

Abstract
Bacterial community structures and their activities in the ocean are tightly coupled with organic matter fluxes and thus control ocean biogeochemical cycles. Bromodeoxyuridine (BrdU), halogenated nucleoside and thymidine analogue, has been recently used to monitor actively growing bacteria (AGB) in natural environments. We labelled DNA of proliferating cells in seawater bacterial assemblages with BrdU and determined community structures of the bacteria that were possible key species in mediating biochemical reactions in the ocean. Surface seawater samples were collected along a north-south transect in the North Pacific in October 2003 and subjected to BrdU magnetic beads immunocapture and PCR-DGGE (BUMP-DGGE) analysis. Change of BrdU-incorporated community structures reflected the change of water masses along a north-south transect from subarctic to subtropical gyres in the North Pacific. We identified 25 bands referred to AGB as BrdU-incorporated phylotypes, belonging to Alphaproteobacteria (5 bands), Betaproteobacteria (1 band), Gammaproteobacteria (4 bands), Cytophaga-Flavobacterium-Bacteroides (CFB) group bacteria (5 bands), Gram-positive bacteria (6 bands), and Cyanobacteria (4 bands). BrdU-incorporated phylotypes belonging to Vibrionales, Alteromonadales and Gram-positive bacteria appeared only at sampling stations in a subtropical gyre, while those belonging to Roseobacter-related bacteria and CFB group bacteria appeared at the stations in both subarctic and subtropical gyres. Our result revealed phylogenetic affiliation of AGB and their dynamic change along with north-south environmental gradients in open oceans. Different species of AGB utilize different amount and kinds of substrates, which can affect the change of organic matter fluxes along transect.
Introduction
A significant ecological role of bacteria in marine environment is now recognized as an important component in the processes of biological production and biogeochemical cycles (Azam, 1998). In the last few decades, bacterial production has been measured by thymidine and/or leucine methods in various marine environments (Fuhrman and Azam, 1980; Kirchman et al., 1985; Cole et al., 1998) and their community composition and distribution have been revealed in detail by culture-independent molecular techniques (DeLong and Karl, 2005; Giovannoni and Stingl, 2005). Recent advances in ecological genomic analysis have been providing powerful tools to access functional genes of as-yet-to-be-cultured microorganisms (Tyson et al., 2004; Venter et al., 2004; DeLong, 2005; Rusch et al., 2007). Thus, it has been required to reveal the link between diversity and functions of natural bacterial assemblages in order to understand their roles in biogeochemical cycles.
Several studies have addressed the relationships between diversity and functions of bacterial communities. A combination of microautoradiography and fluorescence in situ hybridization (Micro-FISH) enabled to measure bacterial substrate uptake, specifically identifying phylotypes of every single-cells under the microscope, which revealed spatio-temporal variability of organic matter utilization of major subgroups of marine bacteria (Cottrell and Kirchman, 2000; 2003; Elifantz et al., 2005). Micro-FISH analysis has revealed that Proteobacteria and Cytophaga-Flavobacterium-Bacteroides (CFB) group bacteria are not only abundant but they also account for most of the heterotrophic bacterial production and consumption of various substances in marine environment. Schafer et al. (2001) identified metabolically active bacterial populations in nutrient-enriched seawaters, by using denaturing gradient gel electrophoresis (DGGE) of reverse transcriptase polymerase chain reaction (PCR) products derived from 16S rRNA. They found that Ruegeria-like bacteria and members of CFB were important contributors to bacterial production and activity in the post-grazing phase of nutrient-enriched experiment.
Bacterial community structure in the open ocean was first reported in the Atlantic and Pacific Oceans (Giovannoni et al., 1990; Schmidt et al., 1991). Following these studies, bacterial diversity in the open ocean has been revealed by many studies, and based on phylogenetic analysis of 16S rRNA genes, several major subgroups of bacteria (i.e. SAR86, SAR116 and SAR11, Roseobacter clusters and CFB group) have been identified in oceanic environments (Giovannoni and Rappe, 2000). Micro-FISH studies showed specific activity of some major subgroups of bacteria in assimilating organic matters (Cottrell and Kirchman, 2000). However, 16S rRNA gene cloning and sequencing studies indicated that each phylogenetic subgroup comprised diverse phylotypes of bacteria presumably having diverse activity. Hence, more detailed phylogenetic identification of actively metabolizing bacteria should give more insights into mechanistic base of spatio-temporal variability of bacterial community structure in open oceans than ever.
We used bromodeoxyuridine (BrdU), halogenated nucleoside that can serve as a thymidine analogue, to determine bacteria with detectable DNA de novo synthesis and thus possibly actively growing bacteria (AGB) in oceanic surface environments. BrdU incorporation and its antibody detection has been recently used for monitoring DNA-synthesizing (presumably actively growing) bacteria in various natural environments such as soils (Yin et al., 2000), rhizospheres (Artursson and Jansson, 2003; Artursson et al., 2005), sewage (Walters and Field, 2006), lake waters and seawaters (Steward and Azam, 1999; Pernthaler et al., 2002; Hamasaki et al., 2004; 2007; Nelson and Carlson, 2005; Pernthaler and Pernthaler, 2005; Warnecke et al., 2005; Hamasaki, 2006). Anti-BrdU antibodies conjugated with magnetic beads have been used for separating BrdU-labelled DNA from total DNA (Borneman, 1999; Urbach et al., 1999). This immunocapture technique combined with 16S rRNA gene fingerprinting and sequencing were used to identify some of AGB in eutrophic and mesotrophic coastal waters (Hamasaki et al., 2007). In our previous study, some of Roseobacter-like bacteria, Gammaproteobacteria and CFB-like bacteria were determined as AGB in coastal waters. Bacterial community structure and their growth may change along environmental gradients such as salinity, temperature and nutrient levels.
Here we report community structure analyses and identification of AGB in the western North Pacific to address the question which species contribute bacterial production in open ocean environments and whether they change along a north-south environmental gradient. The purpose of this study is to determine AGB community structure and its spatial variability in oceanic environments.
Results
Environmental characteristics
Water temperature and salinity in our studied area increased from north to south, ranging from 9.8°C to 28.1°C and from 32.7 PSU to 35.1 PSU respectively (Table 1). These data showed that four northern stations (from ST01 to ST04) were located in the North Pacific Subarctic Gyre, two southern stations (ST06 and ST07) were located in the North Pacific Subtropical Gyre and ST05 was in a transitional area. Concentrations of dissolved oxygen (DO) and inorganic nutrients (SiO2, NO2 and NO3 and NH4) at the northern stations were higher than at the southern stations. Dissolved oxygen concentrations varied from 209.0 to 296.1 μM with the maximum values at 48°N (ST03). Nutrient concentrations ranged from < 0.1 to 27.5 μM for SiO2, < 0.1 to 17.3 μM for NO2 plus NO3 with the maximum value at 48°N (ST03), and from < 0.05 to 0.60 μM for NH4 with the maximum values at 44°N (ST01). Bacterial abundance and chlorophyll a (chla) concentration ranged from 4.0 × 105 to 5.9 × 105 cells ml−1 and from 0.07 to 1.31 μg l−1 respectively.
Table 1.
Environmental characteristics of sampling stations in the western North Pacific.
| Station | Latitude | Longitude | WT (°C) | Sal (PSU) | DO (μM) | NO2 + NO3 (μM) | NH4 (μM) | SiO2 (μM) | Chla (μg l−1) | BA (x105 cells ml−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| ST01 | 44°00N | 155°00E | 11.8 | 32.96 | 281.2 | 5.4 | 0.60 | 11.3 | 0.73 | 4.0 |
| ST02 | 47°00N | 160°00E | 10.4 | 32.71 | 295.5 | 9.4 | 0.52 | 17.7 | 0.76 | 4.7 |
| ST03 | 48°30N | 165°00E | 9.8 | 32.94 | 296.1 | 17.3 | 0.39 | 27.5 | 0.52 | 4.8 |
| ST04 | 45°00N | 165°00E | 10.2 | 32.99 | 290.0 | 9.3 | 0.12 | 17.8 | 1.31 | 4.9 |
| ST05 | 40°00N | 165°00E | 17.2 | 33.74 | 251.1 | 0.36 | 0.10 | 5.1 | 0.47 | 5.9 |
| ST06 | 35°00N | 165°00E | 25.8 | 34.47 | 212.0 | < 0.1a | < 0.05a | 1.4 | 0.25 | 4.8 |
| ST07 | 28°00N | 165°00E | 28.1 | 35.02 | 209.0 | < 0.1a | 0.09 | < 0.1a | 0.07 | 5.4 |
Lower than each detection limit.
WT, water temperature; Sal, Salinity; DO, dissolved oxygen; BA, bacterial abundance.
Community structures of total and BrdU-incorporated bacteria
We optimized the procedure of BUMP-DGGE analysis, in which specific immunoseparation of BrdU-labelled DNA from total DNA was confirmed. The optimized procedure gave no PCR amplification products, when seawater samples without BrdU labelling were subjected to BrdU immunocapture in the same conditions as BrdU-labelled samples (data not shown).
The DGGE profiles of PCR-amplified 16S rRNA genes from total DNA clearly represented change of bacterial community structures along a north-south transect. A similar trend was also observed in the profiles from BrdU-incorporated DNA (Fig. 1). The cluster analysis of DGGE profiles (Fig. 2) distinguished bacterial community structures of northern and transitional stations (ST01–05) from those of southern stations (ST06 and ST07). Also, different clusters were formed between total and BrdU-incorporated community structures within either northern or southern stations. The DGGE profiles of total community changed little (less than 10%) after 10 h incubation (see T0 and T10 in Fig. 2).
Fig 1.
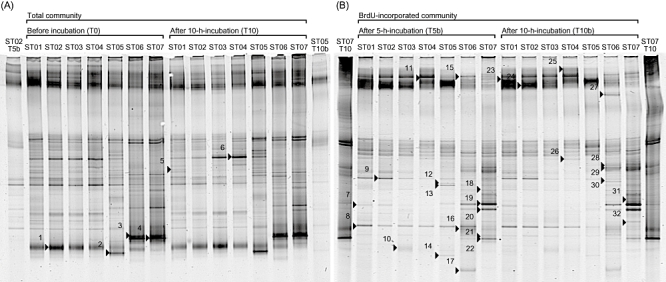
DGGE images of 16S rRNA genes amplified from total and BrdU-incorporated communities in the western North Pacific in October 2003. (A) Total communities before (T0) and after 10 h incubation (T10) and (B) BrdU-incorporated communities after 5 h (T5b) and 10 h incubation (T10b). The lanes of both ends of each image are used for markers to refer band positions for gel-to-gel comparison. Each excised and sequenced band is marked and numbered.
Fig 2.
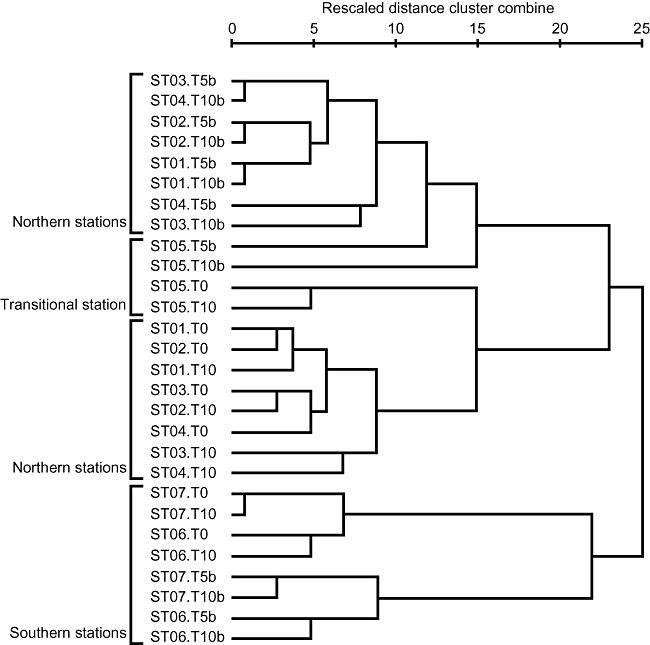
Relationships of community structures between total and BrdU-incorporated bacteria and among stations along a north-south transect in the western North Pacific. The tree was constructed by using the between-group average linkage method for clustering with the software SPSS Base 11.5 J (SPSS, Chicago, IL).
The DGGE profiles of BrdU-incorporated community showed slight difference between the profiles of 5 h incubation (T5b in Fig. 1) and that of 10 h incubation (T10b in Fig. 1). Some DGGE bands appeared in either community in T5b or T10b. The DGGE bands 12 and 13 were only appeared in T5b but not in T10b, while the band 27 only appeared at ST06 in T10b but not in T5b. Also, the band 8 was found at ST06 in T5b but disappeared in T10b.
Sequencing and phylogenetic analysis
We sequenced partial 16S rRNA genes of 32 bands excised from both total and BrdU-incorporated communities (Fig. 1). The migration position of DGGE bands were compared among all sampling stations and the presence and absence of these 32 phylotypes at each sampling station was determined (Table 2). Twenty-five out of 32 bands were referred to as BrdU-incorporated phylotypes, including five alphaproteobacteria, one betaproteobacterium, four gammaproteobacteria, five CFB group bacteria, six Gram-positive bacteria and four cyanobacteria. Four out of five alphaproteobacteria belonged to Rhodobacterales. One betaproteobacterium belonged to Burkholderiales. Although the closest match of relative of this betaproteobacterium was an uncultured ‘Pseudomonas sp.’ clone in the database, this should be miss identification of the registered clone, and it phylogenetically belonged to Betaproteobacteria (Fig. 3). One out of four gammaproteobacteria belonged to Vibrionales and others belonged to Alteromonadales. As for Gram-positive bacteria, four were high-GC group (three actinobacteria and one unidentified bacterium) and two were low-GC group (Lactobacillales bacteria). Also, we found that two phylotypes closely related to Prochlorococcus and other two phylotypes closely related to Synechococcus. There were five phylotypes (KH03–30B and KH03–7B of alphaproteobacteria, KH03–11B of betaproteobacteria, KH03–42B and KH03–24B of CFB group bacteria) widely distributed from north to south as BrdU-incorporated bacteria. Most of gammaproteobacteria, all Gram-positive bacteria, two alphaproteobacteria and Prochlorococcus were found only at southern and transitional stations. In contrast, five phylotypes (KH03–9B of alphaproteobacteria, KH03–13B, KH03–32B and KH03–38B of CFB group bacteria and KH03-77 of Synechococcus) were found only at northern and transitional stations.
Table 2.
The phylogenetic group of 16S rRNA gene sequencing of excised DGGE bands and the presence and absence at each samples.
| DGGE band | Band name | Accession number | Phylogenetic groupa | ST01 | ST02 | ST03 | ST04 | ST05 | ST06 | ST07 |
|---|---|---|---|---|---|---|---|---|---|---|
| Alphaproteobacteria | ||||||||||
| 18 | KH03–30B | AB307981 | uncultured | B | B | B | B | B | T/B | T/B |
| 7 | KH03–9B | AB307970 | Rodobacterales | T/B | T/B | T/B | T/B | T/B | – | – |
| 9 | KH03–7B | AB307969 | Rodobacterales | T/B | T/B | T/B | T/B | T/B | T/B | T/B |
| 20 | KH03–34B | AB307984 | Rodobacterales | – | – | – | – | B | B | T/B |
| 29 | KH03–57B | AB307994 | Rodobacterales | T | T | – | T | T | T/B | T/B |
| Betaproteobacteria | ||||||||||
| 8 | KH03–11B | AB307971 | Burkholderiales | T/B | T/B | T/B | T/B | T/B | B | B |
| Gammaproteobacteria | ||||||||||
| 5 | KH03-40 | AB307996 | Alteromonadales | T | – | – | – | – | – | – |
| 19 | KH03–33B | AB307983 | Alteromonadales | – | – | – | – | T/B | T/B | T/B |
| 31 | KH03–51B | AB307991 | Alteromonadales | – | – | – | – | – | – | B |
| 32 | KH03–52B | AB307992 | Alteromonadales | – | – | – | – | – | B | B |
| 16 | KH03–22B | AB307978 | Vibrionales | – | – | – | – | – | B | B |
| CFB group | ||||||||||
| 11 | KH03–13B | AB307972 | Flavobacteriales | T/B | T/B | T/B | T/B | T | – | – |
| 23 | KH03–24B | AB307980 | Flavobacteriales | T/B | – | – | T/B | T/B | B | B |
| 24 | KH03–32B | AB307982 | Flavobacteriales | T/B | T/B | T/B | T/B | – | T | T |
| 25 | KH03–38B | AB307987 | Flavobacteriales | T/B | T/B | T/B | T/B | – | – | – |
| 26 | KH03–42B | AB307988 | uncultured | T/B | T/B | B | B | T/B | B | – |
| Gram-positive bacteria | ||||||||||
| 12 | KH03–17B | AB307975 | Lactobacillales | – | – | – | – | B | B | B |
| 13 | KH03–18B | AB307976 | Lactobacillales | – | – | – | – | B | T/B | T/B |
| 30 | KH03–47B | AB307990 | Actinobacteria | – | – | – | – | – | T/B | B |
| 17 | KH03–23B | AB307979 | Actinobacteria | – | – | – | – | – | B | – |
| 27 | KH03–46B | AB307989 | Actinobacteria | – | – | – | – | – | B | – |
| 28 | KH03–56B | AB307993 | uncultured | – | – | – | – | – | – | B |
| Cyanobacteria and chlroplasts | ||||||||||
| 1, 10 | KH03–77, 15B | AB307999, AB307973 | Chroococcales | T | T | T/B | T/B | T | – | – |
| 2, 14 | KH03–78, 21B | AB308000, AB307977 | Chroococcales | – | – | – | – | T/B | – | – |
| 3, 21 | KH03–71, 37B | AB307997, AB307986 | Prochlorales | – | – | – | – | – | T/B | T/B |
| 4, 22 | KH03–73, 35B | AB307998, AB307985 | Prochlorales | – | – | – | – | – | T/B | T/B |
| 15 | KH03–16B | AB307974 | Plastid | – | – | – | – | B | T/B | T/B |
| 6 | KH03–36 | AB307995 | Plastid | T | T | T | T | T | T | – |
Class or Order of the closest isolates. The band matched to no isolates with > 95% similarity represented as ‘uncultured’ for convenience.
T, presence of DGGE band in total communities (T0 and T10); B, in BrdU-incorporated comminities (T5b and T10b); T/B, in both communities; –, DGGE band was not detectable.
Fig 3.
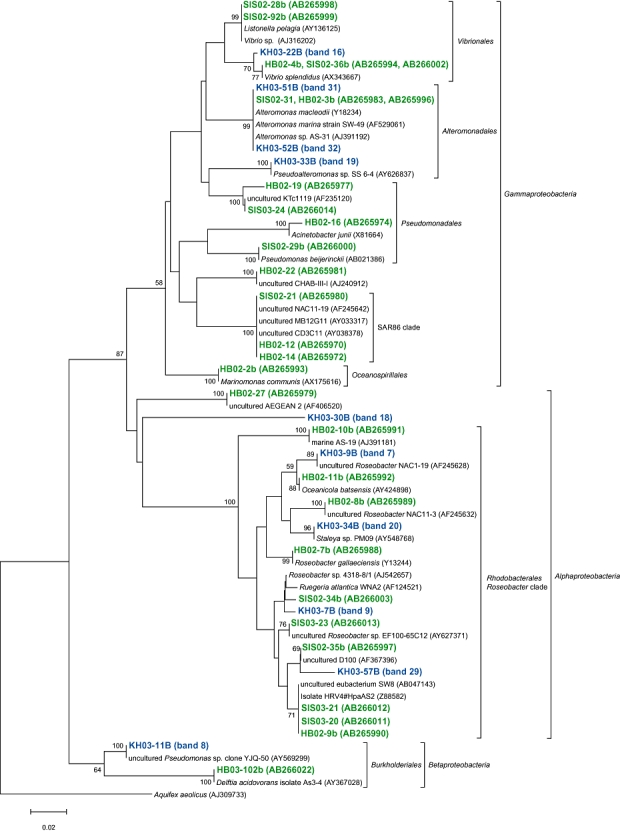
A neighbour-joining tree of 16S rRNA gene sequences of actively growing bacteria from members of the Alphaproteobacteria, Betaproteobacteria and Gammaproteobacteria retrieved from the coastal (Inland Sea of Japan; Hamasaki et al., 2007) and the oceanic (western North Pacific) sites. The band names of green and blue letters represent phylotypes retrieved from coastal and oceanic environments respectively. Bootstrap values > 50% are indicated. The scale bar represents 2% estimated sequence divergence.
Discussion
In our previous study, the BUMP-DGGE analysis revealed phylogenetic affiliations of AGB in coastal waters. This approach allows workers to determine phylotypes contributing to bacterial production at a given time and locale. Furthermore, it is also possible to test growth performance of specific bacteria bearing genes important in biogeochemical cycles when the functional genes instead of 16S rRNA gene are amplified after BrdU-labelling and immunocapturing. For example, the protocol should help to address a question whether light-dependent growth of aerobic phototrophic bacteria and proteorhodopsin-containing bacteria contribute to pelagic bacterial production (Schwalbach et al., 2005; Sieracki et al., 2006). In this study, the same method was successfully applied to open ocean waters where bacterial growth rate was lower and thus BrdU incorporation was less than coastal waters. As ambient substrate availability for growth of bacteria should be much lower in the oceanic waters than the coastal ones, we added much lower concentration of BrdU for labelling to oceanic waters (20 nM) than the coastal ones (1 μM). Hence, our methodology was sensitive enough to detect BrdU-incorporated bacteria in nutrient-poor oligotrophic environments such as the North Pacific Subtropical Gyre, although longer labelling time (> 5 h) was required for it than nutrient-rich coastal waters.
To our knowledge, this is the first report on identifying detailed phylotypes of AGB inhabiting oligotrophic open ocean environments. Micro-FISH studies using 3H-thymidine and 3H-leucine showed active growth of SAR11 bacteria in the North Atlantic Ocean (Malmstrom et al., 2004). Also, BrdU immunofluorescence method in combination with 16S rRNA FISH showed that some phylogenetic groups of bacteria such as Roseobacter clade, Alteromonas clade and SAR86 group are actively growing in the North Sea (Pernthaler et al., 2002). BUMP-DGGE analysis gave more detailed affiliation of AGB. Although several studies using RT-PCR-DGGE have revealed detailed phylotypes of metabolically active bacteria (Moesender et al., 2001; Schafer et al., 2001; Troussellier et al., 2002), it has never been performed in open ocean environments.
Distinct bacterial community structures between the northern and the southern stations in both total and BrdU-incorporated communities reflected the changes of associated water masses from subarctic to subtropical gyres. Environmental characteristics (Table 1) such as water temperature, salinity and inorganic nutrients clearly indicated the change of water masses. Also, surface seawater (10 m depth) DOC concentrations were 60.0–62.2 μM at ST01–04 (subarctic), 63.9 μM at ST05 (transitional), and 70.9 and 70.8 μM at ST06 and 07 (subtropical) respectively (H. Ogawa, pers. comm.). Dissolved organic matter in the highly productive subarctic area reportedly showed lower carbon concentrations but contained higher labile fractions than those in the oligotrophic subtropical area (Ogawa and Tanoue, 2003). Although it is too early to generalize such distinct patterns of bacterial community structure, change of BrdU-incorporated community structure from one to another gyre could lead to the change of organic matter fluxes owing to variable characteristics of nutrient acquisition by different phylotypes of bacteria. Bacterial productivity measured in the same sampling sites was different between the two gyres (Hamasaki, 2006).
BUMP-DGGE can be a powerful method to address questions in structuring mechanism of bacterial community (e.g. bottom-up versus top-down, growth versus mortality). Cluster analysis of DGGE profiles revealed that total community structures were distinguished from BrdU-incorporated community structures (Fig. 2), suggesting total community was not only structured by the growth of bacteria but also structured by other factors such as grazing and viral lysis in our studied samples. Another possibility is that differences between total and BrdU-incorporated communities may be due to differential growth of various phylotypes of bacteria on fine temporal scales such as daily or half-daily scales. The DGGE bands in total communities reflect major bacteria with high abundance, whereas those in BrdU-incorporated communities reflect bacteria with high growth rate. Bacteria with high growth rates but low abundance due to high mortality rates or temporally limited growth may not be detected in the total communities but in the BrdU-incorporated communities. We found such phylotypes mostly in subtropical stations. High water temperature might support instantaneous rapid growth but the final yield of their biomass might be limited by low substrate availability in subtropical oligotrophic open oceans. For example, phylotypes closely related to Alteromonas and Vibrio (KH03–51B, 52B and 22B) were only detected in BrdU-incorporated communities at ST06 and 07. Most of the phylotypes identified as Gram-positive bacteria detected at ST05–07 were only found in BrdU-incorporated communities. Also, no betaproteobacteria were detected in the total communities at ST06 and ST07, although one phylotype was found at all stations in BrdU-incorporated communities. Some rapidly growing bacteria might be subjected to intense grazing by protists or viral lysis and thus prevented from becoming abundant enough to be detected in PCR-DGGE.
We found that bacteria in Roseobacter clade appeared as AGB in both subarctic and subtropical stations. Especially, their presence in the subtropical stations suggested their active growth in oligotrophic open oceans. Roseobacter clade includes aerobic anoxygenic photosynthetic bacteria (AAPB). This group of bacteria reportedly accounted for up to about 10% of marine prokaryotes (Sieracki et al., 2006) and had more advantages than other bacteria in oligotrophic environments because of their potential for photosynthetic energy acquisition (Kolber et al., 2000; Karl, 2002). As we have no data whether Roseobacter-related bacteria found in this study are AAPB or not, more research should be required to reveal the contribution of AAPB to bacterial carbon production in oligotrophic open oceans.
Occurrence of Betaproteobacteria and Gram-positive bacteria was unexpected in this study because they were mostly reported as abundant group in wastewater, soil or subsurface of deep sediments (Hugenholtz et al., 1998). Most DGGE bands related to Gram-positive bacteria were not found in total communities but in BrdU-incorporated communities, suggesting that they are actively growing but relatively minor in abundance. Bouvier and del Giorgio (2007) reportedly showed that Betaproteobacteria and Gram-positive bacteria (Actinobacteria) had a potential to rapidly grow in an open ocean pelagic environment but were minor in the assemblages due to high viral mortality. Betaproteobacterium detected in this study (KH03–11B) were closely related (99.5% similarity) to an isolated strain of ‘Pseudomonas saccharophila’, recently reclassified into Pelomonas saccharophila (Xie and Yokota, 2005). This bacterium has been reported to fix nitrogen (Barraquio et al., 1986). Identity of nitrogen-fixing microorganisms and their contribution to nitrogen supply to oligotrophic open oceans has been attracted much interest (Capone et al., 1997). ‘Pelomonas saccharophila’-like betaproteobacterium could be an as-yet-unknown nitrogen-fixing prokaryote in open oceans.
Cyanobacteria were appeared in BrdU-incorporated community at transitional and southern stations (Table 2). Vaulot and colleagues (1995) reported that Prochlorococcus replicated DNA in late afternoon and divided cells in night. If their growth was synchronized by such a day-night cycle, it would depend on the time point of sampling (morning or afternoon) whether these bacteria appear as ‘BrdU-incorporated’ or not. However, we found no correlation between the time points of sampling and BrdU incorporation in this study. BrdU incorporation by Prochlorococcus was detected from the samples collected both in the morning (8–9 am at the ST06) and afternoon (6–7 pm at the ST07). The incorporation by Synechococcus was also detected from the samples collected both in the early morning (3–4 am at the ST05) and afternoon (5–6 pm at the ST03 and ST04).
We compared AGB phylotypes found in this study with those found in our previous study (Hamasaki et al., 2007), constructing phylogenetic trees representing all AGB ever found by BUMP-DGGE analysis, to see whether common or specific AGB were present between the coastal and the oceanic waters studied in the present and the previous works (Figs 3 and 4). The coastal waters were obtained from nine sampling stations with an environmental gradient from eutrophic to mesotrophic waters (Hamasaki et al., 2007). We have found that the species or strains of AGB found in the coastal sites were basically different from those in the oceanic sites, even though some of them belonged to the same subgroups of bacteria. Cyanobacteria and Gram-positive bacteria only appeared in the oceanic sites, while bacteria belonging to Pseudomonadales, Oceanospirillales and SAR86-cluster only appeared in the coastal sites. Also, AGB belonging to Roseobacter-cluster, Burkholderiales, Vibrionales, Alteromonadales and CFB were commonly present in both sites. However, the most phylotypes belonging to the same subgroups of bacteria in both sites were not exactly the same as each other if 16S rRNA gene sequences were compared. There were more than 3% differences in those partial 16S rRNA gene sequences (V3 variable regions). Roseobacter-related bacteria were previously detected as AGB in the Inland Sea of Japan and North Sea coastal environments using BrdU methodology (Pernthaler et al., 2002; Hamasaki et al., 2007). Also, it has been reported that this group of bacteria are well adapted to nutrient-rich conditions such as coastal waters and algal blooms (Gonzalez et al., 2000; Riemann et al., 2000; Zubkov et al., 2001; Malmstrom et al., 2004; Moran et al., 2004; Grossart et al., 2005). Phylotypes of Roseobacter-related AGB found in this study were different with > 3% mismatch in partial 16S rRNA gene sequences (V3 variable regions) from those found in the Inland Sea of Japan coastal sites. Although the sample coverage is not enough to conclude it, there might be different types of Roseobacter-related bacteria adapted to oligotrophic open oceans (Fig. 3). Further investigation should be required to test the hypothesis.
Fig 4.
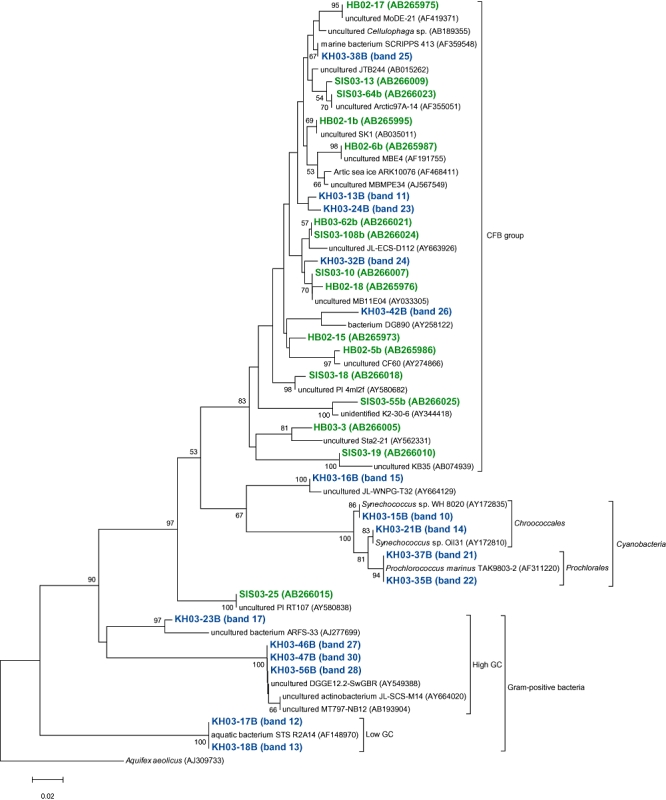
A neighbour-joining tree of 16S rRNA gene sequences of actively growing bacteria from members of the CFB group bacteria, Gram-positive bacteria and Cyanobacteria retrieved from the coastal (Inland Sea of Japan; Hamasaki et al., 2007) and the oceanic (western North Pacific) sites. The band names of green and blue letters represent phylotypes retrieved from coastal and oceanic environments respectively. Bootstrap values > 50% are indicated above the branches. The scale bar represents 2% estimated sequence divergence.
We found that Roseobacter-related and CFB group phylotypes were major AGB from subarctic to subtropical transect in the western North Pacific. Also AGB community structures changed along the transect, where phylotypes of Gammaproteobacteria and Gram-positive bacteria additionally appeared in subtropical stations. Bacterial phylotypes identified as AGB in this and previous studies are responsible for bacterial production and thus possible key players in marine biogeochemical carbon cycles. These phylotypes should be targeted to monitor their dynamics and ecological functions in future works.
Experimental procedures
Sampling and BrdU labelling
Surface seawater samples were collected with a clean bucket at seven stations along a north-south transect in the western North Pacific in October 2003 during the KH-03–2 cruise of R/V Hakuho-maru (Table 1). About 35 l of surface seawater samples was pre-filtered through a 200 μm nylon mesh to remove mesozooplankton, and subjected to further procedure within 1 h. Eleven litres of the pre-filtered samples with BrdU (20 nM in final concentration; Sigma-Aldrich, St Louis, MO) was incubated in dark bottles at in situ water temperature for 5 and 10 h. After incubation, bacterial cells were collected with 0.22 μm pore size Sterivex cartridge filters (Millipore, Billerica, MA) with a peristaltic pump. Immediately after filtration, the Sterivex filters were stored at −20°C until further analysis. Chla concentration was measured with Turner-Designs fluorometer after the extraction with N,N-dimethylformamide (Suzuki and Ishimaru, 1990; Welschmeyer, 1994). Bacterial abundance was enumerated by direct counting with the use of epifluorescence microscopy (Olympus BX-51, Olympus, Tokyo, Japan) after staining with 4′,6-diamidino-2-phenylindole (1.0 μg mlin−1 in final concentration; Molecular Probes, Eugene, OR) (Porter and Feig, 1980). Nitrate + nitrite, ammonium, phosphate and silicate were measured by continuous flow system (AACS-II, Bran + Luebbe GmbH, Norderstedt, Germany). The concentration of DO was determined by Winkler titration method.
BUMP-DGGE analysis
The analysis has been performed according to the procedures previously described (Hamasaki et al., 2007). Briefly, the Sterivex filter was subjected to xanthogenate-SDS DNA extraction and 1 μg of the extracted DNA was used for BrdU immunocapture. The total DNA and immunocaptured BrdU-labelled DNA were used as templates by PCR amplification of 16S rRNA genes using the eubacterial-specific primer 341F-GC with a 40-bp GC clamp (57 mer; 5′-CGC CCG CCG CGC GCG GCG GGC GGG GCGGGG GCA CGG GGG GCC TAC GGG AGG CAG CAG-3′) and the universal primer 907R (20 mer; 5′-CCG TCA ATT C[A/C]T TTG AGT TT-3′) (Schafer and Muyzer, 2001). For DGGE, the PCR products (about 250 ng) were loaded onto 8% polyacrylamide gels in 0.5× TAE with a denaturing gradient from 20% to 70% from the top to the bottom. Electrophoresis was performed as 85 V for 16 h at 60°C in a hot-bath DGGE unit (CBS Scientific, San Diego, CA). The Jaccard coefficient was calculated for cluster analysis of DGGE profiles, and the distance matrix was analysed by using the between-group average linkage method for clustering with the software SPSS Base 11.5 J (SPSS, Chicago, IL).
Sequencing and phylogenetic analysis
Excised DGGE bands were sequenced directly from PCR products which were re-amplified with the primer set used above. Prior to sequencing, the PCR products were analysed by DGGE to confirm the band positions relative to the original sample. Bidirectional sequencing was performed with an automatic sequencer 3730xl (Applied Biosystems, Foster City, CA) after sequencing reaction with a BigDye terminator sequencing kit (Applied Biosystems, Foster City, CA) using 341F and 907R primers. Sequences were aligned to known sequences in the DNA Data Bank of Japan (DDBJ) database using blast (Altschul et al., 1997). Phylogenetic relationships were inferred from multiple alignments using CLUSTAL W (Thompson et al., 1994) and draw the phylogenetic tree by neighbour-joining method with mega 3.1 software (Kumar et al., 2004). All sequences were checked by the CHECK_CHIMERA program available through the Ribosomal Database Project (Maidak et al., 2001). The nucleotide sequences of DGGE bands have been deposited in the DDBJ nucleotide sequence database under accession numbers from AB307969 to AB308000.
Acknowledgments
We would like to thank the officers and crew of the R/V. Hakuho-maru for their assistance and support for sample collection during the KH-03-2 cruise. We also thank H. Ogawa, Ocean Research Institute, The University of Tokyo, for providing information of DOC and critical comments on the manuscript. This research was supported by JSPS Research Fellowships for Young Scientists (No. 05J05361) to A.T., and grant-in-aids (No. 17201004, 18310011 and 18201003) from JSPS to K.H.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artursson V, Jansson JK. Use of bromodeoxyuridine immunocapture to identify active bacteria associated with arbuscular mycorrhizal hyphage. Appl Environ Microbiol. 2003;69:6208–6215. doi: 10.1128/AEM.69.10.6208-6215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artursson V, Finlay RD, Jansson JK. Combined bromodeoxyuridine immunocapture and terminal-restriction fragment length polymorphism analysis highlights differences in the active soil bacterial metagenome due to Glomus mosseae inoculation or plant species. Environ Microbiol. 2005;7:1952–1966. doi: 10.1111/j.1462-2920.2005.00868.x. [DOI] [PubMed] [Google Scholar]
- Azam F. Microbial control of oceanic carbon flux: the plot thickens. Science. 1998;280:694–696. [Google Scholar]
- Barraquio WL, Pader BC, Watanabe I, Knowles R. Nitrogen fixation by Pseudomonas saccharophila Doudoroff ATCC 15946. J Gen Microbiol. 1986;132:237–241. [Google Scholar]
- Borneman J. Culture-independent identification of microorganisms that respond to specified stimuli. Appl Environ Microbiol. 1999;65:3398–3400. doi: 10.1128/aem.65.8.3398-3400.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier T, del Giorgio PA. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environ Microbiol. 2007;9:287–297. doi: 10.1111/j.1462-2920.2006.01137.x. [DOI] [PubMed] [Google Scholar]
- Capone DG, Zehr JP, Paerl HW, Bergman B, Carpenter EJ. Trichodesmium, a globally significant marine cyanobacterium. Science. 1997;276:1221–1229. [Google Scholar]
- Cole JJ, Findlay S, Pace ML. Bacterial production in fresh and saltwater ecosystem: a cross-system overview. Mar Ecol Prog Ser. 1998;43:1–10. [Google Scholar]
- Cottrell MT, Kirchman DL. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high- molecular-weight dissolved organic matter. Appl Environ Microbiol. 2000;66:1692–1697. doi: 10.1128/aem.66.4.1692-1697.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell MT, Kirchman DL. Contribution of major bacterial groups to bacterial biomass production (thymidine and leucine incorporation) in the Delaware estuary. Limnol Oceanogr. 2003;48:168–178. [Google Scholar]
- DeLong EF. Microbial community genomics in the ocean. Nat Rev Microbiol. 2005;3:459–469. doi: 10.1038/nrmicro1158. [DOI] [PubMed] [Google Scholar]
- DeLong EF, Karl DM. Genomic perspectives in microbial oceanography. Nature. 2005;437:336–342. doi: 10.1038/nature04157. [DOI] [PubMed] [Google Scholar]
- Elifantz H, Malmstrom RR, Cottrell MT, Kirchman DL. Assimilation of polysaccharides and glucose by major bacterial groups in the Delaware estuary. Appl Environ Microbiol. 2005;71:7799–7805. doi: 10.1128/AEM.71.12.7799-7805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman JA, Azam F. Bacterioplankton secondary production estimates for coastal waters of British Columbia, Antarctica, and California. Appl Environ Microbiol. 1980;39:1085–1095. doi: 10.1128/aem.39.6.1085-1095.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Rappe MS. Evolution, diversity, and molecular ecology of marine prokaryotes. In: Kirchman DL, editor. Microbial Ecology of the Oceans. New York, USA: Wiley-Liss; 2000. pp. 47–84. [Google Scholar]
- Giovannoni SJ, Stingl U. Molecular diversity and ecology of microbial plankton. Nature. 2005;437:343–348. doi: 10.1038/nature04158. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Britschgi TB, Moyer CL, Field KG. Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990;345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez JM, Simo R, Massana R, Covert JS, Casamayor EO, Pedoros-Alio C, Moran MA. Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl Environ Microbiol. 2000;66:4237–4246. doi: 10.1128/aem.66.10.4237-4246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossart HP, Levold F, Allgaler M, Simon M, Brinkhoff T. Marine diatom species harbour distinct bacterial communities. Environ Microbiol. 2005;7:860–873. doi: 10.1111/j.1462-2920.2005.00759.x. [DOI] [PubMed] [Google Scholar]
- Hamasaki K. Comparison of bromodeoxyuridine immunoassay with tritiated thymidine radioassay for measuring bacterial productivity in oceanic waters. J Oceanogr. 2006;62:793–799. [Google Scholar]
- Hamasaki K, Long RA, Azam F. Individual cell growth rates of marine bacteria, measured by bromodeoxyuridine incorporation. Aquat Microb Ecol. 2004;35:217–227. [Google Scholar]
- Hamasaki K, Taniguchi A, Tada Y, Long RA, Azam F. Actively growing bacteria in the Inland Sea of Japan, identified by combined bromodeoxyuridine immunocapture and denaturing gradient gel electrophoresis. Appl Environ Microbiol. 2007;73:2787–2798. doi: 10.1128/AEM.02111-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl DM. Hidden in a sea of microbes. Nature. 2002;415:590–591. doi: 10.1038/415590b. [DOI] [PubMed] [Google Scholar]
- Kirchman DL, K'nees E, Hodson RE. Leucine incorporation and its potential as a measure of protein synthesis by bacteria in natural aquatic systems. Appl Environ Microbiol. 1985;49:599–607. doi: 10.1128/aem.49.3.599-607.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolber ZS, Van Dover CL, Niederman RA, Falkowski PG. Bacterial photosynthesis in surface waters of the open ocean. Nature. 2000;407:177–179. doi: 10.1038/35025044. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: an integrated software for Molecular Evolutionary Genetics Analysis and Sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Maidak BL, Cole JR, Liburn TG, Parker CT, Saxman PR, Farris RJ, et al. The RDP-II (Ribosomal Database Project) Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrom RR, Kiene RP, Kirchman DL. Identification and enumeration of bacterial assimilating dimethylsulfoniopropionate (DMSP) in the North Atlantic and Gulf of Mexico. Limnol Oceanogr. 2004;49:597–606. [Google Scholar]
- Moesender MM, Winter C, Herndl GJ. Horizontal and vertical complexity of attached and free-living bacteria of the eastern Mediterranean Sea, determined by 16S rDNA and 16S rRNA fingerprints. Limnol Oceanogr. 2001;46:95–107. [Google Scholar]
- Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP, et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature. 2004;432:910–913. doi: 10.1038/nature03170. [DOI] [PubMed] [Google Scholar]
- Nelson CE, Carlson CA. A nonradioactive assay of bacterial productivity optimized for oligotrophic pelagic environments. Limnol Oceanogr Methods. 2005;3:211–220. [Google Scholar]
- Ogawa H, Tanoue E. Dissolved organic matter in the oceanic waters. J Oceanogr. 2003;59:129–147. [Google Scholar]
- Pernthaler A, Pernthaler J. Diurnal variation of cell proliferation in three bacterial taxa from coastal North Sea waters. Appl Environ Microbiol. 2005;71:4638–4644. doi: 10.1128/AEM.71.8.4638-4644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernthaler A, Pernthaler J, Schattenhofer M, Amann R. Identification of DNA-synthesizing bacterial cells in coastal North Sea plankton. Appl Environ Microbiol. 2002;68:5728–5736. doi: 10.1128/AEM.68.11.5728-5736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KG, Feig YS. The use of DAPI for identifying and counting aquatic microflora. Limnol Oceanogr. 1980;25:943–948. [Google Scholar]
- Riemann L, Steward GF, Azam F. Dynamics of bacterial community composition and activity during a mesocosm diatom bloom. Appl Environ Microbiol. 2000;66:578–587. doi: 10.1128/aem.66.2.578-587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:398–431. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer H, Muyzer G. Denaturing gradient gel electrophoresis in marine microbial ecology. In: Paul JH, editor. Methods in Microbiology. Vol. 30. New York, USA: Academic Press; 2001. pp. 425–468. [Google Scholar]
- Schafer H, Bernard L, Courties C, Lebaron P, Servais P, Pukall R, et al. Microbial community dynamics in Mediterranean nutrient-enriched seawater mesocosms: changes in the genetic diversity of bacterial populations. FEMS Microbiol Ecol. 2001;34:243–253. doi: 10.1111/j.1574-6941.2001.tb00775.x. [DOI] [PubMed] [Google Scholar]
- Schmidt TM, DeLong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. Appl Environ Microbiol. 1991;173:4371–4378. doi: 10.1128/jb.173.14.4371-4378.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbach MS, Brown M, Fuhrman JA. Impact of light on marine bacterioplankton community structure. Aquat Microb Ecol. 2005;39:235–245. [Google Scholar]
- Sieracki ME, Gilg IC, Their EC, Poulton NJ. Distribution of planktonic aerobic anoxygenic photoheterotrophic bacteria in the northwest Atlantic. Limnol Oceanogr. 2006;51:38–46. [Google Scholar]
- Steward GF, Azam F. Bromodeoxyuridine as an alternative to 3H-thymidine for measuring bacterial productivity in aquatic samples. Aquat Microb Ecol. 1999;19:57–66. [Google Scholar]
- Suzuki R, Ishimaru T. An improved method for the determination of phytoplankton chlorophyll using N,N-dimethylformamide. J Oceanogr. 1990;46:190–194. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troussellier M, Schafer H, Batailler N, Bernard L, Courties C, Lebaron P, et al. Bacterial activity and genetic richness along an estuarine gradient (Rhone River plume, France) Aquat Microb Ecol. 2002;28:13–24. [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Urbach E, Vergin KL, Giovannoni SJ. Immunological detection and isolation of DNA from metabolically active and bacteria. Appl Environ Microbiol. 1999;65:1207–1213. doi: 10.1128/aem.65.3.1207-1213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaulot D, Marie D, Olson RJ, Chisholm SW. Growth of Prochlorococcus, a photosynthetic prokaryote, in the Equatorial Pacific Ocean. Science. 1995;268:1480–1482. doi: 10.1126/science.268.5216.1480. [DOI] [PubMed] [Google Scholar]
- Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, et al. Environmental genomes shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Walters SP, Field KG. Persistence and growth of fecal Bacteroidetes assessed by bromodeoxyuridine immunocapture. Appl Environ Microbiol. 2006;72:4532–4539. doi: 10.1128/AEM.00038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke F, Sommaruge R, Sekar R, Hofer JS, Pernthaler J. Abundance, identify, and growth state of Actinobacteria in mountain lakes of different UV transparency. Appl Environ Microbiol. 2005;71:5551–5559. doi: 10.1128/AEM.71.9.5551-5559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welschmeyer NA. Fluorometric analysis of chlorophyll a in the presence of chlorophyll b and pheopigments. Limnol Oceanogr. 1994;39:1985–1992. [Google Scholar]
- Xie CH, Yokota A. Reclassification of Alcaligenes latus strains IAM 12599T and IAM 12664 and Pseudomonas sacchatophila as Azohydromonas lata General nov., comb. nov., Azohydromonas australica sp. nov. and Pelomonas saccharophila General nov., comb., respectively. Int J Syst Evol Microbiol. 2005;55:2419–2425. doi: 10.1099/ijs.0.63733-0. [DOI] [PubMed] [Google Scholar]
- Yin B, Crowley D, Sparovek G, De Melo WJ, Borneman J. Bacterial functional redundancy along a soil reclamation gradient. Appl Environ Microbiol. 2000;66:4361–4365. doi: 10.1128/aem.66.10.4361-4365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubkov MV, Fuchs BM, Archer SD, Kiene RP, Amann R, Burkill PH. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ Microbiol. 2001;3:304–311. doi: 10.1046/j.1462-2920.2001.00196.x. [DOI] [PubMed] [Google Scholar]