

Abstract
The (New Zealand black (NZB) × New Zealand white (NZW))F1 (NZB/W) mouse strain spontaneously develops an autoimmune disease characterized by anti-dsDNA antibody production and glomerulonephritis. Although evidence suggests that production of pathogenic autoantibodies is T-cell dependent, the immunological defects that lead to activation of these autoreactive T cells are unknown. In particular, it has not been resolved whether autoreactive T cells become activated in these mice because of a generalized defect in T-cell tolerance induction. Previous work has demonstrated that thymic and peripheral tolerance to strongly deleting antigens are intact in NZB/W mice. In this study we investigate whether these mice possess a more subtle T-cell tolerance defect. To this end, we have produced NZB/W mice carrying a transgene encoding beef insulin (BI) which is expressed at levels close to the threshold for T-cell tolerance induction. In BALB/c mice this transgene produces a profound but incomplete state of BI-specific T-cell tolerance, mediated predominantly by clonal anergy. Comparison of BI-specific tolerance in NZB/W, major histocompatibility complex (MHC)-matched (BALB/c × NZW)F1, and BALB/c BI-transgenic mice clearly demonstrates that T-cell tolerance induction is normal in NZB/W mice. The data suggest that the loss of T-cell tolerance that ultimately supports nephritogenic autoantibody production in NZB/W mice does not result from a generalized defect in T-cell tolerance, and by extension likely results from aberrant activation of specific autoreactive T cells.
Introduction
The (New Zealand black (NZB) × New Zealand white (NZW))F1 (NZB/W) mouse spontaneously develops an autoimmune condition that is considered to be an excellent model of systemic lupus erythematosus (SLE). Autoimmunity in these mice is characterized by production of immunoglobulin G (IgG) autoantibodies with high affinity for dsDNA and nucleosomes resulting in a severe, rapidly progressive glomerulonephritis beginning at approximately 5 months of age.1 An extensive body of evidence indicates that production of these pathogenic autoantibodies is T-cell dependent. For example, pathogenic anti-dsDNA antibodies have the characteristics of an antigen-driven response.2 Congenitally athymic NZB/W nude mice fail to develop glomerulonephritis3 and administration of anti-CD4 monoclonal antibodies (mAb) to NZB/W mice significantly delays the onset of disease.4,5
Despite recent reports that pathogenic autoantibodies and nucleosomes may be recognized by T cells from these and related mouse strains,6,7 the nature of antigens recognized by the autoreactive T-cell population remains in dispute.8 Further, the immunological defect that leads to activation of these autoreactive T cells is unknown. In particular, it has not been resolved whether autoreactive T cells become activated in these mice because of a generalized defect in T-cell tolerance induction. Studies suggest that clonal deletion of autoreactive T cells in the thymus9,10 and exogenous superantigen stimulated T cells11 are normal in NZB/W mice. However, these studies use strongly deleting antigens and do not rule out the possibility that these mice have a more subtle T-cell tolerance defect. The observation that NZB and NZB/W mice are resistant to high zone tolerance induction following administration of soluble antigens,12,13 a mechanism of tolerance that is thought to be mediated by clonal anergy,14,15 is consistent with this possibility.
We recently examined T-cell tolerance in NZB mice by backcrossing a transgene encoding beef insulin (BI) onto the NZB background. In non-autoimmune BALB/c mice the levels of BI produced by the transgene are close to the threshold for T-cell tolerance induction16,17 and induce a profound but incomplete state of T-cell tolerance that is mediated predominantly by clonal anergy16 and does not require the presence of a thymus.18 Comparison of T-cell tolerance in NZB and BALB/c BI transgenic (BITg) mice clearly demonstrated that NZB T cells were at least as tolerant to BI as BALB/c T cells.19
Although NZB mice are autoimmune, producing anti-red blood cell (RBC), -lymphocyte, and -ssDNA antibodies, these mice do not produce the high affinity IgG anti-dsDNA antibodies associated with lupus nephritis in NZB/W mice.1 Further, studies show that both major histocompatibility complex (MHC) and background NZW genes contribute to the development of glomerulonephritis in NZB/W mice.20 In this study we examine the possibility that one of the roles of the NZW background genes is to alter T-cell tolerance induction leading to the generation of nephritogenic autoantibodies in NZB/W mice. To examine this question NZB BITg mice were crossed with NZW mice and T-cell tolerance to BI assessed. We show that BI-specific T-cell tolerance induction is normal in these mice, suggesting that the break in T-cell tolerance that leads to activation of the T cells that provide support for nephritogenic autoantibody production in NZB/W mice probably results from abnormal activation of T cells specific for a subset of autoantigens rather than a generalized defect in T-cell tolerance induction.
Materials and methods
Mice
BITg BALB/c mice were produced, as previously described.16 The transgene was produced by site-directed mutagenesis of a human insulin (HI) genomic clone. Although BI differs from HI at A-chain residues 8 and 10, as well as the terminal B-chain residue, only the A chain residues were mutated because B30 is not contained within the antigenic epitope. Consistent with this observation, the transgene induces tolerance that is specific for BI and for simplicity is referred to as a BI transgene.16 Approximately 10–60% of the insulin produced in BITg mice is derived from the BI transgene, which is under control of the human insulin promoter and appropriately regulated under physiological conditions. BITg NZB/W, or (BALB/c × NZW)F1, mice were generated by breeding BITg NZB (N7 backcross generation), or BALB/c, female mice with male NZW mice, respectively. Offspring were screened for the presence of the transgene by polymerase chain reaction (PCR) amplification of tail DNA with nested HI-specific primers.16 All mice were immunized at 4 months of age.
Antibody production
Serum levels of total IgM were measured by sandwich enzyme-linked immunosorbent assay (ELISA)19 with the amount of IgM calculated from a standard curve using a purified IgM mAb of known concentration (provided by Dr M. Shulman, Toronto, Canada). IgM and IgG anti-ssDNA antibodies were measured by ELISA, as described.19 Serum samples were diluted 1 : 100 in phosphate-buffered saline (PBS)/bovine serum albumin (BSA) and ssDNA-specific antibodies were detected using as a secondary reagent goat anti-mouse IgM or IgG specific antibody (CALTAG, San Francisco, CA).
BI-specific antibody production was measured following injection of 50 µg monocomponent BI (Novo-Nordisk Pharmaceuticals, Copenhagen, Denmark), emulsified in a 1 : 1 ratio with complete Freund’s adjuvant (CFA; Difco Laboratories, Detroit, MI), in a single foot pad (f.p.). Antibody production was measured 14 days later by an ELISA using BI-coated plates as described.16 Alkaline phosphatase-conjugated goat anti-mouse IgG1 and IgG2a were used as developing antibodies (CALTAG).
T-cell proliferation and lymphokine assays
Mice were injected with BI in a single f.p. as described above, killed 14 days later, and single-cell suspensions were prepared from the draining popliteal lymph node or spleen (erythrocytes lysed prior to use). BI-specific T-cell proliferation was measured by culturing 5 × 105 cells/well for 3 days in medium containing 0·5% normal mouse serum together with various concentrations of BI. Proliferation was measured by [3H]thymidine incorporation after an 18-hr pulse with 1 µCi/well.
To assay for antigen-driven interleukin-2 (IL-2) or IL-4 production, BI-primed lymph node or splenic cells were cultured, as above, and supernatants harvested at 48 and 72 hr, respectively. IL-2 was measured in the supernatant by a standard bioassay using the IL-2-dependent cell line, CTL.L. Anti-mouse IL-4 mAb (11B11) was added to block proliferation in response to IL-4. Proliferation induced by 100 U IL-4 was completely inhibited by the antibody. For quantitation of IL-4, proliferation of the IL-4-dependent cell line CT.4S was measured following incubation with supernatant in the presence of S4B6, an anti-IL-2 mAb. This antibody completely inhibits proliferation induced by up to 10 U IL-2. For both IL-2 and IL-4, cytokine concentration (units/ml) was calculated from a log–log plot of cytokine-specific proliferation of the relevant cell line versus serial dilutions of a recombinant cytokine preparation (Genzyme, Cambridge, MA).
Interferon-γ (IFN-γ) and IL-10 levels in tissue culture supernatants were measured by ELISA at 48 and 72 hr, respectively. Paired antibodies were purchased from PharMingen, San Diego, CA: purified rat anti-mouse capture mAb, clone JES5-2A5 for IL-10 and clone R4-6A2 for IFN-γ; and biotinylated rat anti-mouse detection mAb, clone SXC-1 for IL-10 and clone XMG1.2 for IFN-γ. Assays were performed as per manufacturer’s recommendations. The concentration of cytokine in each supernatant was calculated from a log–log plot of absorbance versus concentration of recombinant cytokine preparation (Genzyme).
The limit of detection of these assays were: IL-2 0·01 U/ml, IL-4 0·3 U/ml, IFN-γ 45 pg/ml, and IL-10 20 pg/ml.
Statistical analyses
P-values for comparison of differences in cytokine production between the groups of mice were calculated using the Mann–Whitney test. Fisher’s exact test was used to compare the proportion of mice producing detectable levels of cytokine between groups.
Results
BITg NZB/W mice have serological abnormalities consistent with an autoimmune phenotype
We have previously shown that NZB N5 and N6 backcross BITg mice have immunological abnormalities, including increased serum levels of total IgM and IgM anti-ssDNA antibodies that are consistent with the autoimmune phenotype observed in NZB control mice.19 In this experiment NZB N7 backcross BITg mice were crossed with NZW mice. As shown in Fig. 1,4-month-old backcrossed NZB/W mice (BITg and non-Tg analysed together) had significantly increased levels of IgM (3·42 ± 1·37, NZB/W; 1·39 ± 0·39, BALB/c; 1·18 ± 0·31 (BALB/c × NZW)F1 (BALB/W); both P < 0·0001), IgM anti-ssDNA antibodies (0·240 ± 0·156, NZB/W; 0·162 ± 0·035, BALB/c, 0·111 ± 0·030, BALB/W; P = 0·0043 and < 0·0001, respectively), and IgG anti-ssDNA antibodies (0·142 ± 0·197, NZB/W; 0·036 ± 0·032, BALB/c; 0·032 ± 0·026, BALB/W; both P < 0·0001) relative to nonautoimmune BALB/c and (BALB/c × NZW)F1 (BALB/W) mice. The levels of IgM and IgG anti-ssDNA antibodies observed in backcrossed NZB/W mice are similar to those observed in age-matched control NZB/W mice (n = 17; IgM anti-ssDNA, 0·169 ± 0·044; IgG anti-ssDNA, 0·107 ± 0·136; both P > 0·05).
Figure 1.
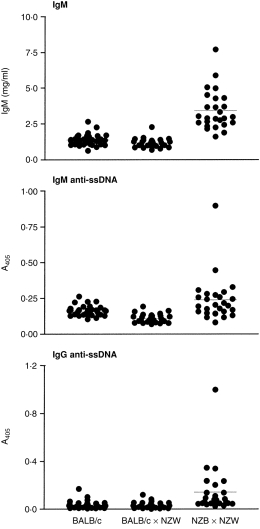
NZB/W BI-transgenic mice have serological abnormalities consistent with an autoimmune phenotype. Serum levels of total IgM, IgM anti-ssDNA antibodies, and IgG anti-ssDNA antibodies were determined by ELISA (1 : 100 dilution serum). Each circle represents an individual mouse. Horizontal lines give the mean for each group.
T-cell tolerance to BI is comparable in BALB/c, BALB/W, and NZB/W BITg mice
T-cell tolerance induction was investigated by comparing T-cell function between autoimmune NZB/W mice and normal BALB/c and BALB/W (MHC-matched) control mice in the presence or absence of the BI transgene. Mice were immunized in a single f.p. with 50 µg BI emulsified in CFA. Splenocytes and draining popliteal lymph node cells were isolated 14 days later and cultured in vitro together with various concentrations of BI. As shown in Fig. 2, the proliferative response to BI, while detectable, was dramatically reduced in BITg mice. The magnitude of this reduction was comparable for autoimmune NZB/W mice and both normal mouse strains. We have previously shown that this reduced proliferation reflects tolerance in the CD4+ T-cell subset and that addition of CD8+ BITg T cells to non-Tg CD4+ T cells has no effect on BI-specific proliferation.19
Figure 2.
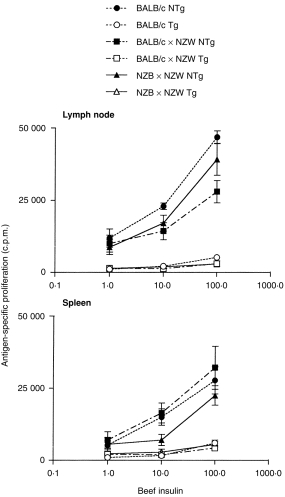
BI-specific T-cell proliferation is equivalently reduced for BITg T cells from normal and autoimmune NZB/W mice. Popliteal lymph node cells, pooled from three to four mice primed 14 days earlier, were cultured for 3 days with increasing concentrations of BI and pulsed overnight with [3H]TdR. Shown are the means of triplicate determinations. Results are representative of three independent experiments.
We next sought to determine whether both T helper 1 (Th1)- and Th2-like T-cell functions had been rendered similarly tolerant in BITg NZB/W mice. Mice were immunized, as above, and antigen-driven secretion of IL-2 and IFN-γ, or IL-4 and IL-10, by BI-primed T cells measured to assess tolerance in Th1 and Th2 cell subsets, respectively. The results for BI-primed lymph node T cells and splenocytes are shown in Fig. 3(a, b), respectively.
Figure 3.
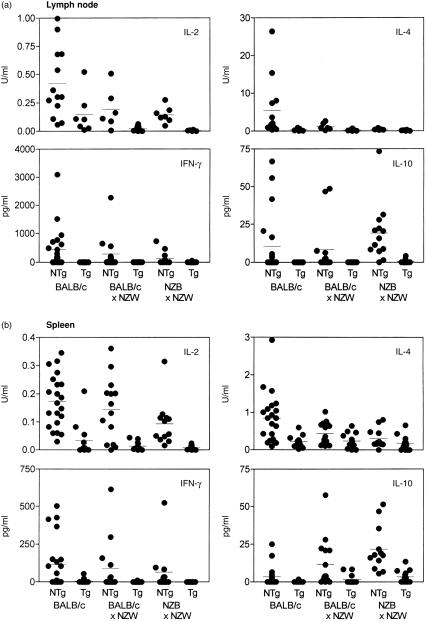
Antigen-driven cytokine production by BI-primed lymph node (a) or splenic (b) T cells is equivalently reduced in normal and autoimmune NZB/W mice. Cells were isolated 14 days after immunization and cultured with BI (100 µg/ml) or media alone. Results shown represent BI-specific cytokine secretion with background cytokine secretion, in the absence of BI, subtracted. Each circle represents the determination for one mouse with horizontal lines showing the mean for each group (Tg, BI transgenic; NTg, non-transgenic littermate control). Cytokine secretion was determined by measurement of cytokine levels in cell supernatants after incubation for 48 hr, for IL-2 and IFN-γ, or 72 hr, for IL-4 and IL-10 (see Materials and methods). All assays were performed in triplicate.
The pattern of cytokines produced by non-Tg lymph node T cells varied somewhat between the mouse strains (Fig. 3a), with NZB/W mice producing significantly more IL-10 than both non-autoimmune mouse strains (BALB/c, P = 0·011; BALB/W, P = 0·012), and less IL-2 and IL-4 than BALB/c mice (P = 0·039 and 0·002, respectively). Despite these different patterns of cytokine secretion, antigen-stimulated BITg T cells from all three mouse strains produced significantly and comparably reduced amounts of every cytokine tested (all P < 0·05) indicating that both Th1- and Th2-like cell functions had been similarly inhibited in the normal and autoimmune mouse strains.
These results were confirmed by analysis of cytokine production by splenic T cells. With the exception of IL-10, supernatants from antigen-stimulated splenic T cells contained lower levels of cytokines than those from lymph node T cells. Nevertheless, similar to lymph node T cells, splenic T cells from non-Tg NZB/W mice produced significantly reduced amounts of IL-2 and IL-4 in response to antigenic stimulation compared to BALB/c mice (P = 0·0093 and 0·0042, respectively) and increased amounts of IL-10 compared to BALB/c (P < 0·0001) and BALB/W (P = 0·048) mice. BALB/W mice demonstrated a cytokine profile intermediate between those of BALB/c and NZB/W mice, producing less IL-4 (P = 0·029) and more IL-10 (P = 0·0064) than BALB/c mice. As observed for lymph node T cells, antigen-stimulated splenic T cells from BITg mice of all three mouse strains demonstrated reduced cytokine secretion of both Th1 and Th2 cytokines relative to their non-transgenic counterparts. This achieved statistical significance in all three mouse strains only for IL-2 (all P < 0·005) and IL-10 (all P < 0·05). IL-4 secretion by splenocytes from BITg mice was significantly reduced compared to those from non-Tg mice only for BALB/c mice (P = 0·0009), while IFN-γ secretion was significantly reduced for both BALB/c (P = 0·012) and NZB/W mice (P = 0·0052). However, the observation that IL-4 and IFN-γ secretion by splenocytes from NZB/W and non-autoimmune BALB/W mice from non-Tg or BITg mice is comparable suggests that a similar state of BI-specific T-cell tolerance is present in the two strains of mice.
T-cell support for autoantibody production following a strong immunogenic stimulus does not differ between normal and autoimmune mice
Although BI transgenic BALB/c mice are tolerant to BI we have previously demonstrated that provision of these mice with a strong immunogenic stimulus, such as f.p. immunization with BI emulsified in CFA, can overcome tolerance, to a limited extent, resulting in BI-specific autoantibody production.16 Support for BI-specific autoantibody production results from activation of low-affinity T cells that appear to have escaped tolerance induction.16 Based upon these results, we reasoned that a subtle T-cell tolerance defect in NZB/W mice might alter T-cell support for BI-specific autoantibody production either quantitatively or qualitatively from that for non-autoimmune BITg mice. We therefore immunized mice, as outlined above, and measured the serum levels of IgG1 and IgG2a BI-specific antibodies 14 days later.
Results from a representative experiment comparing BI-specific antibody production in BALB/c, BALB/W, and NZB/W mice are shown in Fig. 4(a). While IgG1 and IgG2a BI-specific antibody production was decreased only approximately 10-fold in BITg BALB/c mice compared to non-Tg controls, BI-specific antibody production was decreased 300–1000-fold in BITg BALB/W and NZB/W mice. Figure 4(b) shows pooled results from multiple experiments examining BI-specific antibody production in BALB/W and NZB/W mice. BITg NZB/W and BALB/W mice produced significantly lower levels of IgG1 and IgG2a BI-specific antibodies than non-Tg mice (all P < 0·0001). Although non-Tg NZB/W mice have higher levels of IgG2a BI-specific antibodies following immunization than non-Tg BALB/W mice (P = 0·018), the levels of IgG2a BI-specific antibodies in BITg mice do not differ (P = 0·82). Thus, tolerance is not more easily broken in NZB/W mice than non-autoimmune mice.
Figure 4.
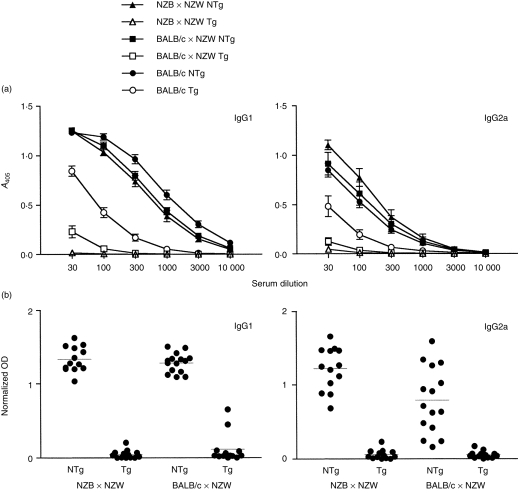
Support of BI-specific antibody production is normal in BITg NZB/W mice. Mice were immunized in a single f.p. with 50 µg BI emulsified in CFA and bled 14 days later. (a) Serial dilutions of serum were analysed for BI-specific IgG1 or IgG2a antibodies by ELISA. The ordinate axes are serial dilution of serum; antibody titres are means from three to eight mice per group. Error bars denote SE. (b) Pooled results from three independent experiments for BI-specific IgG1 or IgG2a antibodies measured at a serum dilution of 1 : 100. Data have been normalized to a standard hyperimmune sera. Each circle represents the determination for a single mouse with horizontal lines showing the mean for each group (NTg, non-transgenic littermates; Tg, BI transgenic; 13–15 mice per group).
Discussion
The current study was undertaken to determine whether NZB/W mice have a generalized defect in T-cell tolerance, which could lead to aberrant activation of autoreactive T cells. We show unequivocally that BI-specific tolerance induction is normal in BITg NZB/W mice. Tolerance to BI in this system is mediated entirely by T-cell tolerance because the levels of BI produced in BITg mice are below those of endogenous mouse insulin,16 and physiological levels of insulin are insufficient to induce tolerance in the B-cell subset.21,22 Furthermore, the low serum concentrations of BI in these mice (< 2 ng/ml) are close to the threshold required for T-cell tolerance induction.16,17 Consequently, the observation that BI-specific T-cell function is similarly reduced in BITg NZB/W and non-autoimmune control mice provides rigorous demonstration that T-cell tolerance mechanisms are intact in NZB/W mice.
Our study does not specifically address the mechanism of tolerance in NZB/W mice. In BALB/c mice T-cell tolerance to BI is mediated predominantly by clonal anergy.16 The BI-specific T-cell response in NZB/W mice is also partially H-2d restricted (data not shown) suggesting that tolerance in these mice is likely to be mediated, at least in part, by a similar mechanism. Nevertheless, the possibility that BI-specific T-cell tolerance results from clonal deletion rather than clonal anergy in BALB/W and NZB/W mice cannot be completely excluded. However, the extent of T-cell tolerance is dependent on the strength of T-cell activation, with stronger signals leading to deletion and weaker signals leading to anergy.23,24 Consequently, this would predict that BALB/W and NZB/W mice are more susceptible rather than less susceptible to T-cell tolerance induction than BALB/c mice.
Given the evidence for normal T-cell tolerance induction in BALB/W and NZB/W mice it is unlikely that NZW genes affect T-cell tolerance induction and thereby contribute to activation of the autoreactive T cells that in turn provide support for nephritogenic autoantibody production in NZB/W mice. Instead we propose that activation of autoreactive T cells in NZB/W mice results from abnormal triggering of T cells by aberrant antigen presenting cell (APC)-derived signals. In this connection, we have recently found that young NZB and NZB/W mice have a dramatically increased proportion of polyclonally activated B cells expressing increased levels of costimulatory molecules, particularly B7.1, which may be sufficient to activate naive T cells.25 Furthermore, a genetic region showing significant linkage with up-regulation of costimulatory molecules in NZB mice encompasses Nba2, a locus previously linked to development of nephritis.20,26 Taken together, the data suggest that activation of autoreactive T cells in NZB/W mice may result from altered antigen presentation rather than intrinsic defective T-cell tolerance mechanisms.
Acknowledgments
The authors would like to thank Dr R. Inman and Dr F. Tsui for critical review of the manuscript, and Novo-Nordisk Pharmaceuticals for supplying beef insulin. This work was supported by a grant from The Arthritis Society of Canada (J.W.). J.W. was the recipient of a Research Scholarship from The Arthritis Society.
Abbreviations
- SLE
systemic lupus erythematosus
- NZB
New Zealand black
- NZW
New Zealand white
- NZB/W
(NZB × NZW)F1
- BALB/W
(BALB/c × NZW)F1
- BI
beef insulin
- HI
human insulin
- BITg
BI transgenic
- non-Tg
non-transgenic
- PCR
polymerase chain reaction
- f.p.
foot pad
References
- 1.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 2.Diamond B, Katz JB, Paul E, Aranow C, Lustgarten D, Scharff MD. The role of somatic mutation in the pathogenic anti-DNA response. Ann Rev Immunol. 1992;10:731. doi: 10.1146/annurev.iy.10.040192.003503. [DOI] [PubMed] [Google Scholar]
- 3.Mihara M, Ohsugi Y, Saito K, et al. Immunologic bnormality in NZB/WF1 mice. Thymus-independent occurrence of B cell abnormality and requirement for T cells in the development of autoimmune disease, as evidenced by an analysis of the athymic nude individuals. J Immunol. 1988;141:85. [PubMed] [Google Scholar]
- 4.Wofsy D, Seaman WE. Successful treatment of autoimmunity in NZB/NZW F1 mice with monoclonal antibody to L3T4. J Exp Med. 1985;161:378. doi: 10.1084/jem.161.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connolly K, Roubinian JR, Wofsy D. Development of murine lupus in CD4-depleted NZB/NZW mice. Sustained inhibition of residual CD4+ T cells is required to suppress autoimmunity. J Immunol. 1992;149:3083. [PubMed] [Google Scholar]
- 6.Ebling FM, Tsao BP, Singh RR, Sercarz E, Hahn BH. A peptide derived from an autoantibody can stimulate T cells in the (NZB × NZW) F1 mouse model of systemic lupus erythematosus. Arthritis Rheum. 1993;36:355. doi: 10.1002/art.1780360311. [DOI] [PubMed] [Google Scholar]
- 7.Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: a major immunogen for pathogenic autoantibody-inducing T cells of lupus. J Exp Med. 1993;177:1367. doi: 10.1084/jem.177.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozzo SJ, Drake CG, Chiang BL, Gershwin ME, Kotzin BL. Evidence for polyclonal T cell activation in murine models of systemic lupus erythematosus. J Immunol. 1994;153:1340. [PubMed] [Google Scholar]
- 9.Kotzin BL, Kappler JW, Marrack PC, Herron LR. T cell tolerance to self antigens in New Zealand hybrid mice with lupus-like disease. J Immunol. 1989;143:89. [PubMed] [Google Scholar]
- 10.Theofilopoulos AN, Singer PA, Kofler R, Kono DH, Duchosal MA, Balderas RS. B and T cell antigen receptor repertoires in lupus/arthritis murine models. Springer Semin Immunopathol. 1989;11:335. doi: 10.1007/BF00197311. [DOI] [PubMed] [Google Scholar]
- 11.Scott DE, Kisch WJ, Steinberg AD. Studies of T cell deletion and T cell anergy following in vivo administration of SEB to normal and lupus-prone mice. J Immunol. 1993;150:664. [PubMed] [Google Scholar]
- 12.Staples PJ, Steinberg AD, Talal N. Induction of immunologic tolerance in older New Zealand mice repopulated with young spleen, bone marrow, and thymus. J Exp Med. 1970;131:1223. doi: 10.1084/jem.131.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskin CA, Taurog JD, Smathers PA, Steinberg AD. Studies of defective tolerance in murine lupus. J Immunol. 1981;127:1743. [PubMed] [Google Scholar]
- 14.Eynon EE, Parker DC. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J Exp Med. 1992;175:131. doi: 10.1084/jem.175.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romball CG, Weigle WO. In vivo induction of tolerance in murine CD4+ cell subsets. J Exp Med. 1993;178:1637. doi: 10.1084/jem.178.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poplonski L, Vukusic B, Pawling J, et al. Tolerance is overcome in beef insulin-transgenic mice by activation of low-affinity autoreactive T cells. Eur J Immunol. 1996;26:601. doi: 10.1002/eji.1830260315. [DOI] [PubMed] [Google Scholar]
- 17.Cabaniols JP, Cibotti R, Kourilsky P, Kosmatopoulos K, Kanellopoulos JM. Dose-dependent T cell tolerance to an immunodominant self peptide. Eur J Immunol. 1994;24:1743. doi: 10.1002/eji.1830240804. [DOI] [PubMed] [Google Scholar]
- 18.Teng Y-T, Gorczynski RM, Iwasaki S, Williams DB, Hozumi N. Evidence for Th2 cell-mediated suppression of antibody responses in transgenic, beef insulin-tolerant mice. Eur J Immunol. 1995;25:2522. doi: 10.1002/eji.1830250917. [DOI] [PubMed] [Google Scholar]
- 19.Wither J, Vukusic B. Autoimmunity develops in lupus-prone NZB mice despite normal T cell tolerance. J Immunol. 1998;161:4555. [PubMed] [Google Scholar]
- 20.Vyse TJ, Kotzin BL. Genetic susceptibility to systemic lupus erythematosus. Ann Rev Immunol. 1998;16:261. doi: 10.1146/annurev.immunol.16.1.261. [DOI] [PubMed] [Google Scholar]
- 21.Adelstein S, Pritchard-briscoe H, Anderson TA, et al. Induction of self-tolerance in T cells but not B cells of transgenic mice expressing little self antigen. Science. 1991;251:1223. doi: 10.1126/science.1900950. [DOI] [PubMed] [Google Scholar]
- 22.Whiteley PJ, Poindexter NJ, Landon C, Kapp JA. A peripheral mechanism preserves self-tolerance to a secreted protein in transgenic mice. J Immunol. 1990;145:1376. [PubMed] [Google Scholar]
- 23.Girgis L, Davis MM, De St Groth BF. The avidity spectrum of T cell receptor interactions accounts for T cell anergy in a double transgenic model. J Exp Med. 1999;189:265. doi: 10.1084/jem.189.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams CB, Allen PM. The study of self-tolerance using murine haemoglobin as a model antigen. Novartis Found Symp. 1998;215:41. doi: 10.1002/9780470515525.ch4. [DOI] [PubMed] [Google Scholar]
- 25.Wither J, Roy V, Brennan L. Activated B cells express increased levels of costimulatory molecules in young autoimmune NZB and (NZB × NZW)F1 mice. Clin Immunol. 2000;94:51. doi: 10.1006/clim.1999.4806. [DOI] [PubMed] [Google Scholar]
- 26.Wither J, Paterson A, Vukusic B. The expanded population of B cells expressing upregulated costimulatory molecules shows linkage to Nba2. Eur J Immunol. 2000;30:356. doi: 10.1002/1521-4141(200002)30:2<356::AID-IMMU356>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]