

Abstract
Interleukin-12 (IL-12) plays a pivotal role in the development of T-helper 1 (Th1) immune response, which may be involved in the pathogenesis of chronic inflammatory autoimmune disorders. In this study we investigated the effects of sulfasalazine, a drug for treating inflammatory bowel disease and rheumatoid arthritis, on the production of IL-12 from mouse macrophages stimulated with lipopolysaccharide (LPS). Sulfasalazine potently inhibited the production of IL-12 in a dose-dependent manner, in part through the down-regulation of nuclear factor κB (NFκB) activation in IL-12 p40 gene. Activation of macrophages by LPS resulted in markedly enhanced binding activities to the κB site, which significantly decreased upon addition of sulfasalazine as demonstrated by an electrophoretic gel shift assay. Importantly, macrophages pretreated with sulfasalazine either in vitro or in vivo reduced their ability to induce interferon-γ (IFN-γ) and increased the ability to induce IL-4 in antigen-primed CD4+ T cells. From these results, sulfasalazine may induce the Th2 cytokine profile in CD4+ T cells by suppressing IL-12 production in macrophages, and sulfasalazine-induced inhibition of IL-12 production in macrophages may explain some of the known biological effects of sulfasalazine.
INTRODUCTION
Interleukin-12 (IL-12), a heterodimeric cytokine composed of two disulphide-linked subunits of 35 000 (p35) and 40 000 MW (p40) encoded by two separate genes, is produced by phagocytic cells and other antigen-presenting cells in response to stimulation by a variety of microorganisms as well as their products.1–3 IL-12 exerts multiple biological activities mainly through T and natural killer cells by inducing their production of interferon-γ (IFN-γ), which augments their cytotoxicity, and by enhancing their proliferation potential. IL-12 production is critical for the development of T-helper 1 (Th1) cells and the initiation of cell-mediated immune responses.4 Recent evidence points to a critical role for IL-12 in the pathogenesis of rodent models of Th1-mediated autoimmune diseases such as type 1 diabetes, multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease and acute graph-versus-host disease. IL-12 has also been implicated in the pathogenesis of sarcoidosis, a multisystem disorder characterized by non-caseating granulomatous inflammation and dominant type 1 cytokine expression.5,6 Thus, pharmacological control of IL-12 production may be a key strategy in modulating specific immune-mediated diseases dominated by type 1 cytokine responses.
Sulfasalazine was synthesized in 1942 to combine an antibiotic, sulfapyridine, and an anti-inflammatory agent, 5-aminosalicylic acid. Sulfasalazine was the first drug with proven efficacy for ulcerative colitis.7 Sulfasalazine has a wide range of biological activities including antibacterial, anti-inflammatory and immunomodulatory activities, and is widely used in rheumatoid arthritis and inflammatory bowel diseases.8 However, the mechanisms by which sulfasalazine exerts its effects remain a matter of debate, and it is also unclear as to which actions are essential for the observed clinical outcome.
In this study we investigated the effect of sulfasalazine on the IL-12 production in mouse macrophages. Here, we demonstrate that sulfasalazine inhibited IL-12 production in activated macrophages. This inhibition was dependent, at least in part, on the down-regulation of nuclear factor κB (NFκB) binding to the p40-κB sequence. Importantly, inhibition of IL-12 production in sulfasalazine-treated macrophages induced a Th2 cytokine profile (increased IL-4 and decreased IFN-γ production) in antigen-primed CD4+ T cells.
MATERIALS AND METHODS
Mice, cell lines and culture medium, and transient transfection
Female DBA/2 mice were obtained from Japan SLC, Inc. (Tokyo, Japan) and used at 6–10 weeks of age. RAW264.7 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) (Gibco BRL, Grand Island, NY) at 37° in a 5% CO2 humidified air atmosphere. Spleen cell populations and macrophages from mice were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS and antibiotics. For transfections, cells were grown in 24-well plates with medium supplemented with 10% FBS for 24 hr and transfected with indicated plasmid in the presence of Superfect, according to the manufacturer’s protocol (Qiagen, Valencia, CA). After 12 hr, cells were washed and refed with DMEM containing 10% FBS. Cells were harvested 24 hr later, luciferase activity was assayed as described,9 and the results were normalized to the LacZ expression.
Monoclonal antibodies, cytokines, and reagents
Anti-IL-12 p40 monoclonal antibodies (mAbs) C17.8 and C15.6 were purified from ascitic fluids by ammonium sulphate precipitation followed by diethylaminoethyl (DEAE)–Sephacel chromatography (Sigma, St. Louis, MO). Anti-IL-12 p35 mAb Red-T/G297–289, anti-IL-10 mAbs JES-2A5 and SXC-1 were obtained from PharMingen (San Diego, CA). Recombinant murine IL-12 was generously provided by Dr S. Wolf (Genetics Institute, Cambridge, MA), and recombinant murine IL-10 were obtained from PharMingen. Lipopolysaccharide (LPS) (from E. coli O111:B4) and sulfasalazine were purchased from Sigma.
IL-12 p40 promoter constructs
The −689/+98 fragment of mIL-12 p40 promoter from pXP210 was subcloned into Kpn I/Xho I sites of pGL3-basic luciferase vector (Promega, Madison, WI). All the deletion mutants were generated by polymerase chain reaction (PCR) using an upstream primer containing Bam HI site. A linker-scanning mutant was generated by a two-step PCR procedure with overlapping internal primers that contain mutated sequences for the NFκB site.
Preparation of splenic macrophages stimulated with LPS or heat-killed Listeria monocytogenes (HKL)
Spleen cells were cultured at 106 cells/ml for approximately 3 hr at 37°. The non-adherent cells were removed by washing with warm DMEM until visual inspection revealed a lack of lymphocytes (>98% of the cell population). The adherent cells were removed from plates by incubating for 15 min with ice-cold phosphate-buffered saline solution and rinsing repeatedly. The isolated adherent cell population was stimulated with 5 μg/ml LPS in the absence or presence of sulfasalazine at 0·05, 0·1, 0·5, 2·5 mm at 1×105 cells per well in 96-well culture plates for 48 hr. For some experiments, the cells were stimulated with HKL at 2×106 bacteria per well.
Purification and induction of cytokine synthesis in CD4+ T cells
Draining axillary, popliteal, and inguinal lymph nodes were removed from mice 9 days after priming with 100 μg keyhole limpet haemocyanin (KLH) (Calbiochem, San Diego, CA) in complete Freund’s adjuvant (CFA) in the footpads. Lymph node cells were depleted of B cells by adherence to goat anti-mouse immunoglobulin-coated dishes for 1 hr at 4°. Non-adherent cells were depleted of CD8+ T cells and other antigen-presenting cells by treating the cells with a mixture of anti-CD8, anticlass II mAbs on ice for 30 min, followed by addition of low toxicity rabbit complement (Pel Freeze, Rogers, AR) and incubation at 37° for 45 min. Purified CD4+ T cells were incubated in 96-well plates at 4×105 cells/well with macrophages and 10 μg/ml KLH. Culture supernatants were harvested after 2 days (for IL-12) or after 4 days (for IFN-γ and IL-4), and assayed by enzyme-linked immunosorbent assay (ELISA).
Cytokine assays
The quantities of IL-12 p40, IL-12 p70 and IL-10 in culture supernatants were determined by a sandwich ELISA using mAbs specific for each cytokine, as previously described.11 The mAbs for coating the plates and the biotinylated second mAbs were as follows: for IFN-δ, HB170 and XMG1.2; for IL-4 BVD4 and BVDG; for IL-12 p40, C17.8 and C15.6; for IL-12 p70, Red-T/G297-289 and C17.8; for IL-10, JES-2A5 and SXC-1. Standard curves were generated using recombinant cytokines. The lower limit of detection was 125 pg/ml for IFN-γ, 3 pg/ml for IL-4, 30 pg/ml for IL-12 p40, 50 pg/ml for IL-12 p70, and 0·2 ng/ml for IL-10.
Electrophoretic gel shift assay
The nuclear extracts were prepared from the cells, as described previously.12 An oligonucleotide containing an NFκB-binding site within the immunoglobulin κ-chain (5′ CCG GTT AAC AGA GGG GGC TTT CCG AG 3′) was used as a probe. Labelled oligonucleotides (10 000 c.p.m.) were incubated for 30 min at room temperature, along with 10 μg of nuclear extracts, in 20 μl of binding buffer (10 mm Tris–HCl, pH 7·6, 500 mm KCl. 10 mm ethylenediamine tetra-acetic acid (EDTA), 50% glycerol, 100 ng of poly(dI-dC), and 1 mm dithiothreitol). The reaction mixture was analysed by electrophoresis on a 4% polyacrylamide gel in 0·5× Tris–borate buffer. Specific binding was confirmed by competition experiments with a 50-fold excess of unlabelled, identical oligonucleotides or cAMP response element-containing oligonucleotides.
Statistical analysis
Student’s t-test was used to determine the statistical differences between various experimental and control groups. A P-value of < 0·01 was considered as significant.
RESULTS
Sulfasalazine inhibited IL-12 production from LPS-activated macrophages
We examined the effect of sulfasalazine on the production of IL-12 by primary macrophages stimulated with LPS. LPS readily induced the production of IL-12 heterodimer as well as the p40 subunit, as expected. However, sulfasalazine significantly inhibited this LPS-induced IL-12 production in a dose-dependent manner (Fig. 1) (P < 0·01 at 0·5 or 5·0 mm sulfasalazine, relative to an untreated group). In contrast, treatment with sulfasalazine had little influence on IL-10 production from LPS-stimulated macrophages, suggesting that the inhibitory effect of IL-12 production by sulfasalazine was not the result of a general dampening of cellular activation.
Figure 1.
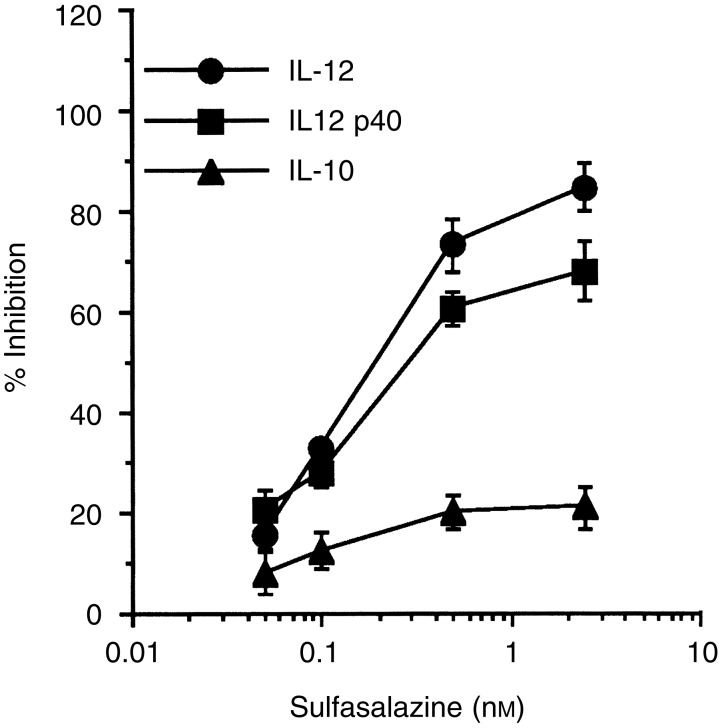
Inhibition of IL-12 production in macrophages by sulfasalazine. Primary macrophages were stimulated with LPS (5 μg/ml) for 48 hr in the absence or presence of varying concentrations of sulfasalazine. Culture supernatants were harvested and cytokine levels were evaluated by ELISA, and results are presented as mean±SD of the percentage response of cytokine production of sulfasalazine-treated macrophages compared with untreated control macrophages stimulated with LPS. Mean cytokine levels in the absence of sulfasalazine were 650 pg/ml (IL-12 p70), 1·9 ng/ml (IL-12 p40) and 1250 ng/ml (IL-10).
Sulfasalazine inhibited NFκB-mediated activation of IL-12 p40 promoter by LPS
An IL-12 p40 subunit was known as the highly inducible and tightly regulated component of IL-12·1 To identify the region involved in these sulfasalazine actions, we generated a series of luciferase reporter constructs containing the p40 promoter sequences from positions −689, −231, and −185 to +98, relative to the transcription initiation site (Fig. 2a). Mouse RAW264.7 monocytic cells were transfected with each of these constructs and stimulated with LPS either in the absence or presence of sulfasalazine, and the luciferase activity was determined. All of these constructs showed strong stimulation with LPS in the absence of sulfasalazine but impaired stimulation with sulfasalazine (Fig. 2b). In particular, deleting sequences to −185 (p40/185) did not diminish the LPS-dependent promoter activities and the inhibitory effect of sulfasalazine was still observed, suggesting that the target site for sulfasalazine should reside within this region. To directly test the role of a κB site found between −121 and −131 of the p40 promoter in the sulfasalazine-mediated inhibitory actions, we introduced a linker scanning mutation into the κB site within the context of the −689/+98 construct (p40/LS). The LPS-dependent promoter activation was still observed with p40/LS, although significantly reduced (Fig. 2b), consistent with the previous findings in which the κB site was shown to be important for the LPS induction of p40 promoter.13 However, addition of sulfasalazine to LPS-stimulated cells induced little repressive effects with p40/LS, clearly indicating that the inhibitory effect of sulfasalazine on IL-12 production was mediated through the κB site.
Figure 2.

Analysis of sulfasalazine-mediated transcriptional repression of p40 promoter constructs activated by LPS. (a) Schematic representation of the mouse p40 promoter constructs as well as a linker-scanning mutant for NFκB site are as shown, along with Ets and NFκB binding sites. The nucleotide sequence numbers for each construct are shown. (b) Transient transfection of RAW264.7 cells with the p40 promoter constructs, followed by stimulation with LPS either in the absence or presence of sulfasalazine (0·5 or 2·5 mm). The results are expressed as induction fold over the value obtained with the unstimulated RAW264.7 cells transfected with the −689/+98 construct, which was given an arbitrary value of 1. The data are representative of three similar experiments.
NFκB binding to the κB site is inhibited by sulfasalazine
To gain more insight into the mechanisms of sulfasalazine-mediated inhibition of p40-κB function, we analysed the κB binding activity present in nuclear extract of unstimulated or LPS-stimulated macrophages, either in the absence or presence of sulfasalazine. As expected, nuclear extracts from LPS-stimulated macrophages exhibited strong κB-binding activity in the electrophoretic mobility shift assays using a labelled oligonucleotide containing a consensus immunoglobulin-κB site (Fig. 3). The binding was specific as it was competed with an unlabelled, identical oligonucleotide, but not with unrelated, non-specific oligonucleotide, and was absent with nuclear extracts from non-stimulated cells. Nuclear extracts from macrophages stimulated by LPS in the presence of sulfasalazine showed much diminished κB-binding activities in a dose-dependent manner (Fig. 3).
Figure 3.
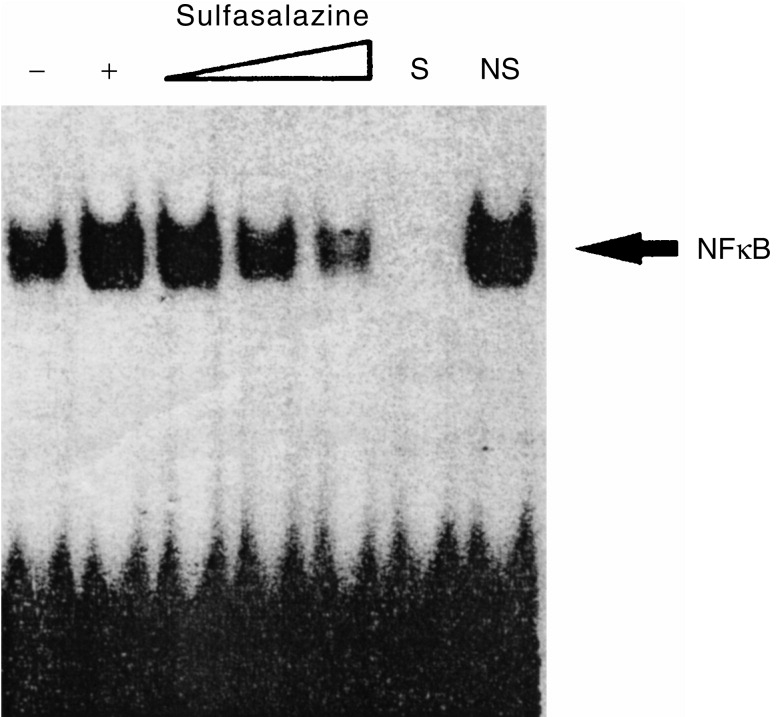
Sulfasalazine-mediated inhibition of κB binding by NFκB. Nuclear extracts prepared from macrophages stimulated by LPS either in the absence or presence of sulfasalazine (0·5, 2·5 and 5·0 mm each) were examined for κB binding activity in the electrophoretic mobility shift assays using a labelled oligonucleotide containing a consensus immunoglobulin-κB site, as indicated. S and NS indicate the presence of an unlabelled, identical oligonucleotide and non-specific oligonucleotide, respectively. The specific NFκB complexes are as indicated.
Pretreatment of macrophages with sulfasalazine enhanced IL-4 production and inhibited IFN-γ production by antigen-primed CD4+ T cells
Because IL-12 has been known to potently inhibit IL-4 and enhance IFN-γ in CD4+ T cells, we asked if the cytokine profiles of CD4+ T cells responding to antigen presented by sulfasalazine-treated macrophages would be altered. Direct effects of sulfasalazine on T cells were eliminated by pretreatment of macrophages from naive mice in vitro with sulfasalazine for 12 hr and washing them to remove sulfasalazine, before culture with syngeneic CD4+ T cells purified from lymph nodes of KLH-primed mice and the antigen KLH. Figure 4(a) shows that IL-12 production in cultures of sulfasalazine-treated macrophages was significantly decreased in comparison with untreated macrophages. In the absence of sulfasalazine treatment, stimulation with KLH resulted in the development of T cells producing high levels of IFN-γ. However, pretreatment of macrophages with sulfasalazine for 12 hr greatly inhibited their capacity to induce IFN-γ production by CD4+ T cells (Fig. 4b) and significantly increased IL-4 production (Fig. 4c). No cytokine production by CD4+ T cells was detected in the absence of macrophages, demonstrating that the sulfasalazine-treated macrophages regulated the cytokine production by CD4+ T cells. Thus, pretreatment of macrophages with sulfasalazine enhances their capacity to preferentially induce Th2 and inhibit Th1 cytokine synthesis.
Figure 4.

Macrophages pretreated with sulfasalazine inhibit IFN-γ and enhance IL-4 production by antigen-primed CD4+ T cells. Macrophages (1×105/well) were pretreated with either 0·05 or 0·5 mm sulfasalazine. After 12 hr, the cells were washed and incubated with KLH-primed CD4+ T cells (5×105/well) and KLH (10 μg/ml). Supernatants were harvested after 2 days for IL-12 (a) or after 4 days for IFN-γ (b) and IL-4 (c), and assayed by ELISA. The data shown represent the mean±SD of triplicate determinations of three separate experiments.
Macrophages from mice treated in vivo with sulfasalazine also induced Th2 cytokine profile by antigen-primed CD4+ T cells
To demonstrate that sulfasalazine had a consequential effect on macrophages in an in vivo setting, mice were injected intraperitoneally (i.p.) with 1 mg sulfasalazine per mouse. After 24 hr, splenic macrophages were purified from sulfasalazine-treated mice, or from saline-injected control mice. Figure 5 shows that macrophages from sulfasalazine-treated mice produced much lower amounts of IL-12 in response to either LPS or HKL than macrophages from saline-injected control mice. To determine whether cytokine production by antigen-primed CD4+ T cells would differ in the presence of antigen presented by macrophages from mice treated in vivo with sulfasalazine, macrophages were purified from DBA/2 mice 24 hr following i.p. injection with sulfasalazine (1 mg), and cultured with CD4+ T cells from KLH-primed mice and KLH. Figure 6 demonstrates that production of IL-12 in cultures containing antigen-primed CD4+ T cells and macrophages from sulfasalazine-treated mice was greatly reduced compared with those from saline-injected control mice. Moreover, macrophages from mice injected with sulfasalazine induced significantly lower amounts of IFN-γ and greater amounts of IL-4 than macrophages from control mice. These results show that in vivo treatment with sulfasalazine alters the capacity of macrophages to induce IFN-γ and IL-4.
Figure 5.
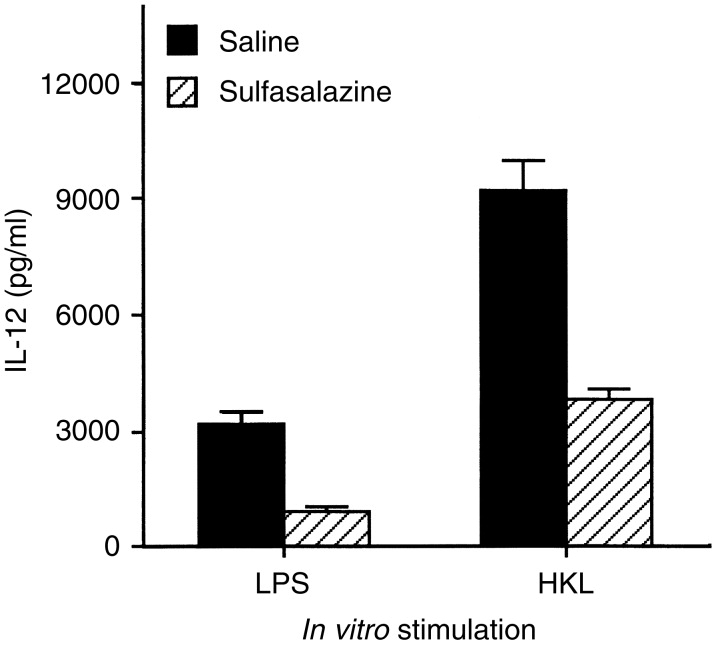
Decreased levels of IL-12 production in macrophages exposed to sulfasalazine in vivo. Mice were treated in vivo with sulfasalazine (1 mg, i.p.). After 24 hr, macrophages were purified and stimulated with LPS or HKL. Culture supernatants were harvested 48 hr later and IL-12 content was assayed by ELISA. The values represent the mean±SD of triplicate determinations of three separate experiments.
Figure 6.

Regulation of cytokine production in antigen-primed CD4+ T cells by macrophages purified from mice treated in vivo with sulfasalazine. DBA/2 mice were treated in vivo with either sulfasalazine (1 mg, i.p.) or saline. After 24 hr, macrophages were purified and incubated with KLH-primed CD4+ T cells and KLH (10 μg/ml). After 4 days, culture supernatants were harvested 4 days and assayed for IL-12 (a), IFN-γ (b) and IL-4 (c) content by ELISA. The values represent the mean±SD of triplicate determinations of two separate experiments.
DISCUSSION
Inhibiting the action of IL-12 has been shown to prevent development and block progression of disease in experimental models of autoimmunity.14 These findings have raised great interest in identifying inhibitors of IL-12 production for the treatment of Th1-mediated diseases such as type 1 diabetes, multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease and acute graft-versus-host disease. In this study we demonstrated that sulfasalazine, a well-known drug used in the treatment of inflammatory bowel disease and rheumatoid arthritis, inhibited IL-12 production in macrophages in a dose-dependent manner. Moreover, pretreatment of macrophages with sulfasalazine resulted in a reduced capacity to induce IFN-γ and an increased capacity to induce IL-4 in CD4+ T cells, suggesting that sulfasalazine-mediated inhibition of IL-12-production resulted in the induction of the Th2 cytokine profile in CD4+ T cells. The lack of inhibition of IL-10 secretion by sulfasalazine indicated that the effect of IL-12 was not the result of a general dampening of cellular events.
The mechanism by which sulfasalazine inhibits IL-12 production in LPS-stimulated macrophages seems to be through the down-regulation of NFκB-mediated activation and the binding to the p40-κB site. This point was supported by several lines of evidence. First, an IL-12 p40 subunit was known as the highly inducible and tightly regulated component of IL-12.1 The inhibitory effect of sulfasalazine on a series of 5′ deletions of the p40 promoter was retained within −185 bp upstream of the transcription initiation site (Fig. 2b), suggesting that sulfasalazine may interfere with the inducible binding of NFκB at position −121/−131 bp in the p40 promoter. Linker scan mutation of the p40-κB site abrogated the inhibitory effect of sulfasalazine on the p40 promoter, indicating that this site plays a role in transcriptional repression of the p40 gene (Fig. 2b). In addition, sulfasalazine significantly decreased the binding activities to the κB site in LPS-activated macrophages, as demonstrated by the electrophoretic mobility assays (Fig. 3). The NFκB-mediated inhibition of IL-12 production is in accord with previous observations that 1,25-dihydroxyvitamin D3 and retinoids inhibited IL-12 production by down-regulating NFκB activation and binding to the κB sequence of the p40 gene.15,16
Further work will be required to elucidate the mechanism(s) by which sulfasalazine inhibits NFκB in LPS-activated macrophages. One mechanism regulating NFκB activation is its cytoplasmic association with a family of ankyrin-containing molecules, the IκBs.17 In most cells, IκBα is the predominant inhibitory molecule, and activation and translocation of NFκB are therefore contingent upon release from IκBα.18,19In vivo, IκBα is rapidly degraded in response to numerous stimuli including phorbol, bacterial LPS, and the inflammatory cytokines IL-1α and tumour necrosis factor-α (TNFα).20,21 Previous reports indicated that sulfasalazine inhibited TNFα-induced activation of NFκB in SW620 colon cells by preventing nuclear translocation of NFκB caused by the inhibition of IκBα phosphorylation and subsequent degradation.22
Although sulfasalazine may affect cytokine production in T cells in several ways, we believe that inhibition of IL-12 production in macrophages is a major mechanism by which sulfasalazine affects cytokine production in T cells, particularly as IL-12 is extremely potent in inhibiting IL-4 and enhancing IFN-γ in T cells.23–25 In our cultures, the effect of sulfasalazine on cytokine production in CD4+ T cells was indirect because the T cells in these culture were never directly exposed to the sulfasalazine. Similar studies showed that β2-adrenergic compounds, including salbutamol, inhibited IL-12 production from human monocytes or dendritic cells by increasing intracellular cAMP levels, leading to inhibit the development of Th1 cells while promoting Th2 cell differentiation.26 Corticosteroids have been known to enhance the capacity of macrophages to induce IL-4 synthesis in CD4+ T cells by inhibiting IL-12 production.27 Constantin et al. reported that in vitro modulation of the cytokine network by methotrexate, increasing Th2 cytokines and decreasing Th1 cytokines, could explain its anti-inflammatory actions in vivo during the treatment of rheumatoid arthritis.28
In conclusion, we have shown that sulfasalazine inhibited IL-12 production in macrophages through the down-regulation of NFκB activation, leading to the induction of a Th2 cytokine profile in antigen-primed CD4+ T cells. These results suggest that sulfasalazine-induced inhibition of IL-12 production in macrophages may explain some of the known biological effects of sulfasalazine, including its anti-inflammatory activity.
Acknowledgments
This work was supported by Grants from the Korea Science and Engineering Foundation (HRC to T. S. Kim and S.-Y. Im) and from the Korean Research Foundation made in the Program Year 1996 (to T. S. Kim). We would like to thank Dr G. Trinchieri (Wistar Institute, Philadelphia, PA) for providing IL-12 p40 promoter constructs, and Dr J. W. Lee (Chonnam National University) for helpful discussion.
Abbreviations
- HKL
heat-killed Listeria monocytogenes
- IFN-γ
interferon-γ
- IL
interleukin
- KLH
keyhole limpet haemocyanin
- LPS
lipopolysaccharide
- NF
nuclear factor
- Th1
T helper type 1.
References
- 1.Kang K, Kubin M, Cooper KD, Lessin SR, Trinchieri G, Rook AH. IL-12 synthesis by human Langerhans’ cells. J Immunol. 1996;156:1402. [PubMed] [Google Scholar]
- 2.Benjamin D, Sharma V, Kubin M, et al. IL-12 expression in AIDS-related lymphoma B cell lines. J Immunol. 1996;156:1626. [PubMed] [Google Scholar]
- 3.Lawrence C, Nauciel G. Production of interleukin-12 by murine macrophages in response to bacterial peptidoglycan. Infect Immun. 1998;66:4947. doi: 10.1080/00222936700770321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trinchieri G. Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol. 1998;70:83. doi: 10.1016/s0065-2776(08)60387-9. [DOI] [PubMed] [Google Scholar]
- 5.Trinchieri G. Proinflammatory and immunoregulatory functions of interleukin-12. Int Rev Immunol. 1998;16:365. doi: 10.3109/08830189809043002. [DOI] [PubMed] [Google Scholar]
- 6.Gately MK, Renzetti LM, Magram J, et al. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 7.Klotz U, Maier K, Fischer C, Heinkel K. Therapeutic efficacy of sulfasalazine and its metabolites in patients with ulcerative colitis and Crohn’s disease. N Engl J Med. 1980;303:1499. doi: 10.1056/NEJM198012253032602. [DOI] [PubMed] [Google Scholar]
- 8.Smedegard G, Bjork J. Sulfasalazine: mechanism of action in rheumatoid arthritis. Br J Rheumatol. 1995;34(Suppl. 2):2. [PubMed] [Google Scholar]
- 9.Ausubel FM, Brent R, Kingston RE, et al. Current Protocols in Molecular Biology. New York: Greene Assoc; 1996. [Google Scholar]
- 10.Ma X, Chow JM, Gri G, et al. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med. 1996;183:147. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim TS, Dekruyff RH, Rupper R, Maecker HT, Levy S, Umetsu DT. An ovalbumin–IL-12 fusion protein is more effective than ovalbumin plus free recombinant IL-12 in inducing a T helper cell type 1-dominated immune response and inhibiting antigen-specific IgE production. J Immunol. 1997;158:4137. [PubMed] [Google Scholar]
- 12.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-κB half-site. Mol Cell Biol. 1995;15:5258. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caspi RR. IL-12 in autoimmunity. Clin Immunol Immunopathol. 1998;88:4. doi: 10.1006/clin.1998.4540. [DOI] [PubMed] [Google Scholar]
- 15.D’ambrosio D, Cippitelli M, Cocciolo MG, et al. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-κB downregulation in transcriptional repression of the p40 gene. J Clin Invest. 1998;101:252. doi: 10.1172/JCI1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Na S-Y, Kang BY, Chung SW, et al. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFκB. J Biol Chem. 1999;274:7674. doi: 10.1074/jbc.274.12.7674. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 18.Brown K, Park S, Kanno T, Franzoso G, Siebenlist U. Mutual regulation of the transcriptional activator NF-κB and its inhibitor, IκB-α. Proc Natl Acad Sci USA. 1993;90:2532. doi: 10.1073/pnas.90.6.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice NR, Ernst MK. In vivo control of NF-κB activation by IκBα. EMBO J. 1993;12:4685. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beg AA, Baldwin AS. The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev. 1993;7:2064. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 21.Cordle SR, Donald R, Read MA, Hawiger J. Lipopolysaccharide induces phosphorylation of MAD3 and activation of c-Rel and related NF-κB proteins in human monocytic THP-1 cells. J Biol Chem. 1993;268:11803. [PubMed] [Google Scholar]
- 22.Wahl W, Liptay S, Adler G, Schmid RM. Sulfasalazine: a potent and specific inhibitor of nuclear factor κB. J Clin Invest. 1998;101:1163. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall JD, Secrist H, Dekruyff RH, Wolf SF, Umestu DT. IL-12 inhibits the production of IL-4 and IL-10 in allergen-specific human CD4+ T lymphocytes. J Immunol. 1995;155:111. [PubMed] [Google Scholar]
- 24.Umetsu DT, Gieni R, Dekruyff RH. Effects of IL-12 in memory CD4+T lymphocyte responses. Ann NY Acad Sci. 1996;795:88. doi: 10.1111/j.1749-6632.1996.tb52658.x. [DOI] [PubMed] [Google Scholar]
- 25.Gerosa F, Paganin C, Peritt D, et al. Interleukin-12 primes human CD4+ and CD8+ T cell clones for high production of both interferon-gamma and interleukin-10. J Exp Med. 1996;183:2559. doi: 10.1084/jem.183.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panina-Bordignon P, Mazzeo D, Lucia PD, et al. β2-agonists prevent Th1 development by selective inhibition of interleukin 12. J Clin Invest. 1997;100:1513. doi: 10.1172/JCI119674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dekruyff RH, Fang F, Umetsu DT. Corticosteroids enhance the capacity of macrophages to induce Th2 cytokine synthesis in CD4+ lymphocytes by inhibiting IL-12 production. J Immunol. 1998;160:2231. [PubMed] [Google Scholar]
- 28.Constantin A, Loubet-Lescoulie P, Lambert N, et al. Antiinflammatory and immunoregulatory actions of methotrexate in the treatment of rheumatoid arthritis: evidence of increased interleukin-4 and interleukin-10 gene expression demonstrated in vitro by competitive reverse transcriptase-polymerase chain reaction. Arthritis Rheum. 1998;41:48. doi: 10.1002/1529-0131(199801)41:1<48::AID-ART7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]