

Abstract
Identification of unique features of cancer cells is important for defining specific and efficient therapeutic targets. Mutant p53 is present in nearly half of all cancer cases, forming a promising target for pharmacological reactivation. In addition to being defective for the tumor-suppressor function, mutant p53 contributes to malignancy by blocking a p53 family member p73. Here, we describe a small-molecule RETRA that activates a set of p53-regulated genes and specifically suppresses mutant p53-bearing tumor cells in vitro and in mouse xenografts. Although the effect is strictly limited to the cells expressing mutant p53, it is abrogated by inhibition with RNAi to p73. Treatment of mutant p53-expressing cancer cells with RETRA results in a substantial increase in the expression level of p73, and a release of p73 from the blocking complex with mutant p53, which produces tumor-suppressor effects similar to the functional reactivation of p53. RETRA is active against tumor cells expressing a variety of p53 mutants and does not affect normal cells. The results validate the mutant p53–p73 complex as a promising and highly specific potential target for cancer therapy.
Keywords: cancer therapy, p73 family, tumor suppressors
Functions of the p53 tumor suppressor are commonly lost in cancer, which unleashes accelerated selection of most aggressively growing cells. Meanwhile, tumor cells usually acquire increased sensitivity to reintroduced wild-type p53, suggesting a strategy for anticancer therapy Because ≈50% of tumors express high levels of mutant p53 proteins, approaches have been developed aiming reactivation of mutant p53 by small-molecule drugs to induce selective apoptosis of cancer cells. However, there are >2,000 different types of mutant p53 proteins in cancer (1), which imposes a serious problem for the development of versatile p53-reactivating drugs. The p53 family members p63 and p73 add to the tumor-suppressor function of p53, because they share many common transcriptional targets with p53 (2) and their activities contribute to the p53-dependent apoptosis and chemosensitivity of cancer cells (3, 4). Each of these genes specifies diverse isoforms, which include longer, transcriptionally active (TA) and shorter, delta N species. The TA isoforms act in a manner similar to p53, responding to certain stresses by activation of genes involved in growth arrest and apoptosis (5). The DN isoforms generally counteract the activities of TA isoforms and of p53, either by competing for common DNA-binding elements or by blocking the TAp63 and TAp73 isoforms through direct protein–protein interactions (6). Although the p73 isoforms can oligomerize with each other and with the p63 isoforms, neither can form heterooligomers with the wild-type p53 (7, 8). Still, a number of p53 mutant proteins can associate with p63 and p73, blocking their transcriptional activity and thus contributing to the malignant phenotype of cancer cells (9–11). Activation of p73 in human cancer can produce substantial cytotoxic effect even in the cells lacking p53 (12–16), which allows considering p73 as a separate anticancer drug target. We decided to search for small molecules that release suppressor activities of the p53 family members blocked by mutant p53. Unlike previously reported drugs that activate p73 independently on the p53 status (13), the quest compounds would act with high specificity, affecting only mutant p53-expressing cancer cells. We describe here a compound that specifically suppresses cancer cells through the mutant p53-dependent activation of p73. The compound could represent a type of specific anticancer drug.
Results
Identification of Small-Molecule RETRA.
Transcriptional activity is considered to be the major determinant of the p53 tumor-suppressor function. We decided to search for small molecules that reactivate the transcriptional activity of p53 in a mutant p53-bearing cancer cell line. We introduced by lentiviral transfer into epidermal carcinoma cell line A431, which is bearing His-273 p53 mutant, a reporter construct LC5 carrying a cassette for expression of β-galactosidase under control of a minimal CMV promoter coupled to multiple p53-binding elements (17) [supporting information (SI) Fig. S1]. A control reporter construct LC0 that does not contain the p53-binding elements was used to determine the background level of reporter activity. We tested the responsiveness of the reporter cell lines A431/LC5 and A431/LC0 to reestablished p53 activity after infection with increasing amounts of lentivirus construct expressing wild-type p53. There was a dose-dependent induction of the β-galactosidase reporter in the A431/LC5 and virtually no effect in the A431/LC0 cells (Fig. S2).
The reporter cell line A431/LC5 was used as a readout in a high-throughput screening of a chemical library, which contained 46,250 individual drug-like compounds. The reporter cells seeded to the 386-well plates were incubated overnight with individual compounds, and the reaction was measured in the o-nitrophenyl β-d-galactoside (ONPG) test with a plate reader. Compounds inducing >2-fold increase in the reporter expression were considered as initial hits. Further assays have confirmed a dose-dependent induction of the reporter for 22 of the 56 initial hits. None of the selected compounds induced the reporter in the control cell line A431/LC0, suggesting the importance of the p53-binding elements for reporter activation.
To identify the compounds that activate the reporter exclusively in the mutant p53-expressing cell lines and to get rid of those capable of inducing the reporter in p53-neagative and wild-type p53-expressing cells, we applied the remaining hits to several filtering assays that measure the reporter induction in control cell lines. The panel of the control reporter cell lines was constructed by the introduction of the LC5 lentiviral reporter construct into several human cell lines with different status of the p53 gene. None of the compounds induced the reporter in p53-negative cell lines H1299/LC5, Saos2/LC5, and PC3/LC5. However, most of the compounds were found to be more or less active in the reporter induction when applied to the wild-type p53-expressing HEFs/LC5 and A549/LC5 cells. We discarded the compounds from further analysis, because, apparently, they are capable of inducing activation of a nonspecific stress response leading to activation of p53. Only five compounds induced the p53-dependent reporter selectively in the mutant p53-expressing A431/LC5 cell line and not in the wild-type p53-positive A549/LC5 cells (Fig. S2). One of the five compounds (#5493343) displayed reasonably high activity and low IC50 value (4 μM). We have chosen the compound, which we named RETRA for (reactivation of transcriptional reporter activity). RETRA is 2-(4,5-dihydro-1,3-thiazol-2-ylthio)-1-(3,4-dihydroxyphenyl)ethanone hydrobromide for further studies (Fig. 1a). Two similar but less-active compounds were among the initial hits during the screening (Fig. S3), suggesting that RETRA represents a class of active compounds.
Fig. 1.

Identification of RETRA. (a) Structural formula of RETRA [2-(4,5-dihydro-1,3-thiazol-2-ylthio)-1-(3,4-dihydroxyphenyl)ethanone hydrobromide]. (b) Induction of β-galactosidase reporter in A431/LC5 cells after 14-h incubation with 4 μM RETRA, 0.05% DMSO. The upper control segment corresponds to the treatment with diluted DMSO (0.05%). Staining with X-Gal. (c) Induction of β-galactosidase reporter in cell lines expressing mutant p53 Arg273His (A431, SW480, HT29, SW620), Arg248Trp (SW837), Arg280Lys (MDA-MB-231), and Gly266Glu (MDA-MB-435) and treated at IC50 (4 μM) with RETRA for 14 h (ONPG reaction). (d) Induction of β-galactosidase reporter in the p53-null cell lines MDA041, H1299, Saos-2, and PC3 and in the wild-type p53-expressing cells HeLa, A549, and HEFs, treated as above.
RETRA Activates p53-Dependent Transcriptional Reporter in Mutant p53-Bearing Tumor Cell Lines.
To reveal the spectrum of cells susceptible to RETRA, we measured the induction of p53-dependent transcriptional reporter in a cell line panel. In the tested cancer cell lines bearing various types of mutant p53 (His-273, Trp-248, Glu-266, Lys-280), treatment with IC50 concentration of RETRA for 12 h resulted in a variable but consistent induction of the reporter, although there was no increase in β-galactosidase in the wild-type p53-expressing (HeLa, A549, and HEFs) or in the p53-null (MDA041, H1299, Saos2, and PC3) cell lines (Fig. 1 b–d). None of the cell lines carrying the control lentiviral construct LC0 demonstrated any response to RETRA (data not shown).
By Northern blot hybridization in A431 cells treated with RETRA, we observed direct transcriptional activation of certain p53-dependent genes. Induction of the CDKN1A (p21) gene transcript was >10-fold, similar with that of the lacZ reporter transcript. There was a 5- to 7-fold induction of BBC3 (PUMA) proapoptotic gene transcript and no substantial induction of transcripts from the hMDM2, GADD45, and IGFBP3 genes (Fig. 2).
Fig. 2.
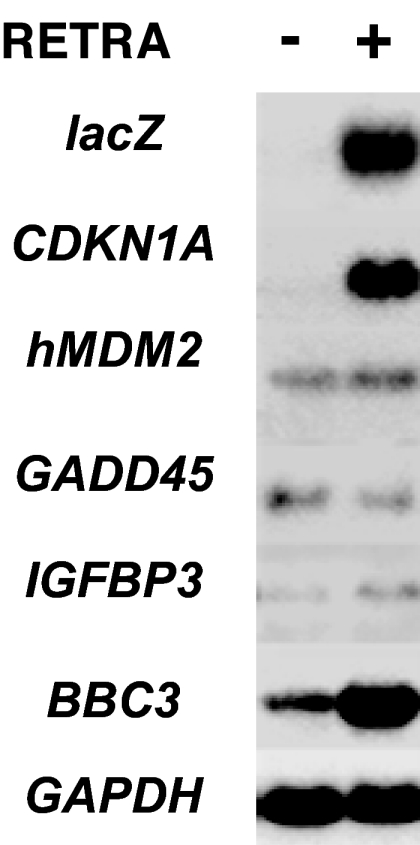
Changes in the levels of transcripts from p53-inducible genes CDKN1A (p21WAF/CIP/SDI), hMDM2, GADD45, IGFBP3, and BBC3 (PUMA). Levels of transcripts from the LacZ reporter and housekeeping gene GAPDH were determined as control. Northern blot analysis of RNAs from untreated A431 cells and the cells treated with 1.5 μg/ml RETRA for 14 h.
To test directly the contribution of mutant p53 to the induction of transcriptional reporter, we introduced His-273 p53 into several p53-negative cell lines. There was noticeable decrease in the basal level of reporter expression after introduction of His-273 mutant and a dose-dependent induction of β-galactosidase in response to RETRA in MDA041 (Fig. 3a), H1299 and PC3 (Fig. S4) cell lines expressing His-273 p53. Noteworthy, in the presence of RETRA, there was a 4-fold increase in reporter compared with baseline, suggesting that mutant p53 contributes to transcriptional activation.
Fig. 3.

Transcriptional activity of RETRA requires expression of mutant p53 and depends on induction of p73. (a) (Upper) RETRA acquires dose-dependent activation of lacZ reporter in p53-null human fibroblasts MDA041 after introduction of recombinant for expression of mutant p53 Arg273His (treatment with RETRA for 14 h, ONPG reaction). (Lower) Level of p53 protein determined by Western blot analysis with monoclonal antibodies DO1. (b) (Upper) Partial inhibition of mutant p53 in A431 cells by expression of shRNA activates the induction of p53-dependent reporter in response to RETRA (ONPG reaction). (Lower) Levels of mutant p53 protein determined by Western blot analysis with DO1 antibodies and levels of CDKN1A (p21) transcripts determined by Northern blot hybridization in the control and p53-shRNA-expressing cells, untreated, or treated with 1.5 mg/ml RETRA. (c) (Upper) Partial inhibition of p63 by shRNA does not affect response of A431/LC5 reporter cells to RETRA; partial inhibition of p73 by shRNA abrogates the induction of A431/LC5 reporter in response to treatment with RETRA. (Lower) levels of transcripts from the p63 gene (RT-PCR) and levels of p73 protein (Western blot analysis with TA/p73-specific antibodies). (d) Changes in the levels of p73 protein in A431, H1299, and A549 cell lines after treatment with 2 μg/ml RETRA or 5 μg/ml 5-fluorouracil for 20 h. Western blot analysis with antibodies specific to TA/p73. The samples were normalized for expression of β-actin and also probed for expression of p53 with monoclonal antibodies DO1. (e) Changes in proportion of mut-p53-bound p73 after treatment of A431 cells with 2 μg/ml RETRA for 20 h. (Left) Western blot analysis of 100 μg per lane of total lysates probed with rabbit TA/p73-specific Ab and α-p53 monoclonal Ab DO-1. (Right) IP of 2 mg of lysates probed with rabbit TA/p74 Ab or with rabbit p53 Ab FL-393.
Effect of RETRA in Mutant p53-Expressing Cell Lines Depends on p73.
In a reciprocal experiment, we tested effects of mutant p53 inhibition in the A431/LC5 reporter cells. Surprisingly, partial inhibition of mutant p53 by expression of shRNAs resulted in a prominent increase of the response to RETRA (Fig. 3b and Fig. S5). To test the possible contribution of p53 family members p63 and p73 to the reporter activation, we separately inhibited p63 and p73 transcripts by introducing the appropriate shRNA-expressing constructs into the A431/LC5 reporter cell line. Down-regulation of p63 did not affect the induction of the p53 reporter in response to RETRA, whereas inhibition of p73 resulted in substantial (3-fold) loss of activity (Fig. 3c). We observed similar inhibition of the response to RETRA in the reconstituted H1299/His-273/LC5 cells after the introduction of p73-specific shRNA (data not shown).
Treatment with RETRA induced a 4- to 5-fold increase in the TA/p73 protein in A431 and virtually no increase in the wild-type p53-expressing A549, whereas treatment with the DNA-damaging drug 5-fluorouracil induced strongly accumulation of p53 in A549 cells but not in the mutant p53-expressing A431 cells (Fig. 3d and Fig. S6). There was no increase in transcripts for TA-p73 isoforms, as revealed by quantitative RT-PCR (data not shown). However, RETRA did not affect levels of TA/p73 protein in the p53-negative H1299 (Fig. 3d), PC3 and Saos2 cells (Fig. S7), which corroborate the results obtained with transcriptional reporters.
As it was previously shown that mutant p53 can bind and sequester p73, the induction of p73 by RETRA could be due to the small molecule-mediated release of active p73 from the complex with mutant p53. To test this possibility, we compared changes in the fraction of p73 bound to mutant p53 in A431 cells after treatment with RETRA. There was a 2- to 3-fold reduction in the fraction of TA/p73 that coimmunoprecipitated with mutant p53 after treatment with RETRA, despite the overall increase in the level of TA/p53 (Fig. 3e). Based on the results of three independent experiments, we have calculated that the amount of free TA/p73 was increased ≈10-fold compared with the untreated control.
RETRA Induces a p73-Dependent Suppression of Mutant p53-Expressing Cells.
We decided to exploit the use of p73 induction by RETRA as a salvage bypassing p53 defects in cancer therapy. Incubation of A431 cells with RETRA resulted in a dose-dependent inhibition of growth, which was enhanced by the expression of shRNA to p53 and partially inhibited by the expression of shRNA to p73 (Fig. 4a). Treatment with RETRA for 12 h reduced dramatically the number of colonies of A431 and SW480 cells, whereas there was almost no effect in A549, H1299, and PC3 cells and much milder effect in A431/sh-p73 cells (Fig. 4b). In A431 cells, treatment with RETRA induced a dose-dependent activation of effector caspases 3 and 7; the effect was partially inhibited by shRNA to p73 (Fig. 4c). In A431 cells with shRNA-mediated inhibition of PUMA expression, there was reduced accumulation of Annexin V-positive apoptotic cells after treatment with RETRA (Fig. S8), suggesting the involvement of this p53/p73-regulated gene product. The results clearly indicate that RETRA is capable of suppressing cancer cells in a mutant p53- and p73-dependent manner.
Fig. 4.

RETRA specifically suppresses mutant p53-expressing cancer cells. (a) Cell viability assay (the XTT-assay) with A431, A431/shp53, and A431/sh-p73 cells treated for 48 h with different doses of RETRA. Five thousand cells were seeded per well of a 96-well plate. (b) Colony-formation assay with A431, A431/sh-p73, SW480, A549, H1299, and PC3 cells treated with RETRA. Semiconfluent cultures were incubated for 24 h with 4 μM RETRA and then plated to four 10-cm Petri dishes, 1,000 cells per dish. Number of colonies was scored on day 12 or day 18 (for SW480 and A549 cells). The results represent the average of two independent experiments. (c) Stimulation of effector caspases 3 and 7 in response to different doses of RETRA in A431, A431-sh-p53, and A431/sh-p73 cells. (d) Tumor-formation assay. We inoculated two groups of mice (10 mice in each group) with 105 A431 cells per inocula, four spots per mouse, and treated by six daily i.p. injections with 0.4 mg of RETRA or with diluted DMSO (the control group). Formation of tumors (>2 mm in diameter) was scored by daily inspection. The diagram shows the results from two independent experiments. (e) Scheme depicting putative mode of action of RETRA, by blocking inhibition of p73 by mutant p53. Activity of p73 is required for induction of p21 and BBC3 (PUMA) as well as for the induction of growth suppression and apoptosis of mutant p53-bearing cancer cells.
Finally, we tested potential therapeutic effects of RETRA in a nude mouse xenograft experiment. We inoculated A431 cells s.c. into nu/nu mice and applied six peritoneal injections with RETRA with daily intervals. A control group of mice with similarly inoculated A431 cells has received injections with diluted DMSO. We scored the number of formed tumors in both groups of mice by daily inspection. The dose of RETRA used was well tolerated by the mice, with no visible toxic effects. In the group of mice treated with RETRA, the time of tumor formation was delayed, and the number of tumors was substantially lower than in the control group (Fig. 4d). The results suggest that RETRA indeed has a therapeutic antitumor potential.
Discussion
Functional reactivation of the p53 tumor-suppressor pathway represent one of the promising strategies for cancer therapy. Because mutated p53 proteins are abundantly expressed in nearly half of cancer cases, an attractive idea was to develop small molecules that could convert inactive mutant p53 into tumor-suppressor form. One such compound named PRIMA-1 has been previously identified in a chemical library screening for small molecules that induce massive apoptosis of mutant p53-expressing cancer cells (18). Because transcriptional activity of p53 is the major determinant of the tumor-suppressor function, we decided to use a transcriptional reporter assay for isolation of additional compounds capable of restoring broken suppressor function in cancer cells. By lentiviral delivery, we introduced a lacZ reporter cassette controlled by a p53-responsive promoter into His-273 mutant p53-bearing carcinoma line A431. We confirmed that the reporter can be activated by introduced wild-type p53. The reporter cell line A431/LC5 was then used as a readout in high-throughput screening of a chemical library, which identified several active compounds. We then selected the compounds that act exclusively in mutant p53-expressing cells by testing all positive hits in a panel of control reporter cell lines. The strongest among several structurally similar compounds was called RETRA and used in further studies. We found that RETRA was capable of inducing p53-dependent transcriptional reporter in a panel of cancer cell lines bearing several types of p53 mutants, although it was active neither in wild-type p53-expressing nor in p53-negative cell lines. However, when the His-273 mutant was introduced into the latter reporter cell lines, a dose-dependent induction of the reporter in response to RETRA was observed, which confirmed the importance of mutant p53 for the effect. Along with the induction of p53-dependent reporter RETRA was capable of activating transcription from the p53-responsive genes CDKN1A (p21) and BBC3 (PUMA), which further suggested the involvement of reactivated mutant p53 in the effect. However, when we decided to check the effect of mutant p53 inhibition by RNAi in the A431 cells, we observed an increase instead of the expected decrease in the reactivity to RETRA. In contrast, inhibition of p73 expression reduced reactivity to RETRA in both A431 and reconstituted H1299/His-273 p53 reporter cell lines, which pointed to a substantial contribution of p73 into the effect of RETRA. Because p53 shares with p73 specific binding elements within certain genes, it was suggested that RETRA could up-regulate p53 reporter through activation of p73. Indeed, treatment of A431 cells with RETRA increased levels of TA/p73 protein and decreased substantially the proportion of p73 bound to mutant p53 (Fig. 4e). Therefore, it seems likely that RETRA assists in releasing p73 from the inhibitory complex with mutant p53, which increases the amount of transcriptionally active p73 capable of inducing certain p53-regulated genes. Introduction of the His-273 p53 mutant into p53-negative cell lines reduces the background level of reporter activity (Fig. 3a and Fig. S4), which is consistent with the ability of mutant p53 to inhibit p73 (10). Meanwhile, the reporter activation by RETRA in the reconstituted cell lines exceeds the basal level 3- to 4-fold, suggesting that certain levels of mutant p53 assist in accumulation of latent p73, which can be released and activated by treatment with the small molecule. Certainly, it would be important to know the mechanism by which RETRA displaces active p73 from its complex with mutant p53. However, because multiple factors are involved in controlling the activity, including complex interaction of p53 family isoforms (6), there is still no straightforward approach to this task: The actual target of the small molecule may not necessarily include components of the complex itself (mutant p53 or TA/p73) but also different DNp73 isoforms or even other modulating factors, such as ASPP proteins (19) or molecular chaperones. Better understanding of the physical and functional interplay among p53 family members is required before revealing the molecular target of RETRA.
Our experimental data suggest that in mutant p53-bearing cancer cells, loss of the p53 tumor-suppressor functions can be partially reversed through a bypass mechanism involving activation of TA/p73. The release of p73 by small-molecule RETRA induces transcriptional activation of several common to transcriptional targets p53 and p73, which leads to mutant p53- and p73-dependent inhibition of growth, reduction of colony formation, and induction of effector caspases. We found that RETRA reduces substantially the formation of tumors when applied to athymic mice injected with mutant p53-bearing A431 cells suggesting that the bypass activation of p73 could be used as an anticancer therapeutic approach. Certainly, at present, RETRA can be considered mainly as a tool for better understanding the potentials of the p53 family members for cancer therapy. A lot of work should yet be done to improve robustness of the effect and to optimize the drug for therapeutic uses. However, we believe that the identified approach for specific activation of p53-related pathways in mutant p53-expressing tumor cells represents an attractive alternative that could help developing therapeutics with minimal side effects, acting against a wide spectrum of human cancer types.
Experimental Procedures
Cell Cultures.
We grew cell lines in DMEM supplemented with 10% FBS. We used human carcinoma cell lines expressing mutant p53 A431, HT29, SW480, SW620 (His-273 mutant p53), MDA-MB-231 (Lys-280 mutant p53), MDA-MB-435 (Glu-266 mutant p53), p53-negative cell lines H1299, PC3, Saos-2, MDA041, wild-type p53 expressing cell lines A549, HeLa and human HEFs (normal fibroblasts from a 12-week human embryo, passage 10–14) and their derivatives with introduced recombinant lentiviral constructs. We created reporter cell lines expressing lacZ under control of p53-responsive promoter by infection of A431, H1299, A549, PC3, Saos-2, and MDA041 cells with recombinant lentiviral construct LC5 as described (17). We generated cell lines expressing p53 mutant with inhibited expression of p53 or p73 by infection with lentiviral constructs pLSLP-sh-p53 and pLSLP-sh-p73 as described (20). We used the HEK293T cell line for preparation of recombinant lentiviral stocks.
Chemicals.
The chemical library DIVERSet, consisting of 46,250 compounds was manufactured by Chembridge. We purchased 5-fluorouracil from Sigma.
Antibodies.
For detection of p53, we used monoclonal antibodies DO1 (Santa Cruz Biotechnology.), dilution 1:1,000 in Western blot analysis, or rabbit polyclonal antibodies FL-393 (Santa Cruz Biotechnology), dilution 1:5,000. For detection of p73, we used affinity-purified rabbit polyclonal antibodies specific for TA-isoforms of p73 (A300-126A, Bethyl Laboratories), dilution 1:2,000 in Western blot analysis.
Recombinant Constructs and Lentiviral Stocks.
We prepared the construct for expressing wild-type mutant p53 Arg273His by placing complementary DNAs under control of human p53 gene promoter in lentiviral vector pLV (20). Lentiviral constructs for expression of shRNA to human p53 (pLSLP-sh-p53) and PUMA (pLSLP-shPUMA1 and 2) were described (20). We used the following regions of p63 and p73 mRNA for designing shRNA oligonucleotides and insertion into pLSLG vector: for p63, 5′-ctaggtagaagtgagcaaa and 5′-gaaaacaatgcccagactcaa; for p73, 5′-gccaagtgactgtgtctgaaa and 5′-cgacatcttttggttctggat. We described lentiviral reporter construct pLC5 for expression of β-galactosidase under the control of a minimal cytomegalovirus promoter and several copies of 20-bp p53-binding segments from the CDKN1A gene (17). We prepared recombinant lentiviral stocks as described (20).
Primary Screening.
We seeded A431/LC5 reporter cells to 384-well plates, 10,000 cells per well. We delivered library compounds 2 h after plating as DMSO solution to a final concentration of 5 μg·ml−1 by using Biomek 2000 (Beckman). The final concentration of DMSO was 0.1%. We determined expression of β-galactosidase 14 h after delivery of the chemicals.
Hit Confirmation and Filtering Assays.
We performed these assays manually in 96-well plates. For the assays on p53-dependent transcriptional reporter induction, we plated 15,000 reporter cells per well 12 h before addition of serial dilutions of individual chemicals. We developed the plates for varying time periods (1–8 h), depending on the cell line used, in standard reaction with ONPG 14 h after application of chemicals and scanned the plates in a Victor plate reader (PerkinElmer) at A405. For the cell viability assay, we plated 5,000 cells per well 12 h before addition of serial dilutions of individual chemicals and developed the plates in reaction with XTT (Sigma) 48 h after the addition of chemicals.
Northern Blot and RT-PCR Analyses.
We performed Northern blot and RT-PCR analyses as described (20). We used the following RT-PCR primers: for monitoring inhibition of transcripts for p63, 5′-gcaccagcacttacttcagaaac and 5′-ctggcaagtctgaaaatccctg; for BBC3 (PUMA) transcripts, 5′-gacctcaacgcacagtac and 5′-gcatctccgtcagtgcac.
Apoptosis Assay.
We performed the effector-caspase activation assay using 30 mM Ac-DEVD-AMC (a fluorigenic substrate specific for caspases 3 and 7) and applied control inhibitor Z-VAD-FMK (2 mM) as described (21).
Tumor-Formation Assay.
Athymic nu/nu mice were injected s.c. with A431 cells, 1 × 105 cells per spot, in four locations per mouse. Simultaneously, the mice received i.p. injection with 0.4 mg of RETRA in 1 ml of PBS and 0.1% DMSO, and the injections were repeated five times with daily intervals. Control group of mice received similar injections with A431 cells and six peritoneal injections with 0.1% DMSO solution. Formation of tumors was scored by daily inspection.
Supplementary Material
Acknowledgments:
We thank Dr. Boris Kopnin for support of the xenograft experiments. The work was supported by National Institutes of Health Grants R01CA10490 and R01AG025278 (to P.M.C.) and R01CA075179 (to A.V.G.), grants from the Russian Basic Research Fund (to P.M.C., E.I.F. and J.E.K.), a grant from Russian Academy of Sciences (to J.E.K.), Howard Hughes Medical Institute Grant 55005603 (to P.M.C.) and grant from the BIRD Foundation (to E.F. and P.M.C.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0802091105/DCSupplemental.
References
- 1.Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC. Tp53 Mutation Spectra and Load: A Tool for Generating Hypotheses on the Etiology of Cancer. Lyon, France: International Agency for Research on Cancer (IARC); 2004. pp. 247–270. [PubMed] [Google Scholar]
- 2.Levrero M, et al. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. J Cell Sci. 2000;113:1661–1670. doi: 10.1242/jcs.113.10.1661. [DOI] [PubMed] [Google Scholar]
- 3.Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 4.Irwin MS, et al. Chemosensitivity linked to p73 function. Cancer Cell. 2003;3:403–410. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 5.Marabese M, Vikhanskaya F, Broggini M. p73: A chiaroscuro gene in cancer. Eur J Cancer. 2007;43:1361–1372. doi: 10.1016/j.ejca.2007.01.042. [DOI] [PubMed] [Google Scholar]
- 6.Coates PJ. Regulating p73 isoforms in human tumours. J Pathol. 2006;210:385–389. doi: 10.1002/path.2080. [DOI] [PubMed] [Google Scholar]
- 7.Muller M, et al. One, two, three—p53, p63, p73 and chemosensitivity. Drug Resist Update. 2006;9:288–306. doi: 10.1016/j.drup.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Davison TS, et al. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem. 1999;274:18709–18714. doi: 10.1074/jbc.274.26.18709. [DOI] [PubMed] [Google Scholar]
- 9.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. 2001;21:1874–1887. doi: 10.1128/MCB.21.5.1874-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Prives C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene. 2007;26:2220–2225. doi: 10.1038/sj.onc.1210311. [DOI] [PubMed] [Google Scholar]
- 11.Strano S, et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem. 2002;277:18817–18826. doi: 10.1074/jbc.M201405200. [DOI] [PubMed] [Google Scholar]
- 12.Bell HS, et al. A p53-derived apoptotic peptide derepresses p73 to cause tumor regression in vivo. J Clin Invest. 2007;117:1008–1018. doi: 10.1172/JCI28920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci USA. 2006;103:11003–11008. doi: 10.1073/pnas.0604507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vayssade M, et al. P73 functionally replaces p53 in Adriamycin-treated, p53-deficient breast cancer cells. Int J Cancer. 2005;116:860–869. doi: 10.1002/ijc.21033. [DOI] [PubMed] [Google Scholar]
- 15.Amin AR, Paul RK, Thakur VS, Agarwal ML. A novel role for p73 in the regulation of Akt-Foxo1a-Bim signaling and apoptosis induced by the plant lectin, Concanavalin A. Cancer Res. 2007;67:5617–5621. doi: 10.1158/0008-5472.CAN-07-0655. [DOI] [PubMed] [Google Scholar]
- 16.Amin AR, et al. SHP-2 tyrosine phosphatase inhibits p73-dependent apoptosis and expression of a subset of p53 target genes induced by EGCG. Proc Natl Acad Sci USA. 2007;104:5419–5424. doi: 10.1073/pnas.0700642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razorenova OV, Ivanov AV, Budanov AV, Chumakov PM. Virus-based reporter systems for monitoring transcriptional activity of hypoxia-inducible factor 1. Gene. 2005;350:89–98. doi: 10.1016/j.gene.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bykov VJ, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan A, Lu X. ASPP: A new family of oncogenes and tumour suppressor genes. Br J Cancer. 2007;96:196–200. doi: 10.1038/sj.bjc.6603525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sablina AA, et al. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strom E, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2:474–479. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.