

Abstract
To date, 11 gene loci that contribute to familial Parkinson's disease (PD) are known. Of these, mutations in six genes have been identified, allowing genetic testing and more accurate phenotypic characterization of genetically defined disease subtypes. In particular, mutations in Parkin, DJ-1, and Pink1 genes are associated with autosomal recessive PD and may also play a major role in early onset PD (EOPD). However, genetic testing for sequence alterations in these genes remains laborious. Therefore, our aim was to develop a flexible, rapid, high-throughput screening procedure using matrix-assisted laser desorption ionization/time of flight technology and homogeneous mass cleave assays. Using this novel approach, we screened all 27 coding exons of the Parkin, DJ-1, and Pink1 genes in 31 patients with EOPD, a total of 367,195 nucleotides. Four positive controls with known autosomal recessive PD mutations that had previously been screened by denaturing high performance liquid chromatography in combination with sequencing were also tested. All known alterations were detected by matrix-assisted laser desorption ionization/time of flight mass spectrometer, as well as additional polymorphisms in formerly unscreened regions. Overall, two previously described mutations in three patients with EOPD, 27 known polymorphisms with 386 occurrences, and eight unknown variants with 21 occurrences were detected. In total, we identified 410 sequence alterations in 31 patients with EOPD. In conclusion, this is the first study using matrix-assisted laser desorption ionization/time of flight mass spectrometry and homogeneous mass cleave assay for high-throughput mutation screening.
Parkinson's disease (PD) is the second most common neurodegenerative disorder in humans. Although in the majority of the patients with PD, the disease-causing factors have not yet been defined, genetic factors play an important role in PD pathogenesis. Among patients with early manifestation of the disease, there is a high incidence of affected first-degree relatives, indicating monogenic factors may play a role in pathogenesis. Over the last few years, a steadily growing number of mutations in genes that contribute to the susceptibility for PD have been detected in these patients. To date, 11 PD disease loci have been discovered, and ongoing studies will most probably reveal further loci in the future.1 Among the known loci, PARK2 (Parkin), PARK6 (PINK1), and PARK7 (DJ-1) contribute significantly to the pathogenesis of autosomal recessive forms of PD (ARPD).2,3,4 In particular, mutations in Parkin play an important role in early onset PD (EOPD) and have been identified in up to 50% of patients with at least one affected sibling.5 It is estimated that mutations in DJ-1 occur in approximately 1 to 2% of the patients with PD and mutations in PINK1 are rather rare.6,7 With this increase in knowledge, accurate and informative genetic counseling advice can now be offered to families with EOPD. Therefore, a flexible and cost-effective high-throughput method is required to link progress in research with clinical routine. In the long run, an early diagnosis may allow protraction of initiation of ARPD already in a presymptomatic state. However, at the moment, the classification of patients with PD according to their underlying genetic defect is extremely important to make phenotype-genotype correlations. This is required to find disease and progression biomarkers, which, in turn, are required for natural history and therapeutic studies, which may finally lead to the development of effective therapeutic strategies.
To establish a new diagnostic tool for these demands, we used the MassArraySystem (Sequenom, Hamburg, Germany) with a matrix-assisted laser desorption ionization/time of flight mass spectrometer (MALDI-TOF MS) to screen the Parkin, DJ1, and Pink1 genes for mutations in patients with PD. In the last decade, MALDI-TOF MS has been rapidly developed for molecular biology purposes with the advantage that it may be used in high-throughput applications.8 To date, molecular haplotyping, expression profiling, DNA methylation analysis, single-nucleotide polymorphism (SNP) genotyping, and mutation screening have become increasingly feasible using MALDI-TOF MS.9 Stanssens and colleagues10 were able to successfully identify new sequence alterations (SNPs) in three exons of the CETP gene using homogeneous mass cleave (hMC) assays in combination with MALDI-TOF MS. We were encouraged by these results, and chose 27 exons in three different genes to evaluate the feasibility of precise and fast detection of mutations with respect to diagnostic needs. In this study, we tested four patients with PD with known mutations in the Parkin, DJ1, and Pink1 genes and 31 genetically unclassified patients with early-onset PD for mutations in the three genes.
Materials and Methods
Patients and Samples
In total, we analyzed 31 uncharacterized patients with PD younger than 55 years and four patients with ARPD with previously known mutations. Diagnosis of PD had been established according to UK brain bank criteria, and patients had given informed consent to genetic analyses of PD. Sixteen patients had already been screened for single genes using other methods. DNA from 12 patients was extracted from blood cells using a standard protocol.11 The remaining samples were isolated using Roche's Magnapure compact instrument (Roche, Penzberg, Germany) following the manufacturer's protocol. No difference in the quality of the hMC assay data were detected based on the different DNA isolation protocols used.
Polymerase Chain Reaction (PCR)
Primers were designed with Primer3 using default settings and an average amplicon length of about 300 to 700 bp (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi; last accessed October 2006). Oligonucleotides were synthesized by Metabion (Martinsried, Germany). Forward and reverse primers were synthesized in T7 and hMC-tagged versions (see Supplemental Table 1 at http://jmd.amjpathol.org). PCR protocols were established for Parkin with 12 exons plus an alternative 3′-end vega-transcript, DJ-1 with seven exons, and PINK1 with eight exons. Large exons were split into overlapping amplicons of approximately 400 nucleotides in length. All but three amplicons could be amplified using the same PCR protocol (described below). GC-rich exons were amplified with Slowdown-PCR, because according to the manufacturer, the use of PCR enhancers such as dimethyl sulfoxide or betaine may alter the MS spectra. Slowdown-PCR was used for exon 1 of Parkin and DJ-1. Exon 1 of PINK1 was amplified by Slowdown PCR with 7-deaza-dGTP.12 Our standard PCR protocol was performed using a commercial PCR kit (HotStart Taq, Qiagen, Hilden, Germany). The PCR assay consisted of 1X buffer containing 15 mmol/L MgCl2, Tris-HCl, KCl, (NH4)2SO4, pH 8.7 at 20°C; 20 ng of DNA, 0.2 U of HotStart Taq-DNA polymerase, 2 mmol/L MgCl2, 200 nmol/L forward and reverse primer, and 200 μmol/L deoxynucleoside-5′-triphosphates (Invitrogen, Germany) in a final volume of 10 μl. An ep384 thermal cycler (Eppendorf, Germany) was used for DNA amplification. As each DNA region was amplified twice using one hMC primer and the corresponding T7-tag elongated forward or reverse primer, 192 different amplicons were placed into one 384 microtiter plate.
Processing Homogeneous Mass Cleave Reactions
All unincorporated deoxynucleoside-5′-triphosphates were dephosphorylated using 0.3 U of shrimp alkaline phosphatase (37°C for 20 minutes, 85°C for 5 minutes). Each amplicon was split into two in vitro transcription reactions performed in new 384-well plates. The transcription mix (5X T7-polymerase buffer, C- or T-cleavage mix, respectively, 5 mmol/L dithiothreitol, 20 U/reaction T7 RNA/DNA-polymerase, 5.4 ng of RNase A) was added to 2 μl of PCR/SAP mix and each amplicon was transcribed in a unidirectional manner according to the location of the T7-tag. Both plates were incubated at 37°C for 3 hours. After the addition of water, the assay was conditioned using Clean-resin (Sequenom) and incubated for 10 minutes to remove ions before MS analysis.
MALDI-TOF MS Measurement
Data management followed the preconditions of SNP Discovery software version 1.2.3.43 2002 (Sequenom) and MS control using MassArray Typer 3.1 RT (Sequenom). The sequences for weight comparison were taken from Ensembl (www.ensembl.org; last accessed June 2007). Fifteen nanoliters of each cleavage reaction were spotted on SpectroCHIPs G384 with a nanodispenser (Sequenom). Chips were air-dried and measured in an Autoflex MALDI-TOF MS (Sequenom).
Sequence Analysis
Several variations were validated using conventional dideoxy sequencing. CEQ GenomeLab DTCS Quick Start kit with CEQ sequencing reaction buffer (Beckman Coulter, Krefeld, Germany) was used following the manufacturer's instructions. We performed both MALDI-TOF MS and sequence analysis using the same PCRs. After the cycle-sequencing reaction, the assays were conditioned with CleanSeq magnetic beads (Agencourt Bioscience) according to the delivered protocol and analyzed on a Beckman CEQ8000 sequence analyzer.
Results
Overall Performance
The aim of this study was to establish a flexible system to detect mutations in genes responsible for ARPD with a high resolution and high sample throughput. Due to its high resolution, MALDI-TOF MS is an ideal tool to measure mass differences with a resolution up to 2 to 5 da. Mass shifts generated by single-nucleotide exchanges in sequences vary between 9 and 40 da per nucleotide and thus mutations and polymorphisms can readily be detected with MALDI-TOF MS. Each amplicon is transcribed in both orientations to generate single strands. Both strands are cleaved after C and T, respectively, generating mass spectra for each of the four nucleotides. Deviations from the calculated masses are used to predict sequence changes. An overview of the general workflow is presented in Figure 1.
Figure 1.
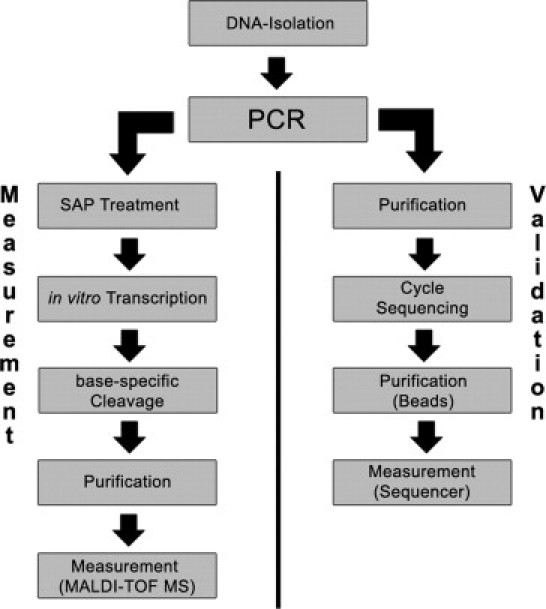
Schematic overview of the workflow. A schematic summary of hMC assay and MALDI-TOF MS measurement is shown on the left. See Materials and Methods section for details. After identification of a sequence alteration, validation was performed using standard (Sanger) sequencing (right).
We designed PCR primers for all exons and adjacent intron sequences for Parkin, PINK1 and DJ1 (see Materials and Methods for details) that were suitable for subsequent hMC analysis.13 Overall, we screened 11,845 nucleotides per patient and performed 2418 PCRs within 2 weeks with the support of pipetting devices and a 384 microtiter plate format. The measurement of the products with MALDI-TOF MS on 16 384 wafers took about 2 days. Altogether, only 47 of 4836 reactions failed (0.97%), and an additional 42 reactions could not be analyzed due to bad spectral quality (0.87%). As the cleavage reactions are redundant in this system, only three sets of cleavage reactions of 1209 were not interpretable (0.25%). Generally, we observed T-cleavage reactions to be more stable, and they caused fewer quality problems compared with C-cleavage reactions.
Detecting Single Base Changes and Mutations
We were able to detect single base changes (substitutions) correctly. Detection of single-nucleotide insertions and deletions is possible as well. However these did not occur in our set of samples. Among the detected sequence changes, we found several previously described mutations (Table 1), common SNPs (Table 1) and novel sequence variations (Table 2). In exon 7 of the Parkin gene, a C to T transition causing the amino acid exchange R275W was found in two patients (Figure 2). Also in PINK1, we detected a previously described G to A mutation in exon 7, which leads to an E to L substitution at position 475 in the protein.15 Several other changes other than common SNPs were confirmed. However, their clinical significance still requires further evaluation (Table 2).
Table 1.
Previously Described Mutations and Known SNPs and Disease-Unrelated Variations Found Among Patients with EOPD
| Frequency |
||||||
|---|---|---|---|---|---|---|
| Gene | Exon | Nucleotide | Amino acid | ho | he | Accession Number/Reference |
| Mutations | ||||||
| Parkin | Exon 7 | 823C>T | R275W | 1× | 1× | 14 |
| Pink1 | Exon 7 | 1426G>A | E476L | 1× | 15 | |
| SNPs | ||||||
| Parkin | IVS2 | +24T>C | intronic | 5× | 7× | rs2075923 |
| Parkin | IVS4 | −20T>C | intronic | 29× | 2× | rs4709583 |
| Parkin | Exon 4 | 500G>A | S167N | 1× | rs1801474 | |
| Parkin | IVS8 | −68C>G | intronic | 10× | 14× | rs3765475 |
| Parkin | IVS8 | −35G>A | intronic | 10× | 13× | rs3765474 |
| Parkin | IVS8 | +48C>T | intronic | 2× | 11× | rs10945756 |
| Parkin | Exon 10 | 1146G>C | V380L | 1× | 4× | 14 |
| Parkin | Exon 11 | 1281G>A | N394D | 1× | 14 | |
| Parkin | Exon 12 | 2797A>G | 3′UTR | 3× | rs3734464 | |
| Parkin | Exon 12 | 3186C>T | 3′UTR | 1× | rs16892481 | |
| Pink1 | Exon 1 | 189C>T | L63L | 8× | 15 | |
| Pink1 | Exon 1 | 344A>T | Q115L | 1× | 15 | |
| Pink1 | IVS2 | −7A>G | intronic | 30× | rs2298298 | |
| Pink1 | IVS5 | −5A>G | intronic | 26× | 4× | rs3131713 |
| Pink1 | Exon 5 | 1018G>A | A339T | 2× | rs3738136 | |
| Pink1 | Exon 8 | 1562A>C | T521N | 2× | 13 | rs1043424 |
| Pink1 | Exon 8 | 1783A>T | 3′UTR | 26× | 4× | rs686658 |
| Pink1 | Exon 8 | 1927C>G | 3′UTR | 27× | 3× | rs513414 |
| Pink1 | Exon 8 | 1941T>C | 3′UTR | 2× | 13× | rs1043443 |
| Pink1 | Exon 8 | 2011G>T | 3′UTR | 13× | 14 | rs512550 |
| Pink1 | Exon 8 | 2161C>G | 3′UTR | 5× | 3× | rs8064 |
| Pink1 | Exon 8 | 2310T>G | 3′UTR | 2× | 12× | rs1043502 |
| Pink1 | Exon 8 | 2512T>C | 3′UTR | 6× | 8× | rs2078073 |
| Pink1 | Exon 8 | 2515_2516insCTT | 3′UTR | 6× | 16× | rs3077908 |
| DJ-1 | Exon 1 | −53G>A | 5′UTR | 6× | rs17523802 | |
| DJ-1 | Exon 1 | −14T>C | 5′UTR | 12× | 5× | rs226249 |
| DJ-1 | Exon 5 | 293G>A | R98Q | 1× | 2× | 16, 17 |
Table 2.
Unknown Variations for Which the Clinical Significance Has Not Yet Been Defined and Which Were Not Found in Public Databases or Publications
| Variation |
Frequency |
||||
|---|---|---|---|---|---|
| Gene | Exon | Nucleotide | Amino acid | ho | he |
| Parkin | Exon 12 | 2059C>T | 3′UTR | 4× | 6× |
| Parkin | Exon 12 | 2227A>T | 3′UTR | 1× | |
| Parkin | Exon 12 | 2659C>T | 3′UTR | 1× | |
| Parkin | Exon 12 | 2753C>T | 3′UTR | 1× | |
| Parkin | Exon 12 | 2887A>G | 3′UTR | 1× | |
| Parkin | Exon 12 | 3177A>C | 3′UTR | 1× | |
| Parkin | Exon 12 | 3187G>A | 3′UTR | 1× | |
| Pink1 | IVS6 | +43C>T | Intronic | 1× | 4× |
Figure 2.

Detection of substitutions with hMC-derived mass spectra in the heterozygous and homozygous state. Mass spectra of the T reverse reaction of a wild type (top), heterozygous (middle), and homozygous (bottom) C253T mutation in exon 7 of Parkin. The sequence in the left part of the figure indicates the variation and the masses of the indicative fragments (underlined). The substitution from G to A in the reverse reaction results in a mass shift of −16 da. The peaks correspond to the mutant (mut) and wild-type (wt) fragment, respectively.
Detecting Complex Sequence Variations
Complex sequence variations encompassing several nucleotides are usually not recognized by the MassCleave software using standard settings, but are indicated as several other single-nucleotide sequence variations. For instance, we detected a three-nucleotide insertion (2515InsCTT) in the 3′ UTR of the PINK1 gene in 22 patients. SNP Discovery indicated this special three nucleotide insertion as several other single base polymorphisms. However, this polymorphism was detected correctly after recalculation with an adjusted variation length, ie, “threshold compomer distance” of three bases (Figure 3). In four cases, there was a heterozygous SNP directly adjacent to the three nucleotide insertion, which was readily detected with standard settings, since unambiguous fragments are produced independently of the insertion. Furthermore, we were able to determine the haplotype of both variations present on the same DNA strand based on the acquired spectra.
Figure 3.
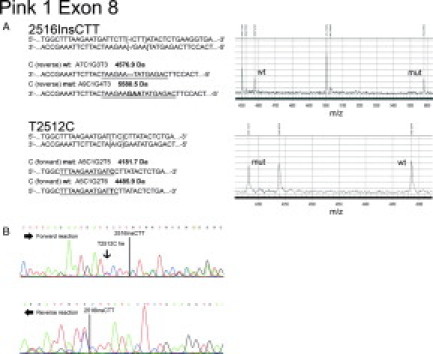
Detection of complex sequence changes with hMC-derived mass spectra. A: Sequence variations (left) and corresponding mass spectra (right) for a heterozygous three-nucleotide insertion (1215InsCTT, upper panel) and an adjacent heterozygous SNP (T2512C, lower panel) in exon 8 of the PINK1 gene. Fragments indicative of the respective sequence variation are underlined. The peaks correspond to the mutant (mut) and wild-type (wt) fragments, respectively. B: Dideoxy sequencing reaction of the same DNA. Forward (upper) and reverse (lower) reactions are shown. The positions of both sequence variations are indicated. Interpretation of the sequencing reaction is hardly possible due to the shift of the trace data after the insertion.
Validation of Sequence Alterations by Dideoxy Sequencing
Common SNPs (Table 1), which occurred in more than three patients, were validated by sequencing and confirmed in three randomly chosen positive homozygous and heterozygous samples with the intention of validating a maximum number of SNPs with minimal sequencing effort. Patients with identical spectra were considered to have the same variations. Additionally, three patients without that particular spectrum were also sequenced as negative controls. In contrast, all mutations known to cause ARPD (Table 1) or previously unknown variations (Table 2) were confirmed by automated sequence analysis in all samples concerned.
The ranking of SNPs in the SNP Discovery software is based on scores assessed due to the spectral quality. These scores are indicators of whether the acquired spectrum supports a putative sequence alteration and each spectrum receives a score between −1 and +1 depending on spectral quality and significance. Scores are also assigned for homozygosity of the calculated sequence variation. These scores are multiplied by a weight factor based on the spectral quality and added up to give a sample score. The sample score of a SNP varies between 0 and +10, where the value corresponds to the evidence supporting the denoted alteration. By default, sequence variations with a score of at least 2.0 are selected by the software. To include sequence variations from spectra with lower quality, we used a reduced overall score of 1.5. We discovered that SNPs indicated by the software below that limit could not be verified by sequencing and were therefore excluded (provided that at least three reactions were successful). Only one single alteration with a score lower than 1.5 could be confirmed by sequencing. However, for this alteration, only two of the three reactions were of sufficient quality. Furthermore, SNPs with a score above 1.5 but with at least one contradictory spectrum score of −1 were also shown to be false positives. Taken together, three different sequence variations in seven cases could not be verified, corresponding to an error rate of 1.70% (7/410). With respect to the total number of nucleotides analyzed, we observed only seven incorrect nucleotides out of a total of 367,195, which corresponds to an error rate of 0.0019%. The three SNPs that could not be verified had a score around 1.5 with some spectra with negative scores and were therefore close to our inclusion criteria. The status of variations, either heterozygous or homozygous, was predicted correctly in 96.23% of cases, following sequencing and manual analysis (in 93.08% on automatic analysis). Difficulties occurred particularly with amplicons that generated several fragments of equal molecular weights, and this interfered with calculation of the sequence changes. Consequently, this estimate of accuracy could easily be exceeded by using special amplicon designs that generate fragments with different molecular weights.10
Discussion
To date, no efficient, fast, and precise diagnostic tools for mutation analysis of ARPD have been discovered. Previous studies concerning ARPD focused only on single loci, which is a highly limited strategy for routine diagnosis of ARPD mutations. Here, we present the first screening setup for all three known ARPD genes, Parkin, DJ-1 and PINK1, using MALDI-TOF mass spectrometry and hMC-Array system. We screened four patients with PD with known sequence variations and were able to confirm all these variations. We also analyzed 31 additionally uncharacterized patients with EOPD and found a total of 410 variations that mainly represented common SNPs. Two mutations in three different samples that could be either heterozygous or homozygous were found (Table 1). One of these patients had a homozygous mutation in Parkin exon 7 that would be likely to cause EOPD, whereas the other two samples were heterozygous for their mutations. In fact, heterozygosity may also contribute to EOPD, as was shown for Parkin.18 Furthermore, large deletions that span entire exons, such as those that have previously been described for the Parkin gene, will not be detected with our approach, and therefore, the approach should be complemented by quantitative PCR or multiple ligation-dependent probe amplification.2,19
Experiences with hMC-Array and MALDI-TOF MS
The software used for this screening was originally designed for SNP Discovery in large populations. With several alterations, we were able to use this software for mutation screening However, future software releases may further facilitate this process. Since SNP Discovery was developed for detection of single-nucleotide exchanges, we also had difficulties recognizing larger sequence variations, as seen with the three nucleotide polymorphism described above (Figure 3). However, even with standard settings, the software indicates these changes as well as several other possible single-nucleotide exchanges. After recalculation with proper adjustments, this polymorphism was correctly calculated. So even if the correct nature of the sequence change is not readily revealed in every case, it is indicated and can be analyzed in more detail and will not be missed. Detection of the same sequence variation demonstrates the high resolution and power of the technology. The detection of a heterozygous SNP next to a heterozygous three-nucleotide insertion is challenging using direct sequencing (Figure 3), whereas MALDI-TOF MS spectra circumvent these restrictions and both changes were resolved. Furthermore, it was possible to extract the haplotype with both alterations residing on the same allele. Taken together, mutation detection was actually improved compared with other screening methods, since MALDI-TOF MS spectra are acquired within seconds and avoid the effort required to sequence the entire gene. Sequencing of known variations might not be necessary due to exact allocation of position, base, and kind of alteration, and this would therefore reduce validation efforts. Contrary to Stanssens et al,10 we detected three false-positive sequence alterations in all affected samples (1.7% of all alterations). Although they were close to our detection limits, the fact that false-positive sequence variations were interpreted as mutations may lead to a false diagnosis and treatment. However, in our opinion, validation of sequence changes by sequencing is still a necessity whenever new variations occur. Data for all other variations were coherent.
The predictive power of hMC cleavage products for any particular nucleotide is strongly correlated with the number of reactions resulting in fragments with a minimum of four nucleotides containing the interrogated nucleotide position. In our experience, at least two reactions are necessary to receive valid results. Below that limit, the selection of possible sequence alterations spreads both in position information and type of base changes making more validation steps necessary. Since each strand is effectively cleaved in every reaction after a different type of base (C and T and forward as well as reverse), there are theoretically fragments longer than three nucleotides in at least in two reactions in every sample. Generally, MALDI-TOF MS with hMC-Assay is able to detect 98% of all possible SNPs in a 500-bp fragment.10 However, in practice, some reactions might not produce interpretable results due to low quality MS spectra, fragments of masses beyond the detection range or several fragments of the same mass generating silent peaks. In agreement with Stanssens and colleagues,10 we also found a minority of SNPs that could not be allocated to a particular position but may occur in a limited number (generally two or three) of close nearby positions for the same alteration due to distinct base pair combinations. In these cases, sequencing is also a necessity.
Comparison of hMC with Alternative Screening Methods
Several screening methods are commonly used for the identification of sequence variations that might associate with a given disease. The MALDI-TOF MS system used in this study provides some major advantages compared to standard screening approaches like pyrosequencing, denaturing performance liquid chromatography, denaturing gradient gel electrophoresis, or single-strand conformational polymorphism. Pyrosequencing has several known inherent problems. The theoretical maximum of the reading length of one pyrosequencing run is estimated to lay beyond 300 nucleotides, and this is close to the size of PCR products analyzable with MALDI-TOF MS.20 Detection of unknown mutations and insertions/deletions of a different nucleotide may lead to out-of-phase sequencing reactions that can make interpretation of sequences very difficult.21 The same authors also discussed problems in determining the number of incorporated nucleotides in homopolymeric regions of more than five or six identical nucleotides due to nonlinear light responses. Software algorithms can partially compensate this effect. Another group experienced difficulties in the discovery of unknown mutations and variations due to the fact that only a few positions of “mutation hot spots” can be analyzed simultaneously.22 Chen et al23 noticed that pyrosequencing has not yet reached the level of accuracy needed in a diagnostic laboratory. In comparison, MALDI-TOF MS is able to detect previously unknown variations in a given amplicon, regardless of the location or type of sequence alteration (substitution, insertion, or deletion). We identified several unknown sequence polymorphisms by MALDI-TOF MS. The predicted base changes and zygosity could be verified in each case by capillary sequencing. In addition, base changes in homopolymeric stretches can be identified with confidence, since problems due to small cleavage fragments only occur in one cleavage reaction out of four, and therefore conclusions may be drawn from each reaction. As shown with an associated SNP, only three nucleotides upstream of a three-base insertion polymorphism, several alterations in the same amplicon can be identified next to each other. Pyrosequencing is comparable to MALDI-TOF MS in performance, especially in highly parallel systems.24 However, pyrosequencing is much more time-consuming for data acquisition and is associated with much higher costs.
The workflow of the MALDI-TOF MS SNP discovery is rather complex, with several post-PCR processing steps including in vitro transcription, RNase cleavage, conditioning, and finally acquisition of MS spectra, providing several potential failure points. The use of a much simpler and therefore more robust system is denaturing high performance liquid chromatography, in which the PCR product is analyzed without further processing on a reverse phase HPLC column under denaturing conditions. However, screening with denaturing high performance liquid chromatography is elaborate and difficult to standardize, since temperature conditions for each amplicon have to be established individually.25 Furthermore, denaturing high performance liquid chromatography has low resolution and does not supply any information regarding type, position, or status of the underlying nucleotide substitution. Therefore, validation by sequencing is required to determine alterations within sequences, especially for previously unknown variations. For the MALDI-TOF MS analysis presented here, we performed all reactions following a standard protocol with robotic support, thus reducing the necessary manpower to a minimum. Furthermore, the hMC protocol proved to be robust in performance, with only 47 of 4836 reactions yielding low-quality spectra. Single spectra are acquired within seconds and analysis using the SNP Discovery software is fast and accurate. Every sequence variation is denoted with its position, base type and status (Tables 1 and 2), saving time for sequence validation in most cases.
Sanger sequencing is considered to be the gold standard to identify sequence alterations. Compared to the MALDI-TOF MS protocol described here, direct sequencing of the target genes for EOPD would also be feasible using commonly available eight or 96 capillary sequencers at costs comparable to the hMC protocol. This would also directly yield information regarding position and type of sequence alteration. Assuming an average read length of 800 nucleotides in a 96-capillary sequencer, this would result in ∼38 kb of sequence information when both strands are sequenced. Since the majority of human exons are smaller than 800 nucleotides, the overall sequence information per run will in fact be less. With an average size of PCR products of 300 to 400 nucleotides, which is more likely to be the size of an exon, the analysis of a single 384-well SpectroCHIP results in 58 to 72 kb of sequence information from both strands. The throughput is further increased, since depending on the mass spectrometer, two to 10 chips can be analyzed in a single run. However, validation of identified sequence changes or mutations is not obsolete when direct sequencing is used, since due to good diagnostic practice each mutation should be verified on an independent PCR product.
Conclusion
We have established the first mutation screening for previously unknown mutations using an hMC assay in a patient study and detected two disease-causing mutations in three of 31 patients with EOPD. We show that SNP Discovery is a valid possibility for mutation screening whenever a flexible low-cost method for high-throughput genotyping is needed. The hMC assay allows the sequence analysis even in complex situations and is therefore a highly efficient method for unbiased screening of patient groups.
Acknowledgements
We thank Sequenom Inc. and especially Dr. Susanne Müller for assistance during data acquisition.
Footnotes
Supported by Interdisciplinary Center of Clinical Research Tübingen IZKF project no. 1553-0-0 (to C.S.) and by the ReForM program of the Medical Faculty of the University of Regensburg.
C.S. and M.W. contributed equally to this work.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Supplementary data
References
- 1.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 2.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 3.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 4.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 5.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 6.Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology. 2004;62:389–394. doi: 10.1212/01.wnl.0000113022.51739.88. [DOI] [PubMed] [Google Scholar]
- 7.Pankratz N, Foroud T. Genetics of Parkinson disease. NeuroRx. 2004;1:235–242. doi: 10.1602/neurorx.1.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Boom D, Ehrich M. Discovery and identification of sequence polymorphisms and mutations with MALDI-TOF MS. Methods Mol Biol. 2007;366:287–306. doi: 10.1007/978-1-59745-030-0_16. [DOI] [PubMed] [Google Scholar]
- 9.Gut IG. DNA analysis by MALDI-TOF mass spectrometry. Hum Mutat. 2004;23:437–441. doi: 10.1002/humu.20023. [DOI] [PubMed] [Google Scholar]
- 10.Stanssens P, Zabeau M, Meersseman G, Remes G, Gansemans Y, Storm N, Hartmer R, Honisch C, Rodi CP, Bocker S, van den Boom D. High-throughput MALDI-TOF discovery of genomic sequence polymorphisms. Genome Res. 2004;14:126–133. doi: 10.1101/gr.1692304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachmann HS, Siffert W, Frey UH. Successful amplification of extremely GC-rich promoter regions using a novel “slowdown PCR” technique. Pharmacogenetics. 2003;13:759–766. doi: 10.1097/00008571-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Hartmer R, Storm N, Boecker S, Rodi CP, Hillenkamp F, Jurinke C, van den Boom D. RNase T1 mediated base-specific cleavage and MALDI-TOF MS for high-throughput comparative sequence analysis. Nucleic Acids Res. 2003;31:e47. doi: 10.1093/nar/gng047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- 15.Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, Tavella A, Marconi R, Nicholl DJ, Chien HF, Fincati E, Abbruzzese G, Marini P, De Gaetano A, Horstink MW, Maat-Kievit JA, Sampaio C, Antonini A, Stocchi F, Montagna P, Toni V, Guidi M, Dalla Libera A, Tinazzi M, De Pandis F, Fabbrini G, Goldwurm S, de Klein A, Barbosa E, Lopiano L, Martignoni E, Lamberti P, Vanacore N, Meco G, Oostra BA. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]
- 16.Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann Neurol. 2003;54:283–286. doi: 10.1002/ana.10675. [DOI] [PubMed] [Google Scholar]
- 17.Hedrich K, Schafer N, Hering R, Hagenah J, Lanthaler AJ, Schwinger E, Kramer PL, Ozelius LJ, Bressman SB, Abbruzzese G, Martinelli P, Kostic V, Pramstaller PP, Vieregge P, Riess O, Klein C. The R98Q variation in DJ-1 represents a rare polymorphism. Ann Neurol. 2004;55:145–146. doi: 10.1002/ana.10816. [DOI] [PubMed] [Google Scholar]
- 18.Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- 19.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mashayekhi F, Ronaghi M. Analysis of read length limiting factors in Pyrosequencing chemistry. Anal Biochem. 2007;363:275–287. doi: 10.1016/j.ab.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11:3–11. doi: 10.1101/gr.11.1.3. [DOI] [PubMed] [Google Scholar]
- 22.Sivertsson A, Platz A, Hansson J, Lundeberg J. Pyrosequencing as an alternative to single-strand conformation polymorphism analysis for detection of N-ras mutations in human melanoma metastases. Clin Chem. 2002;48:2164–2170. [PubMed] [Google Scholar]
- 23.Chen DC, Saarela J, Nuotio I, Jokiaho A, Peltonen L, Palotie A. Comparison of GenFlex tag array and pyrosequencing in SNP genotyping. J Mol Diagn. 2003;5:243–249. doi: 10.1016/S1525-1578(10)60481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosaki K, Udaka T, Okuyama T. DHPLC in clinical molecular diagnostic services. Mol Genet Metab. 2005;86:117–123. doi: 10.1016/j.ymgme.2005.07.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.