

Abstract
Immunization with T-dependent antigens induces a rapid differentiation of B cells to plasmacytes that produce the primary immunoglobulin M (IgM) and IgG antibodies with low affinities for the immunogen. It is proposed that the IgG antibody forms immune complexes with the residual antigen which provide an important stimulus for the formation of germinal centres (GC) and the activation of somatic mutation. This hypothesis was tested by passive administration of hapten-specific antibody into mice shortly after the immunization with nitrophenyl (NP) coupled to chicken gamma globulin (NP-CGG) in an environment of limited T-cell help. Athymic mice that received normal T helper cells at 72 hr after the administration of antigen produced low levels of anti-NP antibody and the splenic GC formation was delayed until day 12 after the antigen administration. The analysis of VDJ segments from NP-reactive GC B cells showed very few mutations in the VH genes. Passive injection of anti-NP IgG1 monoclonal antibody – but, not IgM – stimulated the GC formation up to normal levels and the somatic mutation activity in the GC B cells was fully restored. In addition, GC B cells in the recipients of IgG1 antibody demonstrated a change in the usage of germline-encoded VH genes which was not apparent among the primary antibody-forming cells. These results suggest the existence of a specific feedback mechanism whereby the IgG antibody regulates the GC formation, clonotypic repertoire and somatic mutation in GC B cells.
INTRODUCTION
Primary immunization with conventional antigens induces the differentiation of B cells to immunoglobulin M (IgM) antibody-forming cells (AFC) within 48 hr1,2 and, in the case of T-cell-dependent responses, a switch to IgG antibody production that occurs between the 3rd and 5th days after the immunization.3–5 The early primary IgM and IgG antibodies, which are produced by AFC in the T-cell-rich areas of lymphoid tissues6,7 are encoded by the unmutated (germline-encoded) V genes and bind to the antigen with relatively low affinities.8,9 These antibodies may form immune complexes with the residual antigen which are thought to regulate the ongoing immune responses. In particular, the complexes of antigen with IgG antibodies localize avidly and selectively in the network of follicular dendritic cells and are thought to stimulate the formation of germinal centres (GC) within the B-cell follicles.10,11 GC are the site of differentiation to memory B-cell lineage12,13 which is highlighted by somatic hypermutation of the rearranged IgV genes and by selection of cells with increasing affinity for the antigen.14 The GC formation and somatic hypermutation are detectable, respectively, on day 5 and day 7 after the immunization15,16,17 which coincides in time with the emergence of IgG antibody and the formation of immune complex. Direct evidence for a causal relationship between these two events has been provided by studies involving immunization with preformed antigen–antibody complexes. Administration of immune complexes induced faster and/or stronger GC formation compared to immunization with the antigen alone.18,19 As a corollary, the B-cell memory developed sooner after the priming with antigen–IgG antibody complexes – but not with complexes containing IgM – than after the antigen priming.19 We have subsequently shown an increased rate of somatic mutation in antigen-specific GC B cells following the immunization with preformed immune complexes.20
However, an administration of preformed, insoluble antigen–IgG complex may not approximate the mechanisms that take place during the antibody isotype switch in vivo. The physiological events may be modelled better with passive administration of IgG antibody at an early stage of the primary response. Numerous studies have shown that administration of specific IgG antibodies – but not IgM – at the time of immunization, or shortly thereafter, inhibits the primary AFC.21–23 To our knowledge, the effects of such treatment on GC B cells have not been examined. We reasoned that passive antibody administration, which may suppress the AFC differentiation, could nonetheless produce immune complexes in vivo that would stimulate the GC pathway. The present study was designed to test this hypothesis using a well-characterized model of antibody response to the hapten (4-hydroxy-3-nitrophenyl) acetyl(NP) coupled to a protein carrier, chicken gamma globulin (NP-CGG).
The kinetics of primary AFC response and GC formation after immunization with NP-CGG has been described.6,24 The somatic hypermutation becomes active on day 6 to day 7 after the immunization and reaches a plateau by days 12–14, reaching an average of three unique point mutations per rearranged VH gene.15 The NP-reactive B cells use antigen receptors with H chains encoded by several germline V genes of the V186.2/V3 subfamily of the J558 gene family, however, the response in mice with an Ighb allotype is dominated by B-cell clones expressing the VH 186.2 segment.25,26 This experimental model has nonetheless a slight drawback for studies on passive IgG antibody in that the isotype switch occurs very early. The Cγ1 switch transcripts are detectable at 48 hr after immunization with NP-CGG27 and the IgG-producing cells appear on days 3–4.5 The rapid production of endogenous IgG antibody leaves rather a small window of time for manipulation of the response with exogenous IgG. To circumvent this problem, we carried out the present studies in NP-CGG-immunized athymic (nude) mice that received T helper cells with a delay of 3 days. The anti-NP antibody responses in these animals were markedly delayed and diminished and the effects of passively administered antibody were quite dramatic. The recipients of anti-NP IgG1 – but not of IgM – developed a robust GC reaction and their somatic hypermutation activity approached that seen in normal mice. Moreover, the treatment with passive antibody produced a shift in the VH gene repertoire in the GC B cells whereas the AFC remained dominated by the V186.2-expressing cells. These results reveal a feedback regulation of GC B-cell repertoire by IgG antibody.
MATERIALS AND METHODS
Mice
Normal and thymus-deficient, nude (nu/nu) C57BL/6J mice (2–3 months old) were purchased either from Jackson Laboratory (Bar Harbor, ME) or from Taconic (Germantown, NY). All mice were maintained in a restricted animal facility in sterile microisolator cages (Lab Products, Inc., Maywood, NJ) on a 12-hr day/night cycle. The mice were handled according to a protocol #1198005 approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine.
Antigen, antibody and immunization
NP (Cambridge Research Biochemicals, Cambridge, UK) was conjugated to CGG (Sigma, St. Louis, MO) as described.28 The final conjugation ratio was NP21:CGG. Mice were immunized intraperitoneally (i.p.) with a single dose of 50 μg NP-CGG precipitated in alum. Anti-NP monoclonal antibody (mAb) IgG1 was purified by protein G affinity chromatography (ImmunoPure IgG kit, Pierce, Rockford, IL) from supernatants of a transfectoma clone pEVHCγ1/λ1,29 kindly provided by Dr G. Kelsoe (Duke University, Durham, NC). Anti-NP mAb IgM was purified from culture supernatants of B1-8 hybridoma25 by ammonium sulphate precipitation followed by affinity chromatography on agarose coupled with goat anti-mouse IgM (Sigma). Both IgM and IgG1 antibodies are encoded by unmutated, canonically rearranged V186.2/DFL16.1/JH2 heavy-chain genes and unmutated λ1 light-chain gene. The IgG1 mAb and the monomeric IgM mAb bind to NP with the dissociation constant Kd=1·2×10−6 m as determined by fluorescence quenching. Anti-NP mAb were injected intravenously (i.v.) at 0·7–0·8 mg/mouse.
Antibody measurement
Serum levels of NP-specific antibody IgM, IgG1 and IgG3 which exhibit heteroclitic binding to the NP analogue (4-hydroxy-5-iodo-3-nitrophenyl) acetyl (4-hydroxy-3-iodo-5-nitrophenylacetic acid, NIP), were determined by standard enzyme-linked immunosorbent assay (ELISA) techniques, using NIP–bovine serum albumin (BSA) conjugate as antigen24 in solid phase and isotype-specific goat anti-mouse immunoglobulin labelled with horseradish peroxidase (Southern Biotechnology Associates, Inc., Birmingham, AL) as secondary antibodies followed by a tetramethylbenzidine hydrogen peroxide substrate kit (Bio-Rad Laboratories, Richmond, CA). The antibody titres were expressed as reciprocal end-point dilutions of the sera.
Preparation of CD4+ T cells and cell transfers
Splenocyte suspension was prepared by mashing spleens from normal, unimmunized C57BL/6J mice in RPMI-1640 medium supplemented with 25 mm HEPES (Gibco BRL, Gaithersberg, MD) and 0·5% BSA (Sigma Chemical Co.). T-cell-enriched splenocytes were prepared by filtration through nylon wool columns (Wako Bioproduct, Richmond, VA) using the manufacturer’s protocol. Non-adherent cells were treated once with mAb 3.155 plus rabbit complement to eliminate CD8+ T cells. Cells were resuspended in 0·4 ml of phosphate-buffered saline (PBS) containing 1% (v/v) of normal mouse serum and injected i.v. into C57BL/6J nu/nu mice, ∼1·5×107 cells/mouse.
Immunochemical staining of GC and AFC and microdissection of cells
Frozen splenic sections were prepared and stained as previously described.30 The specific, NP-reactive GC were identified by dual staining with peanut agglutinin (PNA) coupled to horseradish peroxidase (EY Laboratories, Inc., San Mateo, CA) in combination with either a biotinylated goat anti-mouse λ chain antibody (Fisher Biotech, Pittsburgh, PA) or with biotinylated NIP–BSA. Cells (approximately 50–100) from individual PNA+/λ+ or PNA+/NIP+ GC were recovered using a sharpened micropipette controlled by micromanipulator.
To assess the magnitude of GC reaction, the PNA-stained areas within B-cell follicles were counted and the number was expressed as the percentage of all follicles in the splenic section (mean from two sections).
NP-specific AFC were visualized as plasmacytes stained with biotinylated NIP–BSA followed by strepavidin-conjugated to alkaline phosphatase (Fisher). These cells formed typical foci close to the borders of B-cell follicles (10–50 AFC/focus). Clusters of AFC were microdissected from the most dense areas of individual foci (10–20 AFC/sample) using a micromanipulation-controlled micropipette.30
Amplification and sequencing of VDJ DNA recovered from individual GC and AFC foci
Amplification and sequencing of VDJ DNA were carried out as previously described.20 The two pairs of primers for nested PCR are as follows. Primers 5′-CCTGACCCAGATGTCCC TTCTTCTCCAGCAGG-3′ and 5′-GGGTCTAGAGGTGT CCCTAGTCCTTCATGACC-3′, corresponding to the V186.2 genomic DNA 5′ transcription start site sequence and to the intron JH2 sequence, respectively, were used for the first round polymerase chain reaction (PCR). The primers for the second round PCR were 5′-TCTAGAATTCAGGTCCAACTGCAGCAGCC-3′ complementary to the initial 20 nucleotides of the V186.2/V3 gene family (with an additional recognition sequence for Pst I) and 5′-ACGGATCCTGTGAGAGTGGTGCCT-3′, complementary to the JH2 gene segment with a Bam HI recognition site. Positive PCR products were cloned using pBluescriptSKII plasmid (Stratagene) and DH5αEscherichia coli, and screened by hybridization with VH gene-specific, 32P-labelled oligonucleotides (see below). DNA extracted from positive colonies was sequenced using the Sequenase Version 2·0 DNA sequencing kit (Amersham, Piscataway, NJ) in a Sequi-Gen Sequencing System (Bio-Rad) according to the recommended procedure.
Screening of VH repertoire by differential hybridization
Essentially all transformed colonies hybridized with the probe (5′-GTAGCCAGAAGCCTTGCAGGA-3′) corresponding to the region of genomic DNA (amino acid position 21–27) that is shared by the V186.2/V3 group of 22 genes of the J558 family.31 A proportion of these colonies also hybridized with the probe (5′-TACCACCACTATTAGGATCAATCCT-3′) which identifies a DNA region (amino acid position 50–57) specific for the V186.2 gene sequence. The proportion (%) of V186.2+ B cells in the population was determined as:
This method permits a large repertoire sampling (≡103 clones/experiment) and accurately determines the ‘dominance’ of V186.2 gene-expressing B cells in response to NP.20
Frequency of mutations
An error rate of 8·5×10−6 misincorporations per base pair (bp) per PCR cycle is expected from the Expand High Fidelity polymerase according to the manufacturer. Thus, approximately five VDJ fragments (1715 bp) recovered from tissue by 80 cycles of amplification would be expected to contain one mutation attributable to the polymerase error (i.e. 0·2 mutations/VDJ). We found no mutations after sequencing six clones recovered from two independent amplifications of B1–8 hybridomas cells (V186.2, DFL16-1 and JH2). Thus we assume that mutations in excess of the theoretical average value (0·2 mutations/VDJ) have resulted from a somatic process. Mutation rates are based on scores of base substitutions in the VH sequences only. Shared mutations within a set of clonally related sequences (according to the complementarity-determining region (CDR) 3 region were counted as one mutational event.
Experimental design
Five groups of athymic (nu/nu) mice were immunized with NP-CGG/alum on day 0 (Table 1). Three days later, groups I–III received freshly isolated splenic CD4+ lymphocytes from normal, syngeneic donors. Group I represents the control response group for the experiment. The mice in groups II and III were injected with IgM and IgG1 anti-NP mAb, respectively, within 1–2 hr after the adoptive T-cell transfer (Table 1). Negative controls included immunized mice that did not receive any T cells with (group IV) and without (group V) passive IgG1 antibody.
Table 1.
Experimental protocol
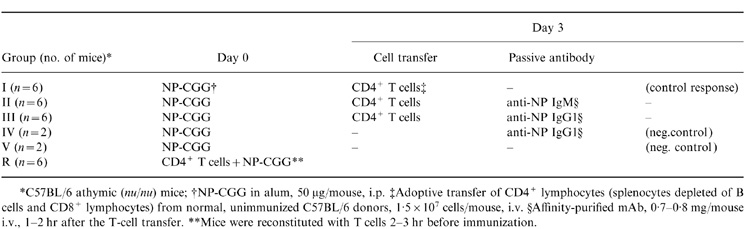 |
The responses of nu/nu mice with delayed T-cell help were compared with those of fully reconstituted mice (Table 1, group R) that received CD4+ T cells prior to the immunization with NP-CGG. The anti-NP response in group R is comparable with that of normal, euthymic animals5 and it has been included in this study as a reference response. Three mice from each group were killed and examined for splenic GC and AFC on days 8 and 12 after the immunization.
RESULTS
Antibody response and germinal center formation
The athymic mice that were supplemented with helper T cells 3 days after the immunization with NP-CGG (see the experimental design, Table 1, group I control) demonstrated a slow and diminished anti-NP response as expected. Serum anti-NP antibody of IgM, IgG1 and IgG3 isotypes became detectable by the 2nd week after the immunization and their titres, particularly for the IgG isotypes, were 1–2 log lower than those in the normal (R, reference) response group (Fig. 1, group I control versus group R). Likewise, there were no GC detectable in the spleens of these animals until day 12 after the immunization (Fig. 2).
Figure 1.

NP-specific serum antibody titres (reciprocal of end-point dilution) of IgM (a), IgG1 (b) and IgG3 (c) isotypes at various days after immunization with NP-CGG/alum. Symbols indicate mean values of six mice (days 4 and 8) and three mice (day 12) from the following groups (see Table 1): group I, control with antigen alone (○); group II, antigen+anti-NP IgM (▪); group III, antigen + anti-NP IgG (▴); and standard response to NP-CGG (○, dotted line).
Figure 2.

Germinal centre formation in athymic mice at day 8 and day 12 after immunization with NP-CGG/alum. Mice were reconstituted with T cells on day 3 and injected passively with anti-NP mAb as follows: None (group I), no antibody; IgM (group II), IgM; IgG1 (Group III), IgG1. GCs are expressed as per cent of PNA+ splenic follicles. Each column/point represents mean percentage value from three mice±SD.
Administration of anti-NP IgM (Table 1, group II), which generated passive IgM serum titres of ≡1:20 000 (Fig. 1a), had no demonstrable effect on the active (endogenous) IgG1 (Fig. 1b) or IgG3 (Fig. 1c) antibody responses and it influenced neither the kinetics nor magnitude of the GC formation (Fig. 2). However, the injection of anti-NP IgG1 (Table 1, group III) stimulated a robust GC response that was comparable to that in normal responder mice in its kinetics and magnitude (Fig. 2). The serum levels of passive IgG1 in these experimental mice mimicked the levels of actively produced anti-NP IgG1 in normal animals (Fig. 1b) whereas the endogenous IgM and IgG3 serum isotypes remained as low as they were in the group I control (Fig. 1a,c). Thus, it is apparent that passive administration of IgG1 antibody produced a dramatic effect on the GC formation without affecting the serum levels of endogenous antibodies of other isotypes.
It should be noted that the stimulation of GC response by passive IgG1 antibody was T-cell-dependent. Nude mice that were immunized with NP-CGG but did not receive CD4+ splenocytes (Table 1, groups IV + V) failed to develop GC even if they were treated with passive antibody (group IV) (data not shown).
VH gene repertoires in GC and AFC
The heavy chains of anti-NP antibodies in C57BL/6 mice can be encoded by at least 10–20 different, germline-encoded VH segments of the V186.2/V3 gene family.31 If these genes were used stochastically each should be expressed in ≤5–10% of NP-reactive B cells; a disproportional usage of a particular VH gene in >20% of cells may be considered as a dominant expression of that gene in the anti-NP response. We screened >500 transformants from each group for productive rearrangement of the V186.2 segment (see the Materials and Methods) and found that the GC in the control, NP-CGG-immunized mice were dominated by this gene (Table 2, group I). Most of the NP+GC in group I (12 GC out of 16) contained >20% of 186.2 B+ cells and, on average, 53% of GC B cells expressed the 186.2 VH gene. This repertoire dominance was comparable to that in the standard response (Table 2, group R, 51% 186.2+ B cells per GC) and consistent with previous studies from several laboratories including ours.15,16,20 However, mice that received NP-CGG plus passive IgG1 antibody demonstrated a dramatic loss of 186.2 gene dominance (Table 2, group III). Only four of 15 GC contained >20% of 186.2+ B cells, averaging 18% of all NP+ GC B cells, with the remaining 82% of cells expressing any one of the other members of the V186.2/V3 gene family. The identity of these VH genes from selected colonies was determined by DNA sequencing (see below).
Table 2.
VH gene repertoire of transformed colonies with VDJ segments recovered from the germinal centres and from the foci of antibody-forming cells
 |
Interestingly, the VH gene repertoire shift in mice with passive IgG1 antibody appeared to be restricted to the GC developmental pathway because it did not occur among the AFC. We screened the VH gene usage in NP-binding plasmacytes within the antibody foci in the same spleens and found little effect of passive IgG1 on their clonotypic repertoire (Table 2). This study compared the results in antibody-treated mice (group III) with the standard response (group R) because the foci of AFC in the control group I were less well developed. Seven out of nine antibody foci in group III mice were dominated by the 186.2+ clones that represented, on average, 65% of AFC in each focus. Similarly, in the standard response (group R) 11 out of 12 foci were dominated by the V186.2 gene with a mean of 80% 186.2+ AFC/focus.
Somatic hypermutation in rearranged VH genes
The DNA from randomly selected, 186.2+ and 186.2− colonies was amplified and sequenced. The VH segments from all colonies that hybridized with the 186.2-specific probe (total of 89 sequenced) corresponded to the V186.2 gene whereas the 186.2 probe-negative colonies (total of 112) yielded sequences corresponding to germline-encoded analogue genes: V23 (30% of sequences), V24.8 (24%), V165.1 (16%), V102, V671.5 (6–8% each), VC1H4, V593.3, V3 and V4M110 (2–3% each).
The control mice (group I) immunized with antigen only failed to develop GC on day 8 (Fig. 1) and the VH genes recovered from GC on day 12 were mostly unmutated (Table 3 and Fig. 3). The V186.2 and analogue VH gene segments in GC B cells from experimental mice that received antigen plus IgG1 (group III) contained point mutations that were analysed and compared with the mutation patterns in standard responder mice (group R). The average number of mutations per V186.2 gene in passive antibody recipient mice and responder mice increased, respectively, from, 0·8 and 1·2 on day 8 to 1·4 and 1·7 on day 12 (Fig. 3a). These values are within the range of mutation frequencies reported earlier.15,20 However, the patterns of mutations in these two groups were quite different (Table 3). The ratios of replacement (R) and silent (S) mutations in the standard response group were comparable to the ratios expected from random mutagenesis in the IgV genes; they ranged from 2:1 to 3:1 in both framework regions and CDR1+2 which is a typical GC B-cell response preceding clonal selection.17,32 In the immunized mice that received passive IgG1 antibody, however, the R:S ratios in CDR1+2 increased up to 11:1 (Table 3, group III) indicating that the immune complex formation had augmented and/or accelerated the selection of V186.2 mutant B cells.
Table 3.
Patterns of mutations
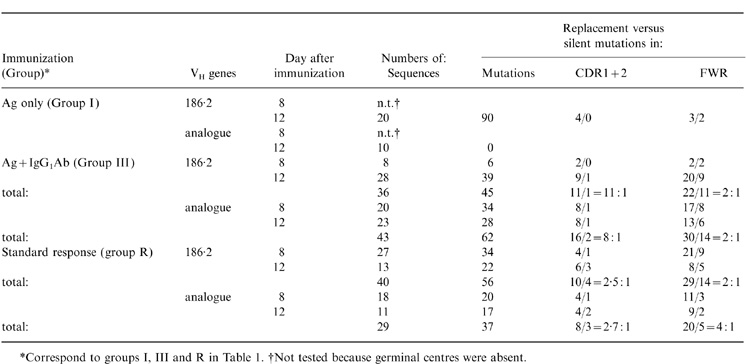 |
Figure 3.

Frequency of point mutations in V186.2 segments (a) and analogue V segments (b) recovered as VDJ rearrangements from germinal centre B cells from control mice (○), mice with passive IgG1 antibody (▴) and standard responder mice (○, broken line) at days 8 and 12 after immunization.
The analysis of the analogue genes is shown in Fig. 3(b). In the course of the standard response (group R), these genes continued to accumulate mutations from means of 1·1/VH on day 8 to 1·6/VH on day 12, which was only slightly lower than the mutation rates in the 186.2 gene (Fig. 3a). Likewise, the low R:S mutation ratios of 2–3 in both CDR and frame-work region (FWR) domains indicated that little, if any, selection of the analogue mutants occurred in these GC (Table 3). In contrast, the mice with immune complexes reached an average of 1·7 mutations/analogue V gene on day 8, a maximum level that did not increase any further (Fig. 3b). The mutation activity in individual GC varied greatly on both day 8 (0 mutations/VH to 4 mutations/VH) and day 12 (0–2·8 mutations/VH). Because of this variation, the apparent decline of average number of mutation from day 8 to day 12 was not significant (0·05<P<0·10). The high level of mutation activity in the analogue VH genes was accompanied by an increased R:S mutation ratios in the CDR (8:1, Table 3) indicating a clonal selection. The comparison of data in Fig. 3(a),(b) suggests that the passive IgG1 antibody exerts differential effects on the mutation process among various B-cell clonotypes in the GC.
DISCUSSION
Our experiments show that the deficient GC formation and somatic mutation in immunocompromised mice immunized with NP-CGG can be fully restored with passive administration of IgG1 antibody against the NP-hapten. The kinetics of GC response and mutation was nearly normalized despite a 3-day delay in T-cell help, suggesting that the passive antibody accelerated these processes. The treatment with passive IgG also produced a shift in the VH gene usage in GC B cells. While the GC in the control groups were occupied mostly by B cells with H chains encoded by the V186.2 gene segment, the GC in mice with passive IgG consisted of diverse cell populations expressing different, albeit related, germline segments of the V186.2/V3 (J558) gene family. These dramatic effects of passive IgG are not limited to the immune response of mice with diminished T-cell help that were used in the present study. Similar changes in the GC B-cell repertoire have been seen in normal animals immunized with complexes of NP-KLH with anti-NP IgG120 and, to a lesser extent in normal mice that received passive antibody within 2 days after immunization (unpublished data from our laboratory).
The GC response in our model system was enhanced with IgG1 antibody but not with IgM, and the VH repertoire shift occurred in the GC B cells and not in the B cells that differentiate into antibody-forming plasmacytes. Both these results are consistent with the earlier studies demonstrating a rapid and selective sequestration of the antigen–IgG antibody complexes in the cellular network of B-cell follicles.10,11 However, our results showing that the regulation of B-cell repertoire is restricted to antibody of IgG isotype should be interpreted with caution. Although it was clearly shown that administration of IgM failed to stimulate the GC response the VH repertoire of B cells in the rudimentary GC in these animals was not tested. It seems unlikely that the passive IgM could influence the B-cell mutation and selection without affecting the GC formation but such a possibility remains. Indeed, Kosco-Vilbois et al.33 have demonstrated an IgM deposition in GC by immunochemical staining. The in vivo effects of immune complexes may depend not only on the antibody isotype but also on antigen–antibody stoichiometry and antibody affinity. It should be noted that both IgG1 and IgM anti-NP mAb used in the present experiments were encoded by identical pairs of unmutated VH/VL genes and express a low affinity for NP, which may explain, in part, the selective enhancement of GC by passive IgG without an apparent feedback inhibition of the endogenous antibody response that was seen in earlier studies using passive antibody purified from immune sera.21–23 Preliminary experiments from our laboratory show that passive administration of high-affinity IgG1 anti-NP mAb is less effective in stimulating the GC formation but capable of inhibiting the antibody and AFC formation in mice immunized with NP-CGG (unpublished data).
Once in the follicle, the antigen–IgG complex may exert its effects on GC B cells directly and/or indirectly through the follicular dendritic cells (FDC). Stimulation of B cells with antigen–antibody complex is much more powerful compared to stimulation with the antigen alone. It has been shown that oligomerization of antigen with antibody lowers the B-cell receptor (BCR) affinity threshold for antigen uptake and presentation by B cells in vitro.34 The complexing of antigen with passive antibody in vivo may likewise increase the avidity of antigen–BCR ligation and strengthen the BCR-mediated signal. Moreover, the immune complex will initiate complement deposition and bridging of BCR with CD21 (the C receptor) on B cells thereby producing costimulation through CD19/CD81.35–37 Stimulation of B cells via this molecular complex appears to be a hundredfold more effective than stimulation with antigen alone.38 Both of these models provide a plausible mechanism for the enhancement of somatic mutation by passive IgG as well as for the shift in VH gene usage in GC B cells. It is thought that the dominance of V186.2 B+ cells in response of IgHb mice to NP is due to the superior germline affinity of their BCR for the hapten.39 The emergence of the diverse, presumably low-affinity clonotypes in mice with passive IgG would be a consequence of more robust stimulation of B cells by the immune complexes.
However, an important role in the process may belong to FDC, the specialized cells of GC carrying receptors for IgG.40 The uptake of antigen–IgG complexes through FcγRI and -III that carry tyrosine-based activation motifs41 may activate the FDC to express an array of costimulating ligands and cytokines that enhance the GC B-cell stimulation, somatic mutation and clonotypic diversity. The potential importance of FDC in this process has been highlighted by the recent experiments of Vora et al.42 on mice with genetically ablated FcγRI and -III showing that the passive follicular deposition of immune complexes in these animals failed to produce changes in the repertoire of anti-NP response.
The administration of IgG produced differential effects on the kinetics of mutations in different B-cell clonotypes. The mutations in the 186.2+ B cells increased steadily similarly to those in the normal responder mice, whereas the analogue B-cell clones appeared to accumulate mutations early (day 8) with no further increase (Fig. 3). A plausible explanation of this result could be found in the presumptive diversity of antigen affinities among these clones.39,40 Activation of somatic mutation requires stimulation through BCR, which is affinity-driven, and help from T cells.43,44 It is likely that the BCR- and TH-mediated signals must be finely balanced in order to provide an optimal stimulus for somatic mutation. Indeed, there is an indirect evident that various NP-reactive clonotypes differ in their TH-dependence for activation.30,45 In the present experiments, the T-cell help was delayed and the threshold for BCR signalling was lowered by the formation of an immune complex. As a result, B cells expressing certain analogue genes might have gained advantage in entering the GC and/or rapid activation of somatic mutation.
Our results support the hypothesis that the GC reaction and somatic mutation can be regulated by feedback from the early IgG-producing AFC. However, they do not formally prove a physiological role of the endogenous immune complexes that are formed in the course of antibody response. Vora et al. recently argued46 against such mechanism based on their observation that in the A strain of mice immunized with arsanil hapten, the GC developed even though the primary AFC response was very low. In fact, the GC formation in their system is also less robust and slower than in C57BL/6 mice immunized with NP, which is consistent with the proposal that the precisely timed sequence of isotype switch in AFC followed by GC maturation47 does represent an important regulatory mechanism in the life of GC B cells.
Acknowledgments
The authors wish to thank Dr Garnett Kelsoe, Duke University, for the IgG1-producing cell line and Ms June Green for expert preparation of the manuscript. This work was supported by NIH grants RO1AG14584 and PO1AG10207.
Abbreviations
- AFC
antibody-forming cells
- CDR
complementarity-determining region
- FDC
follicular dendritic cells
- GC
germinal centre(s)
- PNA
peanut agglutinin
References
- 1.Longevoort HL, Asofsky RM, Jacobson EB, devries T, Thorbecke GJ. Gamma globulin and antibody formation in vitro. II. Parallel observations on histologic changes and on antibody formation in the white and red pulp of the rabbit spleen during the primary response with special reference to the effect of endotoxin. J Immunol. 1963;90:60. [Google Scholar]
- 2.Cerny J, McAlack RF, Sajid MA, Fronton J, Freiedman M. Early accumulation of antibody plaque-forming cells in mouse spleen lacking a pre-existing immune background. J Immunol. 1971;106:1371. [PubMed] [Google Scholar]
- 3.Sterzl J. Factors determining the differentiation pathways of immunocompetent cells. Cold Spring Harb Symp Quant Biol. 1967;32:492. [Google Scholar]
- 4.Henry C, Jerne NK. Competition of 19S and 76S antigen receptors in the regulation of the primary immune response. J Exp Med. 1968;128:133. doi: 10.1084/jem.128.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller C, Stedra J, Kelsoe G, Cerny J. Facultative role of germinal centers and T cells in the somatic diversification of IgVH genes. J Exp Med. 1995;181:1319. doi: 10.1084/jem.181.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacob J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorbecke GJ. Focusing: the dilemma of interpreting sharp images on a blurred background. J Immunol. 1990;145:2779. [PubMed] [Google Scholar]
- 8.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 9.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centers. Nature. 1991;354:389. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 10.Mandel TE, Phipps RP, Abbot A, Tew JG. The follicular dendritic cell: long term antigen retention during immunity. Immunol Rev. 1980;53:29. doi: 10.1111/j.1600-065x.1980.tb01039.x. [DOI] [PubMed] [Google Scholar]
- 11.Klaus GGB, Humphrey JH, Kunkl A, Dongworth DW. The follicular dendritic cell: its role in antigen presentation in the generation of immunological memory. Immunol Rev. 1980;53:3. doi: 10.1111/j.1600-065x.1980.tb01038.x. [DOI] [PubMed] [Google Scholar]
- 12.Coico RF, Bhogal BS, Thorbecke GJ. Relationship of germinal centers in lymphoid tissue to immunologic memory. IV. Transfer of B cell memory with lymph node cells fractionated according to their receptor for peanut agglutinin. J Immunol. 1983;131:2254. [PubMed] [Google Scholar]
- 13.Kraal G, Weissman IL, Butcher EC. Germinal center cells: antigen specificity, heavy chain class expression and evidence of memory. In: Klaus GGB, editor. Microenvironments in the Lymphoid System. Vol. 186. New York: Plenum press; 1985. pp. 145–151. [DOI] [PubMed] [Google Scholar]
- 14.Weiss U, Rajewsky K. The repertoire of somatic antibody mutants accumulating in the memory compartment after primary immunization is restricted through affinity maturation and mirrors that expressed in the secondary response. J Exp Med. 1990;172:1681. doi: 10.1084/jem.172.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob J, Przylepa J, Miller C, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. III. The kinetics of V-region mutation and selection in germinal centers B cells. J Exp Med. 1993;178:1293. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McHeyzer-Williams MG, McLean MJ, Lalor PA, Nossal GJV. Antigen driven B cell differentiation in vivo. J Exp Med. 1993;178:295. doi: 10.1084/jem.178.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy NS, Malipiero UV, Lebecque SG, Gearhart PJ. Early onset of somatic mutation in immunoglobulin VH genes during the primary immune response. J Exp Med. 1989;169:2007. doi: 10.1084/jem.169.6.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laissue J, Cottier H, Hess MW, Stoner RD. Early and enhanced germinal center formation and antibody responses in mice after primary stimulation with antigen-isologous antibody complexes as compared with antigen alone. J Immunol. 1971;107:822. [PubMed] [Google Scholar]
- 19.Kunkl A, Klaus GGB. The generation of memory cell: IV Immunization with antigen-antibody complexes accelerates the development of B-memory cell, the formation of germinal centers and the maturation of antibody affinity in the second response. Immunology. 1981;43:371. [PMC free article] [PubMed] [Google Scholar]
- 20.Nie X, Basu S, Cerny J. Immunization with immune complex alters the repertoire of antigen-reactive B cells in the germinal centers. Eur J Immunol. 1997;27:3517. doi: 10.1002/eji.1830271253. [DOI] [PubMed] [Google Scholar]
- 21.Uhr JW, Möller G. Regulatory effect of antibody on the immune response. Adv Immunol. 1968;8:81. doi: 10.1016/s0065-2776(08)60465-4. [DOI] [PubMed] [Google Scholar]
- 22.Fitch FW. Selective suppression of immune response. Progr Allergy. 1975;19:195. [PubMed] [Google Scholar]
- 23.Voisin GA. Role of antibody classes in the regulatory facilitation reaction. Immunol Rev. 1980;49:3. doi: 10.1111/j.1600-065x.1980.tb00425.x. [DOI] [PubMed] [Google Scholar]
- 24.Jacob J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bothwell ALM, Paskind M, Reth M, Imanishi-Kari T, Rajewsky K, Baltimore D. Heavy chain variable region contribution to the NPb family of antibodies: Somatic mutation evident in a γ 2a variable region. Cell. 1981;24:625. doi: 10.1016/0092-8674(81)90089-1. [DOI] [PubMed] [Google Scholar]
- 26.Bothwell ALM, Paskind M, Reth M, Imanishi-Kari T, Rajewsky K, Baltimore D. Somatic variants of murine immunoglobulin lambda light chains. Nature (Lond) 1982;298:380. doi: 10.1038/298380a0. [DOI] [PubMed] [Google Scholar]
- 27.Toellner K-M, Luther SA, Sze DM-Y, et al. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. J Exp Med. 1998;187:1193. doi: 10.1084/jem.187.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinberger JZ, Green MI, Benacerraf B, Dorf ME. Hapten-specific T cell responses to 4-hydroxy-3-nitropheny acetyl. I. Genetic control of delayed-type hypersensitivity by V and I-A region genes. J Exp Med. 1979;149:1136. doi: 10.1084/jem.149.6.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen D, Simon T, Sablitsky F, Rajewsky K, Cumano A. Antibody engineering for the analysis of affinity maturation of an anti-hapten response. EMBO J. 1988;7:1995. doi: 10.1002/j.1460-2075.1988.tb03038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X, Stedra J, Cerny J. Relative contribution of T and B cells to hypermutation and selection of the antibody repertoire in germinal centers of aged mice. J Exp Med. 1996;183:959. doi: 10.1084/jem.183.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu H, Tarlington D, Muller W, Rajewsky K, Forster I. Most peripheral B cells in mice are ligand selected. J Exp Med. 1991;173:1357. doi: 10.1084/jem.173.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss U, Zoebelein R, Rajewsky K. Accumulation of somatic mutants in the B cell compartment after primary immunization with a T cell-dependent antigen. Eur J Immunol. 1992;22:511. doi: 10.1002/eji.1830220233. [DOI] [PubMed] [Google Scholar]
- 33.Kosco-Vilbois MH, Zentgraf HJ, Bonnefoy J-Y. To ‘B’ or not to ‘B’ a germinal center? Immunol Today. 1997;18:225. doi: 10.1016/s0167-5699(97)01048-7. [DOI] [PubMed] [Google Scholar]
- 34.Batista FD, Neuberger MS. Affinity dependence of the B cell response to antigen: a threshold, a ceiling and the importance of off-rate. Immunity. 1998;8:751. doi: 10.1016/s1074-7613(00)80580-4. [DOI] [PubMed] [Google Scholar]
- 35.Carter RH, Spycher MD, Ng YC, Hoffman R, Fearon DT. Synergetic interaction between complement receptor type 2 and membrane IgM on B lymphocytes. J Immunol. 1988;141:457. [PubMed] [Google Scholar]
- 36.Tsoko GC, Lambris JD, Finkelman FD, Anatassiov ED, June CH. Monovalent ligands of complement receptor 2 inhibit whereas polyvalent ligands enhance anti-Ig induced human B cell intracytoplasmic free calcium concentration. J Immunol. 1990;144:1640. [PubMed] [Google Scholar]
- 37.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105. [PubMed] [Google Scholar]
- 38.Dempsey PW, Allsion MED, Akkarajv S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 39.Dal Porto JM, Haberman AM, Schlomchik HJ, Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol. 1998;161:5381. [PubMed] [Google Scholar]
- 40.Schriever F, Nadler LM. The central role of follicular dendritic cells in lymphoid tissues. Adv Immunol. 1992;51:293. doi: 10.1016/s0065-2776(08)60489-7. [DOI] [PubMed] [Google Scholar]
- 41.Isakov N. ITIMs and ITAMs. Immunol Res. 1997;16:85. doi: 10.1007/BF02786325. [DOI] [PubMed] [Google Scholar]
- 42.Vora KA, Ravetch JV, Manser T. Amplified follicular immune complex deposition in mice lacking the Fc receptor chain does not alter maturation of the B cell response. J Immunol. 1997;159:2116. [PubMed] [Google Scholar]
- 43.Källborg E, Jainandunsing S, Gray D, Leanderson T. Somatic mutation of immunoglobulin V genes in vitro. Science. 1996;271:1285. doi: 10.1126/science.271.5253.1285. [DOI] [PubMed] [Google Scholar]
- 44.Decker DJ, Linton PJ, Zaharevitz S, Biery M, Gingeras TR, Klinman NR. Defining subsets of naive and memory B cells based on the ability of their progeny to somatically mutate in vitro. Immunity. 1995;2:195. doi: 10.1016/s1074-7613(95)80092-1. [DOI] [PubMed] [Google Scholar]
- 45.Maizels N, Bothwell A. The T-cell-independent immune response to the hapten NP uses a large repertoire of heavy chain genes. Cell. 1985;43:715. doi: 10.1016/0092-8674(85)90244-2. [DOI] [PubMed] [Google Scholar]
- 46.Vora KA, Tumas-Brundage KM, Manser T. A periarteriolar lymphoid sheath-associated B cell focus response is not observed during the development of the anti-arsenate germinal center response. J Immunol. 1998;160:728. [PubMed] [Google Scholar]
- 47.Peakman M-C, Maizels N. Localization of splenic B cells activated for switch recombination by in situ hybridization with Ig1 switch transcript and Rad51 Probes. J Immunol. 1998;161:4008. [PubMed] [Google Scholar]