastases of the treated mice (P > 0·05). In vivo depletion demonstrated that both CD4+ and CD8+ T cells were necessary for B16/DC vaccine-induced therapeutic antitumour immunity, but depletion of T cells only partially neutralized the therapeutic effects of the B16/DC.GM vaccine, suggesting that additional effector cells might be involved in B16/DC.GM vaccine-induced tumour rejection. As a cytokine with multiple biological activities, GM-CSF is capable of promoting cytokine production by macrophages and granulocytes, augmenting the phagocytic and cytocidal activity of macrophages and promoting antibody-dependent cellular cytotoxicity of monocytes and neutrophils.43,44In situ combination of the adenovirus-mediated cytosine deaminase gene and GM-CSF gene transfer followed by 5-fluorocytosine administration could lead to a greater number of DC and CD8+ T cells infiltrating into the tumour mass and a more potent antitumour response.45 In this study, pathological examination showed that, in addition to CD4+ and CD8+ T cells, numerous neutrophils and macrophages infiltrated in the B16/DC.GM vaccine-inoculating sites. Therefore, macrophages, granulocytes and other non-specific immune cells may partially contribute to the tumour rejection induced by the B16/DC.GM hybrid vaccine.
In this study, we used a GM-CSF gene-transfected B16 tumour vaccine (B16.GM) as a control to evaluate the efficacy of the B16/DC hybrid vaccine. Contrary to previous reports,8 vaccination of mice with the B16.GM vaccine failed to induce any protective antitumour immunity. This discrepancy may be a result of the different expression vectors used to mediate gene transfer, the expression levels and duration of the target gene in vivo and, potentially, the different immunogenicity of B16 cells transfected with the GM-CSF gene. In our experiments, B16 cells were transduced with the GM-CSF gene via an adenovirus vector. Usually, adenovirus vector mediates temporary gene expression, compared with a retrovirus vector, and thus may lead to different antitumour efficacy of tumour vaccine.34,46 We also tested the efficacy of GM-CSF gene-modified colon carcinoma vaccine with relatively high immunogenicity, and found that GM-CSF gene-modified C26 vaccine could induce more effective tumour rejection (data not shown). Therefore, we presume that the immunogenicity of tumour vaccines plays an important role in inducing antitumour immunity, even if they produce the same immunostimulatory cytokines in vivo. This is supported by the observation that the B16/DC.GM hybrid vaccine, rather than the B16.GM vaccine, could induce tumour rejection against B16 tumours. In addition, there may be a dose–effect correlation between target gene expression levels of tumour vaccines and their vaccination efficacy. Studies of GM-CSF gene-transfected HGH18 murine melanoma tumour vaccines showed that the efficacy of HGH18/GM-CSF vaccines varied with their GM-CSF expression levels.47 A dose–effect correlation was also observed in IL-2 gene-modified mouse S91 melanoma tumour vaccine.48 Optimal immunization was achieved with vaccine producing IL-2 levels of 1000–3000 U/105 cells/24 hr. When IL-2 expression levels were higher or lower than that range, the protection efficacy was diminished. The optimal GM-CSF expression levels of GM-CSF gene-modified tumour vaccines, in correlation with their therapeutic efficacy, have still to be evaluated.
In conclusion, fusion of tumour cells with DCs can generate tumour/DC hybrid tumour vaccine with the potency to induce tumour rejection, providing a promising strategy for cancer immunotherapy. GM-CSF gene-modified DCs exhibit a more potent immunostimulatory activity and tend to be able to generate hybrid tumour vaccine with potentially more potent therapeutic efficacy after fusion with tumour cells. Clinically, DCs can be generated from the blood or bone marrow of cancer patients by culture with GM-CSF, IL-4 and/or TNF-α,49,50 and tumour cells may be available from biopsy samples or surgically removed tumour mass. This DC-based hybrid tumour vaccine awaits validation in clinical trials.
Acknowledgments
This work was supported in part by grants from the National Natural Science Foundation of China (39421009) and National High Biotechnology Project of China (BH03-01-03) to X.C.
REFERENCES
- 1.Finn OJ. Tumor-rejection antigens recognized by T lymphocytes. Curr Opin Immunol. 1993;5:701. doi: 10.1016/0952-7915(93)90124-b. [DOI] [PubMed] [Google Scholar]
- 2.Boon T, Cerottini JC, Van den eynde B, Van der bruggen P, Van pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:37. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175. doi: 10.1016/s0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Ashe S, Brady WA, et al. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71:1093. doi: 10.1016/s0092-8674(05)80059-5. [DOI] [PubMed] [Google Scholar]
- 5.Restifo NP, Esquivel F, Kawakami Y, et al. Identification of human cancers deficient in antigen processing. J Exp Med. 1993;177:265. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen HL, Gabrilovich D, Tampe R, Girgis KR, Nadaf S, Carbone DP. A functionally defective allele of TAP1 results in loss of MHC class I antigen presentation in a human lung cancer. Nature Genet. 1996;13:210. doi: 10.1038/ng0696-210. [DOI] [PubMed] [Google Scholar]
- 7.Baskar S, Ostrand-Rosenberg S, Nabavi N, Nadler LM, Freeman GJ, Glimcher LH. Constitutive expression of B7 restores immunogenicity of tumor cells expressing truncated major histocompatibility complex class II molecules. Proc Natl Acad Sci. 1993;90:5687. doi: 10.1073/pnas.90.12.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte–macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:35. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259:368. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 10.Young JW, Inaba K. Dendritic cells as aduvants for class I major histocompatibility complex-restricted antitumor immunity. J Exp Med. 1996;183:7. doi: 10.1084/jem.183.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuler G, Steinman RM. Dendritic cells as adjuvants for immune-mediated resistance to tumor. J Exp Med. 1997;186:1183. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao XT, Zhang WP, Ma SH, Zhang MH, Wang JL, Ye T. Enhanced antitumor effects of tumor antigen-pulsed dendritic cells by their transfection with GM-CSF gene. Sci China (Ser C) 1997;40:539. doi: 10.1007/BF03183594. [DOI] [PubMed] [Google Scholar]
- 13.Ahuja S, Mummidi S, Malech H, Ahuja SK. Human dendritic cell (DC)-based anti-infective therapy: engineering DCs to secrete functional IFN-gamma and IL-12. J Immunol. 1998;161:868. [PubMed] [Google Scholar]
- 14.Cao X, Zhang W, He L, et al. Lymphotactin gene-modified bone marrow dendritic cells act as more potent adjuvants for peptide delivery to induce specific antitumor immunity. J Immunol. 1998;161:6238. [PubMed] [Google Scholar]
- 15.Mayordomo JI, Zorina T, Storkus WJ, et al. Bone marrow-derived dendritic cells pulsed with synthetic tumor peptides elicit protective and therapeutic antitumor immunity. Nature Med. 1995;1:1297. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 16.Porgador A, Gillboa E. Bone marrow-generated dendritic cells pulsed with a class I-restricted peptide are potent inducers of cytotoxic T lymphocytes. J Exp Med. 1995;182:255. doi: 10.1084/jem.182.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boczkowski D, Nair S, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD. Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J Exp Med. 1996;183:283. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med. 1996;183:317. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeves M, Royal RE, Lam JS, Rosenberg SA, Hwu P. Retroviral transduction of human dendritic cells with a tumor-associated antigen gene. Cancer Res. 1996;56:5672. [PubMed] [Google Scholar]
- 21.Song W, Kong H, Carpenter H, et al. Dendritic cells genetically modified with an adenovirus vector encoding the cDNA for a model tumor antigen induce protective and therapeutic antitumor immunity. J Exp Med. 1997;186:1247. doi: 10.1084/jem.186.8.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo Y, Wu M, Chen H, et al. Effective tumor vaccine generated by fusion of hepatoma cells with activated B cells. Science. 1994;263:518. doi: 10.1126/science.7507262. [DOI] [PubMed] [Google Scholar]
- 23.Gong J, Chen D, Kashiwaba M, Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nature Med. 1997;3:558. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Saffold S, Cao X, Krauss J, Chen W. Eliciting T cell immunity against poorly immunogenic tumors by immunization with dendritic cell-tumor fusion vaccines. J Immunol. 1998;161:5516. [PubMed] [Google Scholar]
- 25.Steinman RM. The dendritic cells system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 26.Hart DNJ. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245. [PubMed] [Google Scholar]
- 27.Banchereau J, Steinman R. Dendritic cells and the control of immunity. Nature. 1998;392:245. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 28.Wiltmer-Pack MD, Olivier W, Valinsky J, Schuler G, Steinman RM. Granulocyte–macrophage colony-stimulating factor is essential for the viability and function of cultured murine epidermal Langerhans’ cells. J Exp Med. 1987;166:1484. doi: 10.1084/jem.166.5.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markowicz S, Engleman EG. Granulocyte–macrophage colony-stimulating factor promotes differentiation and survival of human peripheral blood dendritic cells in vitro. J Clin Invest. 1990;85:955. doi: 10.1172/JCI114525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tazi A, Bouchonnet F, Grandsaigne M, Boumsell L, Hance AJ, Soler P. Evidence that granulocyte–macrophage colony-stimulating factor regulates the distribution and differentiated state of dendritic cells/Langerhans’ cells in human lung and lung cancers. J Clin Invest. 1993;91:566. doi: 10.1172/JCI116236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen C, Ritchie SC, Hendrix R, et al. Regulation of immunostimulatory function and costimulatory molecule (B7 and B7) expression on murine dendritic cells. J Immunol. 1994;152:5208. [PubMed] [Google Scholar]
- 32.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte–macrophage colony-stimulating factor plus interleukin 4 and down-regulated by tumor necrosis factor α. J Exp Med. 1994;179:1109. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi H, Matsubara H, Yokota T, et al. Molecular cloning and characterization of the gene encoding mouse melanoma antigen by cDNA library transfection. J Immunol. 1992;149:1223. [PubMed] [Google Scholar]
- 34.Abe J, Wakimoto H, Yoshida Y, Aoyagi M, Hirakawa K, Hamada H. Antitumor effect induced by granulocyte/macrophage-colony-stimulating factor gene-modified tumor vaccination: comparison of adenovirus- and retrovirus-mediated genetic transduction. J Cancer Res Clin Oncol. 1995;121:587. doi: 10.1007/BF01197775. [DOI] [PubMed] [Google Scholar]
- 35.Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med. 1990;171:1753. doi: 10.1084/jem.171.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuting T, Deleo AB, Lotze MT, Storkus WJ. Genetically modified bone marrow-derived dendritic cells expressing tumor-associated viral or ‘self’ antigens induce antitumor immunity in vivo. Eur J Immunol. 1997;27:2702. doi: 10.1002/eji.1830271033. [DOI] [PubMed] [Google Scholar]
- 37.Zitvogel L, Mayordomo JI, Tjandrawan T, et al. Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell 1-associated cytokines. J Exp Med. 1996;183:87. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, He L, Yuan Z, et al. Enhanced therapeutic efficacy of tumor RNA-pulsed dendritic cells after genetic modification with lymphotactin. Human Gene Ther. 1999:1151. doi: 10.1089/10430349950018148. [DOI] [PubMed] [Google Scholar]
- 39.Maraskovsky E, Brasel K, Teepe M, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arthur JF, Butterfield LH, Roth MD, et al. A comparison of gene transfer methods in human dendritic cells. Cancer Gene Ther. 1997;4:17. [PubMed] [Google Scholar]
- 42.Wan Y, Bramson J, Carter R, Graham F, Gauldie J. Dendritic cells transduced with an adenoviral vector encoding a model tumor-associated antigen for tumor vaccination. Human Gene Ther. 1997;8:1355. doi: 10.1089/hum.1997.8.11-1355. [DOI] [PubMed] [Google Scholar]
- 43.Handman E, Burgess AW. Stimulation by granulocyte–macrophage colony-stimulating factor of Leishmania tropica killing by macrophages. J Immunol. 1979;122:1134. [PubMed] [Google Scholar]
- 44.Kushner BH, Cheung NK. GM-CSF enhances 3F8 monoclonal antibody-dependent cellular cytotoxicity against human melanoma and neuroblastoma. Blood. 1989;73:1936. [PubMed] [Google Scholar]
- 45.Cao X, Ju DW, Wang BM, et al. Adenovirus-mediated GM-CSF gene and cytosine deaminase gene transfer followed by 5-fluorocytosine administration elicit more potent antitumor response in tumor-bearing mice. Gene Ther. 1998;5:1131. doi: 10.1038/sj.gt.3300727. [DOI] [PubMed] [Google Scholar]
- 46.Arca MJ, Krauss JC, Strome SE, Cameron MJ, Chang AE. Diverse manifestations of tumorigenicity and immunogenicity displayed by the poorly immunogenic B16–BL6 melanoma transduced with cytokine genes. Cancer Immunol Immunother. 1996;42:237. doi: 10.1007/s002620050276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armstrong CA, Botella R, Galloway TH, et al. Antitumor effects of granulocyte–macrophage colony-stimulating factor production by melanoma cells. Cancer Res. 1996;56:2191. [PubMed] [Google Scholar]
- 48.Schmidt W, Schweighoffer T, Herbst E, et al. Cancer vaccines: the interleukin 2 dosage effect. Proc Natl Acad Sci USA. 1995;92:4711. doi: 10.1073/pnas.92.10.4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romani N, Gruner S, Brang D, et al. Proliferating dendritic cell progenitors in human blood. J Exp Med. 1994;180:83. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szabolcs P, Moore MA, Joung JW. Expansion of immunostimulatory dendritic cells among the myeloid progency of human CD34+ bone marrow precusors cultured with c-kit ligand, granulocyte-macrophage colony-stimulating factor, and TNF-α. J Immunol. 1995;154:5851. [PubMed] [Google Scholar]