

Abstract
The VanC phenotype for clinical resistance of enterococci to vancomycin is exhibited by Enterococcus gallinarum and Enterococcus casseliflavus. Based on the detection of the cell precursor UDP-N-acetylmuramic acid pentapeptide intermediate terminating in d-Ala-d-Ser instead of d-Ala-d-Ala, it has been predicted that the VanC ligase would be a d-Ala-d-Ser rather than a d-Ala-d-Ala ligase. Overproduction of the E. casseliflavus ATCC 25788 vanC2 gene in Escherichia coli and its purification to homogeneity allowed demonstration of ATP-dependent d-Ala-d-Ser ligase activity. The kcat/Km2 (Km2 = Km for d-Ser or C-terminal d-Ala) ratio for d-Ala-d-Ser/d-Ala-d-Ala dipeptide formation is 270/0.69 for a 400-fold selection against d-Ala in the C-terminal position. VanC2 also has substantial d-Ala-d-Asn ligase activity (kcat/Km2 = 74 mM−1min−1).
In the treatment of bacterial infections, the utility of an antibiotic is typically controlled by the development of resistance to its action by clinically significant pathogens. In hospital settings the Gram-positive enterococci have emerged as ascendant pathogens in bacteremias and in urinary tract, wound, and post-surgical infections (1–3). Vancomycin has been a front line antibiotic against enterococci, but the increased reliance on this drug has led to a greater than 20-fold increase in a 6-year period in resistant isolates, from less than 0.5% of nosocomial isolates in 1989 to 10% by 1995 (4).
Three vancomycin resistance enterococcal (VRE) phenotypes have been defined as VanA, VanB, or VanC based on patterns of resistance to specific drugs and to chromosomal or plasmid loci for the resistance genes. Antibiotic resistance phenotypes of VanA and VanB glycopeptide are distinguished in that VanA shows resistance to both vancomycin and teicoplanin, whereas VanB is resistant to vancomycin but susceptible to teicoplanin [minimal inhibitory concentration (MIC) ≤ 0.5 μg/ml] (5). Otherwise, VanA and VanB phenotypes are molecularly related, arising from the plasmid-encoded expression of five necessary and sufficient genes vanR, S, H, A, X in enterococci such as Enterococcus faecalis and Enterococcus faecium (6, 7). vanS and R encode a two-component sensor/response regulator system and thereby control expression of the three enzymes VanH, A, and X that lead to production and accumulation of depsipeptide d-Ala-d-lactate rather than the prototypic dipeptide d-Ala-d-Ala. Subsequent incorporation of d-Ala-d-lactate at the termini of peptidoglycan (PG) intermediates leads to a 1,000-fold drop in binding affinity of vancomycin (compared with a d-Ala-d-Ala PG terminus) and accounts for phenotypic resistance (8).
The genotypic and molecular characteristics of the third VRE phenotype VanC differs from those of VanA and VanB. VanC presents a modest resistance to vancomycin (MIC = 2–32 μg/ml) with sensitivity retained to teicoplanin (MIC ≤ 0.5 μg/ml) (3, 5). Unlike VanA and VanB, the VanC resistance gene(s) in such organisms as Enterococcus gallinarum (9) and Enterococcus casseliflavus (10, 11) is chromosomal, rather than on plasmid. PG intermediates from VanC enterococci have been analyzed and found not to terminate in either d-Ala-d-Ala (which would predict antibiotic sensitivity) or d-Ala-d-lactate (as in VanA and VanB), but instead in d-Ala-d-Ser (12). This has led to the prediction that VanC, a d-Ala-d-Ala ligase homologue by sequence analysis (9, 11), would encode a d-Ala-d-Ser ligase and that a lipid pentapeptide terminating in d-Ala-d-Ser would lead to a lower affinity for vancomycin in the PG cross-linking steps. In this paper, we report overproduction of an E. casseliflavus VanC ligase (VanC2) (11) in Escherichia coli, its purification, and enzymatic characterization as a d-Ala-d-Ser ligase with 400-fold preference for producing d-Ala-d-Ser instead of d-Ala-d-Ala.
MATERIALS AND METHODS
Materials.
E. casseliflavus ATCC 25788 was purchased from the American Type Culture Collection. Oligonucleotides were from Integrated DNA Technologies (Coralville, IA) and restriction enzymes and polymerases were from New England Biolabs. d-cycloserine, ATP, all amino acids, d-lactate, and buffers were purchased from Sigma. d-[14C]-Ala (0.1 mCi/ml, 0.55 μCi/μmol; 1 Ci = 37 GBq) and d-[14C]-Ser (0.1 mCi/ml, 0.55 μCi/μmol) were from American Radiolabeled Chemicals (St. Louis), and thin-layer chromatography (TLC) cellulose sheets were from Kodak. Phosphinate analog of d-Ala-d-Ala (d-3-[(1-aminoethyl)phosphinyl]-d-2-methylpropionic acid) was a generous gift from P. A. Bartlett and B. A. Ellsworth (Department of Chemistry, University of California, Berkeley).
Cloning, Overexpression, and Purification.
E. coli d-Ala-d-Ala ligase B (DdlB) and VanA were prepared as described (13). vanC2 was cloned by PCR of E. casseliflavus genomic DNA, by using two primers designed from the reported sequences (GenBank accession no. L29638) (11). Primer 1 (CGGTC GAGAG GAAGG AAGAA ACATA TGAAA AAAAT CGCCA TTATT TTTGG) contains a XbaI restriction site, Shine–Dalgano sequence, and upstream sequences of vanC2. Primer 2 (GCGGA TCCCTC ATTTGA CTTCCT CCTTTG CTAAGA CAAG) contains a BamHI restriction site and downstream sequences of vanC2. PCR was performed as reported previously (13). The DNA fragment was subcloned into the XbaI–BamHI site of pET22b (Novagen), transformed into E. coli DH5α, and subsequently into E. coli BL21(DE3). Overexpression and purification were performed by essentially the same method previously described (13) with the exception that in the purification the 20–40% ammonium sulfate soluble fraction was saved. About 15 mg of >90% pure (on SDS/polyacrylamide gel) VanC2 protein was recovered from ≈5 g cell (wet weight) (see Fig. 1). Protein amount was quantified by using Bradford assay with BSA as standard (14).
Figure 1.

Purification of VanC2. Each stage of purification was analyzed on SDS/polyacrylamide gel. The numbers indicate molecular weights (in kDa). CE, supernatant of cell extract; Am, 20–40% soluble fraction of ammonium sulfate precipitation; GF, gel filtration column fraction; Q, Q-Sepharose chromatographic fraction.
Enzyme Assay.
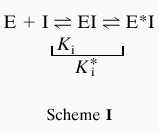
Enzyme assay was performed by TLC using radioactive substrate or by coupled ADP release assay where ADP formation was coupled to NADH reduction by pyruvate kinase and l-lactate dehydrogenase as described (13). The same reaction condition (100 mM Hepes, pH 7.5/10 mM KCl/10 mM MgCl2) has been used throughout this study. Authentic d-Ala-d-Ser chemically synthesized was used to identify the product in TLC assay (data not shown). Analysis of kinetics and equations used are essentially the same as those described previously (13, 15). For calculation of inhibitory parameters of the slow binding phosphinate inhibition Eq. 1 was used based on Scheme I.
 |
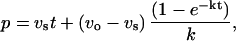 |
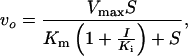 |
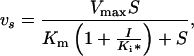 |
1 |
where p = product, vs = velocity at time t, vo = initial velocity, S = concentration of substrate, k = inactivation rate constant, I = inhibitor concentration, Ki and Ki* = inhibition constants as defined in Scheme I.
RESULTS
The vanC2 gene from E. casseliflavus ATCC 25788, previously reported by Navarro and Courvalin (11), was amplified by PCR and subcloned for overexpression in E. coli. As seen in Fig. 1, the 38 kDa VanC2 could be readily purified in quantity (≈15 mg/liter of culture) from E. coli extracts. Because it was recovered at a similar elution time on gel filtration as DdlB, which is a dimer of a 32 kDa polypeptide, VanC2 also appears to be a dimer. The dipeptide ligase activity could be screened either by TLC analysis using radioactive substrates (e.g., d-[14C]-Ala or d-[14C]-Ser) to search for dipeptide products or by a nonradioactive, spectrophotometric assay in which amino acid dependent cleavage of ATP to ADP could be continuously monitored. As shown in Fig. 2A by TLC analysis, d-Ala-d-Ser is made by VanC2 but not by the DdlB or the d-Ala-d-lactate depsipeptide ligase VanA as assessed with d-[14C]-Ser in the presence of unlabeled d-Ala. With 14C-labeled d-Ala, lanes 5–8 show that whereas E. coli DdlB makes d-Ala-d-Ala dipeptide, this product is not detected by autoradiography in incubations containing VanC2 or VanA, arguing that under these condition (pH 7.5) neither VanC2 or nor VanA has substantial d-Ala-d-Ala ligase activity. Fig. 2B shows that VanC2 can also activate d-Asn to produce the d-Ala-d-Asn dipeptide product. The TLC assay was used to establish a broad pH optimum between pH 8 and pH 9.5 (data not shown) for the d-Ala-d-Ser ligase activity of VanC2. To test more broadly for d-Ala-d-X formation, VanC2 was assayed by ADP production in the presence of 10 mM d-Ala (to fill subsite 1 and generate enzyme bound d-alanyl phosphate) and addition of 10 mM d- or 20 mM d,l-amino acids. As noted in Fig. 3A, only d-Ala-d-Ser and d-Ala-d-Asn gave strong product signals in this screen. In particular, essentially no d-Ala-d-Ala product was detectable. Small signals for d-Thr, d-Gln, d-aminobutyrate, and d-homoserine prompted the kinetic analysis presented in Fig. 3B, which show the preference d-Ser > d-Asn > d-Thr > d-Gln > d-homoserine, d-aminobutyrate, d-allo-Thr by Km and Vmax criteria. Catalytic efficiency rates are collected in Table 1 along with some Km and kcat values. The Km for d-Ala1 is 1.8 mM, whereas the Km for d-Ser as the C-terminal substrate is similar at 1.8 mM. There was very little competition for d-Ala at site 2 with d-Ser or reciprocally of d-Ser for d-Ala at site 1 as assessed by the very low rates of d-Ala-dependent or of d-Ser-dependent ATPase activity. Whereas the TLC assay could detect trace amounts of d-[14C]-Ala-d-Ala (0.69 mM−1min−1), there was no detectable [14C]-Ser-Ser when [14C]-Ser and ATP were incubated with VanC2 and assayed by TLC. With regard to kcat, d-Asn is as good (kcat = 532 min−1) a substrate as d-Ser (kcat = 490 min−1), but with a 4-fold higher Km2 (7.2 mM vs. 1.8 mM), the catalytic efficiency for yielding d-Ala-d-Asn is only 25% that for d-Ala-d-Ser. The other β-hydroxy amino acid, d-Thr is down 150-fold in kcat/Km2 compared with d-Ser. Notably, VanC2 is about 400-fold more efficient in using d-Ser in place of d-Ala to make d-Ala-d-Ser rather than d-Ala-d-Ala.
Figure 2.

Enzyme assay by TLC. The reaction mixture (10 μl) contained 0.34 μg of purified VanC2, 5 mM ATP, 10 mM d-amino acids, and radiolabeled substrate (d-[14C]-Ala or d-[14C]-Ser) at a concentration of 0.2 mM plus unlabeled corresponding d-amino acid substrate to make the final concentration 10 mM. Enzyme reactions were performed at room temperature for 16 hr. No Enz indicates no enzyme was included in reaction mixture.
Figure 3.

Enzyme assay by ADP-coupled assay. The reaction mixture (1 ml) in A contained 34 μg VanC2 protein, 5 mM ATP, 10 mM d-Ala plus either 20 mM dl- or 10 mM d-isomers of the indicated amino acids. In B the mixture contained 17 μg VanC2 protein, 10 mM d-Ala plus d-Ser (○), d-Asn (▪), d-Thr (▴), d-Gln (□), d-homoserine (•), d-aminobutyrate (+), d-allo-Thr (◊), d-Asp (▵), or d-Cys (×), respectively.
Table 1.
VanC2 activities
| Activity | Km1, mM | Km2, mM | kcat, min−1 | kcat/Km2, mM−1 min−1 | Km(ATP), mM |
|---|---|---|---|---|---|
| d-Ala-d-Ser | 1.8 | 1.8 | 490 | 270 | 1.9* |
| d-Ala-d-Asn | 7.2 | 532 | 74 | ||
| d-Ala-d-Thr | 1.7 | ||||
| d-Ala-d-Gln | 1.3 | ||||
| d-Ala-d-Ala | 0.69 | ||||
| d-Ala-d-homoserine | 0.55 | ||||
| d-Ala-d-aminobutyrate | 0.29 | ||||
| d-Ala-d-allo-Thr | 0.26 |
Determined using 10 mM d-Ala and 10 mM d-Ser.
Initial studies on inhibitors of VanC2 indicated an IC50 of 0.8 mM for d-cycloserine, a known Ddl and VanA inhibitor (16), and revealed time dependent onset of inhibition by the phosphinate analog of d-Ala-d-Ala (17), consistent with the slow binding mode seen for DdlB and VanA by this compound (16) (Fig. 4). The Ki* for VanC2 was estimated to be 2.7 μM.
Figure 4.

Inhibition of VanC2 by cycloserine and phosphinate mimic of d-Ala-d-Ala. Reaction mixture (1 ml) contained 17 μg VanC2 protein, 5 mM ATP, 10 mM d-Ala, and 2 mM d-Ser plus d-cycloserine (A) or 7.8 μg VanC2 protein, 10 mM d-Ala, and 10 mM d-Ser plus phosphinate inhibitor (B). Both were measured by ADP-coupled assay.
DISCUSSION
Overproduction and purification of the E. casseliflavus VanC2 dipeptide ligase from extracts of E. coli have validated the expectation that VanC2 is indeed a d-Ala-d-Ser ligase. PCR analysis in E. casseliflavus had previously indicated that in addition to VanC2 there was also a d-Ala-d-Ala ligase encoded by a ddl gene (11). Although the PG intermediate of E. casseliflavus has not been analyzed for the presence of d-Ala-d-Ser, this was validated by studies in E. gallinarum carrying VanC1. The analysis of PG intermediates in strain BM 4174 of this bacteria found UDP-N-acetylmuramic acid pentapeptide terminating in d-Ala-d-Ser, but in strain BM 4175 derived from BM 4174 by insertional inactivation of VanC1 the pentapeptide intermediate terminated in d-Ala-d-Ala (9, 12). Analogously, the VanC-mediated resistance in E. gallinarum AIB39 was constitutive and correlated with d-Ala-d-Ser PG termini, whereas in E. gallinarum GS1 vancomycin resistance was inducible, with d-Ala-d-Ala PG termini seen before induction being supplanted by d-Ala-d-Ser PG termini after induction, presumably as a consequence of regulated expression of VanC, to make d-Ala-d-Ser available (18). Thus, there can apparently be differential regulation of d-Ala-d-Ser ligase and d-Ala-d-Ala ligase activation in VanC phenotype resistance.
This work establishes a 400-fold difference in catalytic efficiency of VanC2 for using d-Ser over d-Ala in the second amino acid subsite. The molecular basis of the discrimination will require structural comparison of VanC2 vs. Ddl; we have reported x-ray structures of both wild-type E. coli DdlB (19) and a Y216F mutant that has gained depsipeptide ligase activity (20). Efforts to crystallize VanC2 are now underway. In addition to d-Ser as the nucleophilic cosubstrate, VanC2 also shows the capacity to activate d-Asn with equivalent kcat but 4-fold lessened Km2. The other 18 proteogenic amino acids are not well recognized suggesting that the d-Asn activity is not merely a lack of selectivity. It is not likely that d-Asn is a relevant substrate for VanC2 physiologically in that it was not reported in PG termini from VanC-type resistant enterococci (12). Nor is it likely that d-Asn is generated at appreciable concentration (Km2 for VanC2 = 7 mM) in bacterial metabolism. In fact, the origin of d-Ser (Km2 = 1.8 mM) in enterococci is not yet clear. One possibility is via racemase action. The d-Ser racemase activity of Ala racemases is low (e.g., for Salmonella Alr) (21) but has not been explicitly analyzed with an enterococcal Ala racemase. A Thr α-epimerase has been reported from Pseudomonas putida (22) but is in fact a broad specificity pyridoxal–phosphate dependent amino acid racemase. Intriguingly, Ser has 3-fold higher activity with that epimerase than Thr, and Asn 4-fold higher, raising the prospect that if there were an equivalent racemase in enterococcal strains, it could produce either d-Ser or d-Asn from the l-enantiomers. A separate route, precedented in other Gram-positive bacteria, would be transamination of hydroxypyruvate by a low specificity d-amino acid transaminase (23) and this would also be a potential route to d-Asn. Lastly, there is the formal possibility that a d-Ser dehyratase could run in the reverse direction, but it is highly unlikely that the process involving hydration of the ene-amino acid aminocrotonate is in fact readily reversible.
Given the essentially exclusive ability of VanC2 to make d-Ala-d-Ser and not d-Ala-d-Ala, the strategy of two ligases, VanC2 and Ddl, in VanC phenotypes of VREs bears further analysis. In the constitutively resistant stains, either only VanC2 and not Ddl is expressed or there would need to be a companion VanX, as in the VanA and VanB phenotypes (7), to hydrolyze d-Ala-d-Ala (generated by Ddl action) to prevent its accumulation and carry forward by MurF into PG pentapeptides. Reynolds et al. (12) did detect VanX activity in extracts of E. gallinarum BM 4174 (but not in BM 4175, suggesting negative regulation). However, VanX from VanA VRE showed d-Ala-d-Ser hydrolyzing activity with one-third the activity of d-Ala-d-Ala dipeptidase (24). If VanX is indeed performing a hydrolytic surveillance function in VanC VREs then it suggests d-Ala-d-Ser must be a much poorer substrate for hydrolysis than d-Ala-d-Ala in those enterococci. In the equivalent competition in VanA and VanB resistance phenotypes between d-Ala-d-Ala and d-Ala-d-lactate, we have reported greater than 106-fold selectivity of VanX (24).
In the three phenotypes (VanA, VanB, and VanC) of clinically significant enterococcal resistance to vancomycin there are two types of variant dipeptide ligases that create products that when taken forward produce PG termini with lowered affinity and consequent higher MICs for vancomycin and differentially, teicoplanin: alteration of either the amide linkage or of the C-terminal methyl side chain in the d-Ala-d-Ala terminus of the cell wall (Fig. 5). The depsipeptide ligases VanA and VanB generate d-Ala-d-lactate to replace the amide NH with the ester oxygen, while leaving the C-terminal methyl side chain interact. This action causes a 1,000-fold decrease in vancomycin binding to PG tetrapeptide-d-lactate cell wall intermediates (8) and a correlative three logarithm increase in vancomycin MIC (5, 7). In comparison, the VanC strains are only moderately resistant, 2–32 times the typical MIC of a sensitive strain. For example, when the vanC gene in E. gallinarum BM 4174 was insertionally inactivated, the vancomycin MIC dropped 8-fold from 16 μg/ml to 2 μg/ml (9). This suggested that the change of the C-terminal methyl side chain in d-Ala-d-Ala to the -CH2OH in d-Ala-d-Ser had only a one logarithm effect. This was validated by direct binding assay that indicated that N-acetyl-d-Ala-d-Ser had only a 6-fold lower affinity for vancomycin than N-acetyl-d-Ala-d-Ala (25).
Figure 5.

Vancomycin resistance. See text for details.
It is intriguing that the increase in steric bulk in d-Ala-d-Ser vs. d-Ala-d-Ala on the surface interacting directly with the convex face of vancomycin would result in creating ≈10-fold resistance. This is 80- to 100-fold less disruptive than the substitution of d-Ala-d-Ala with d-Ala-d-lactate (amide vs. ester). On the one hand, it correlates with the higher frequency of VanA and VanB VRE phenotypes over VanC. On the other hand, it forces deeper analysis of the molecular forces between the altered PG termini and vancomycin. The d-Ala-d-lactate PG termini compared with d-Ala-d-Ser termini have lost one of the five hydrogen bonds between N-acyl dipeptide and the antibiotic and there is also the lone pair repulsion of the ester oxygen of d-Ala-d-lactate to the vancomycin backbone carbonyl (26). Most intriguing are recent studies from Williams and his colleagues (27) that differential effects on vancomycin dimerization induced by the different ligands may be a key quantitative determinant in d-Ala-d-Ala vs. d-Ala-d-lactate recognition. These studies should be extended to d-Ala-d-Ser termini. Two additional questions raised by the initial characterization of VanC2 as a d-Ala-d-Ser ligase are: (i) why VanC strains remain completely sensitive to teicoplanin even while constitutively resistant to vancomycin and (ii) whether combination of a VanA and VanC phenotype will emerge with a depsipeptide ligase that makes d-Ala-d-glycerate and a multiplicative, 103 × 10 = 104 resistance to vancomycin compared with sensitive parent enterococcal strains (e.g., Fig. 5, lower right hand corner).
Acknowledgments
We thank Vicki Healy for help and discussion and laboratory members for discussion. We also thank Dr. P. Courvalin (Pasteur Institute) for the vanC2 plasmid. This work was supported in part by National Institutes of Health Grant GM49338.
ABBREVIATIONS
- Ddl
d-Ala-d-Ala ligase
- MIC
minimal inhibitory concentration
- PG
peptidoglycan
- TLC
thin-layer chromatography
- VRE
vancomycin resistance enterococci
References
- 1.Moellering R C., Jr J Antimicrob Chemother. 1991;28:1–12. doi: 10.1093/jac/28.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Schaberg D R. Ann Emergency Med. 1994;24:462–464. doi: 10.1016/s0196-0644(94)70185-7. [DOI] [PubMed] [Google Scholar]
- 3.Gold H S, Moellering R C., Jr N Engl J Med. 1996;335:1445–1453. doi: 10.1056/NEJM199611073351907. [DOI] [PubMed] [Google Scholar]
- 4.Gaynes R P, Edwards J R, Jarvis W R, Culver D H, Tolson J S, Martone W J. Pediatrics. 1996;98:357–361. [PubMed] [Google Scholar]
- 5.Arthur M, Courvalin P. Antimicrob Agents Chemother. 1993;37:1563–1571. doi: 10.1128/aac.37.8.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh C T. Science. 1993;261:308–309. doi: 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 7.Walsh C T, Fisher S L, Park I S, Prahalad M, Wu Z. Chem Biol. 1996;3:21–28. doi: 10.1016/s1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- 8.Bugg T D, Dutka-Malen S, Arthur M, Courvalin P, Walsh C T. Biochemistry. 1991;30:2017–2021. doi: 10.1021/bi00222a002. [DOI] [PubMed] [Google Scholar]
- 9.Dutka-Malen S, Molinas C, Arthur M, Courvalin P. Gene. 1992;112:53–58. doi: 10.1016/0378-1119(92)90302-6. [DOI] [PubMed] [Google Scholar]
- 10.Leclercq R, Dutka-Malen S, Duval J, Courvalin P. Antimicrob Agents Chemother. 1992;36:2005–2008. doi: 10.1128/aac.36.9.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navarro F, Courvalin P. Antimicrob Agents Chemother. 1994;38:1788–1793. doi: 10.1128/aac.38.8.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reynolds P E, Snaith H A, Maguire A J, Dutka-Malen S, Courvalin P. Biochem J. 1994;301:5–8. doi: 10.1042/bj3010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park I S, Walsh C T. J Biol Chem. 1997;272:9210–9214. doi: 10.1074/jbc.272.14.9210. [DOI] [PubMed] [Google Scholar]
- 14.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 15.Park I S, Lin C H, Walsh C T. Biochemistry. 1996;35:10464–10471. doi: 10.1021/bi9603128. [DOI] [PubMed] [Google Scholar]
- 16.Bugg T D, Wright G D, Dutka-Malen S, Arthur M, Courvalin P, Walsh C T. Biochemistry. 1991;30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 17.Duncan K, Walsh C T. Biochemistry. 1988;27:3709–3714. doi: 10.1021/bi00410a028. [DOI] [PubMed] [Google Scholar]
- 18.Sahm D F, Free L, Handwerger S. Antimicrob Agents Chemother. 1995;39:1480–1484. doi: 10.1128/aac.39.7.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan C, Moews P C, Walsh C T, Knox J R. Science. 1994;266:439–443. doi: 10.1126/science.7939684. [DOI] [PubMed] [Google Scholar]
- 20.Fan C, Park I S, Walsh C T, Knox J R. Biochemistry. 1997;36:2531–2538. doi: 10.1021/bi962431t. [DOI] [PubMed] [Google Scholar]
- 21.Esaki N, Walsh C T. Biochemistry. 1986;25:3261–3267. doi: 10.1021/bi00359a027. [DOI] [PubMed] [Google Scholar]
- 22.Lim Y H, Yokoigawa K, Esaki N, Soda K. J Bacteriol. 1993;175:4213–4217. doi: 10.1128/jb.175.13.4213-4217.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yonaha K, Misono H, Yamamoto T, Soda K. J Biol Chem. 1975;250:6983–6989. [PubMed] [Google Scholar]
- 24.Wu Z, Wright G D, Walsh C T. Biochemistry. 1995;34:2455–2463. doi: 10.1021/bi00008a008. [DOI] [PubMed] [Google Scholar]
- 25.Billot-Klein D, Blanot D, Gutmann L, van Heijenoort J. Biochem J. 1994;304:1021–1022. doi: 10.1042/bj3041021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schafer M, Schneider T R, Sheldrick G M. Structure. 1996;4:1509–1515. doi: 10.1016/s0969-2126(96)00156-6. [DOI] [PubMed] [Google Scholar]
- 27.Cho Y R, Maguire A J, Try A C, Westwell M S, Groves P, Williams D H. Chem Biol. 1996;3:207–215. doi: 10.1016/s1074-5521(96)90264-1. [DOI] [PubMed] [Google Scholar]