

Abstract
Mutations in nucleophosmin (NPM1) exon 12 are thought to be the most common genetic event in acute myelogenous leukemia (AML) and to confer favorable clinical prognoses. In this report, we describe a simple molecular test for the detection of NPM1 exon 12 mutations in patients with AML using polymerase chain reaction amplification of genomic DNA followed by the analysis of amplification products by capillary electrophoresis. Mutations were reproducibly detected when present in at least 5% of cells, and all NPM1 exon 12 mutations reported to date in AML could be identified using this method. This method was successfully employed using paraffin-extracted DNA, allowing for the examination of archived clinical specimens, and the assay was validated by the direct sequencing of 33 patient samples. This sensitive test is straightforward to perform and provides important information that can influence both the clinical management and treatment options for many patients with AML.
Acute myelogenous leukemia (AML) is a heterogeneous disease clinically, molecularly, and cytogenetically. Approximately 30% of cases display recurrent cytogenetic abnormalities, typically reciprocal translocations, which help define distinct entities and often confer a favorable prognosis.1 However, approximately 40% to 50% of AML cases have normal karyotypes with variable prognoses. Several genetic changes, including mutations in nucleophosmin (NPM1) and FMS-like tyrosine kinase 3 (FLT-3), have been identified in karyotypically normal AML that can aid in prediction of clinical outcome. NPM1 mutations in AML have been reported to occur in about half of karyotypically normal cases and to confer a more favorable disease outcome, especially in the absence of FLT-3 mutations.2,3,4
Mutations in NPM1 in AML all involve exon 12 and were originally identified due to the resulting mislocalization of the mutant protein to the cytoplasm.5 Although at least 37 different NPM1 exon 12 mutations have been identified in AML, the vast majority of cases harbor type A (75% to 80%), type B (10%), and type D (5%) mutations.6 The type A mutation is an insertion of the tetranucleotide TCTG after position 863 of the coding sequence (GenBank accession no. NM_002520). The remaining cases harbor insertions of different tetranucleotides at the same position or, rarely, insertion/deletion mutations at other locations in exon 12. All mutations cause a frameshift in translation near the C terminus and abolish at least one of two tryptophan residues essential for a nucleolar localization signal. In addition, a CRM1-dependent nuclear export signal is acquired in the new reading frame of all mutations. As a result, the mutant proteins are mislocalized to the cytoplasm.
It is believed that NPM1 mutations are an early event in transformation based on their stability,7 but it is still unclear by what mechanism NPM1 contributes to the development of AML. Wild-type NPM1 is a nucleolar phosphoprotein with multiple functions. It normally acts as a chaperone during shuttling of pre-ribosome particles from the nucleolus to the cytoplasm. Several tumor suppressor proteins, including p53, ARF, and IRF-1, physically interact with and are regulated by NPM1.8,9,10 NPM1 also binds to centrosomes and regulates their duplication during the cell cycle.11 NPM1 could therefore be affecting leukemia development by altering the normal function of a variety of proteins.
The availability of an accurate and rapid test for the presence of NPM1 exon 12 mutations is of importance to help direct the appropriate treatment of patients with AML that have normal cytogenetic studies. Here we describe a simple and sensitive test using polymerase chain reaction (PCR) amplification of genomic DNA and capillary electrophoresis.
Materials and Methods
Samples and DNA Preparation
Leftover cryopreserved peripheral blood and bone marrow patient specimens sent to the ARUP hematological flow cytometry laboratory for leukemia phenotyping were used for these studies. All were diagnostic of AML and contained on average 63% leukemic blasts (21% to 99% range). Of the 33 AML cases, 11 were females and 22 were males, the mean age was 64 (range, 18–103), and 28 were CD34-positive. The research use of these specimens was approved by the University of Utah Internal Review Board (IRB no. 11905). The cell line OCI-AML3 was obtained from the German Collection of Microorganisms and Cell Cultures and grown in α-minimal essential medium (Invitrogen Corp., Carlsbad, CA; catalog no. 32571) supplemented with 20% fetal bovine serum. The SUDHL-4 cell line was obtained from Dr. Kojo Elenitoba-Johnson (University of Michigan, Ann Arbor, MI) and grown in RPMI 1640 medium (Invitrogen Corp., catalog no. 72400) supplemented with 10% fetal bovine serum. Cells were harvested by centrifugation and genomic DNA was extracted using the cultured cells protocol of the Puregene kit (Qiagen, Inc., Valencia, CA; catalog no. 158745). DNA samples were brought to a final concentration of 50 ng/μl.
PCR Amplification and Capillary Electrophoresis
Genomic DNA was amplified with the primers NPM-F, 6-FAM-5′-GATGTCTATGAAGTGTTGTGGTTCC-3′, and NPM-R, 5′-GGACAGCCAGATATCAACTG-3′. Reactions of 20 μl contained 100 ng of genomic DNA, primers (0.2 μmol/L each), deoxynucleoside-5′-triphosphates (0.2 mmol/L each), 1X cloned Pfu buffer (Stratagene, La Jolla, CA), 1.25 units of Pfu Turbo (Stratagene), MgCl2 (3.5 mmol/L final), where 1.5 mmol/L MgCl2 is contributed by the cloned Pfu buffer. After an initial denaturation at 94°C for 2 minutes, DNA was amplified in 35 cycles of 94°C for 20 seconds, 60°C for 20 seconds, 72°C for 20 seconds, and followed by a hold at 72°C for 2 minutes and a cooldown. In initial experiments we amplified the DNA with 1 U of GoTaq Flexi polymerase (Promega Corp., Madison, WI) in 1X Green GoTaq Flexi buffer and MgCl2 (3 mmol/L) using the same primers and cycling conditions (see below). The 6-FAM-labeled PCR products were diluted fivefold in water, and 1 μl was mixed with 9 μl of HiDi formamide (Applied Biosystems, Inc., Foster City, CA) and 0.5 μl of GeneScan ROX 350 internal size standards (Applied Biosystems, Inc.) and heated to 95°C for 2 minutes. The samples were run on an ABI 3130xl Genetic Analyzer using 36-cm capillaries and POP-7 polymer. The samples were injected at 2 kV for 5 seconds and run at 15 kV for 950 seconds at 60°C. PCR products and internal standards were detected using filter set D. Raw data were analyzed with GeneMapper v4.0 software (Applied Biosystems, Inc.). For simplicity, rounded base pair values are used in the text. The original fragment length values are shown in the figures.
DNA Sequence Analysis and Cloning of Individual NPM1 Alleles
For direct DNA sequence analysis 10 μl of PCR product was mixed with 2 μl of ExoSAP-IT (USB Corp., Cleveland, OH) and incubated at 37°C for 45 minutes followed by an enzyme inactivation step at 85°C for 5 minutes. For DNA sequence analysis 6 μl of DNA template was mixed with 8 μl of NPM-R primer (0.8 μmol/L) and sequenced using BigDye Terminator chemistry and an ABI 3100 Genetic Analyzer (Applied Biosystems, Inc.) according to the manufacturer's instructions. For DNA sequence analysis of individual alleles to confirm the presence or absence of length polymorphisms, cloned products were evaluated. For this, NPM1 was amplified as described above with primers NPM-F-xba, 5′-GCATCTAGAGATGTCTATGAAGTGTTGTGGTTCC-3′, and NPM-R-hind, 5′-GCAAAGCTTGGACAGCCAGATATCAACTG-3′. The PCR products were purified with the QIAquick PCR purification kit (Qiagen, Inc.) and digested with XbaI and HindIII (New England Biolabs Inc., Ipswich, MA). The fragments were ligated with T4 DNA ligase (New England Biolabs, Inc.) into pBluescript II KS(+) (Stratagene) digested with the same enzymes. After transformation of the ligations into TOP 10 Escherichia coli cells (Invitrogen Corp.) 1-ml cultures from five colonies of each ligation were grown overnight, and plasmid DNA was isolated with the QIAprep Spin Miniprep kit (Qiagen, Inc.) in a final volume of 20 μl. Six microliters of plasmid DNA were sequenced as above.
Results
Assay Design
Primers were designed to amplify a genomic fragment that contains the coding region of NPM1 exon 12. The human genome contains multiple NPM1-like elements that lack intronic sequences and are the likely result of insertions of reverse-transcribed mRNA. Following a BLAST search of the GenBank database, approximately 30 copies on 17 human chromosomes were identified with a high degree of homology to a portion of the NPM1 cDNA comprising exons 11 and 12. We therefore designed a forward primer within the intron between NPM1 exons 11 and 12, a region unique to the NPM1 gene on chromosome 5 (Figure 1). Moreover, no polymorphisms were identified at these primer sites. Since all mutant cases reported so far are heterozygous, we use the presence of a wild-type peak as an indication of successful DNA extraction and PCR amplification. According to the GenBank sequence of NPM1 we expect a 169-bp wild-type fragment and in mutant cases an additional 173-bp mutant fragment (or a 174-bp fragment in very rare cases of a net 5-bp insertion).
Figure 1.
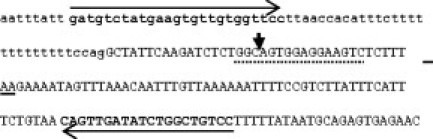
Genomic DNA sequence of the junction of NPM1 intron 11 and exon 12. Intronic residues are in lower case. The NPM1 wild-type stop codon is underlined. The vertical arrow indicates the site of tetranucleotide insertion for the most common mutation types. The type A mutation results in the duplication of the preceding TCTG sequence. Regions of PCR primer binding are shown in bold. The dotted line indicates the area where insertions and insertion/deletions in AML have been observed.6
Amplification using TaqDNA polymerase shows a high proportion of +1 fragment peaks following capillary electrophoresis of PCR products (Figure 2A). This peak variation was strongly suppressed by the use of a high-fidelity DNA polymerase (Figure 2B), consistent with polymerase slippage likely due to a track of 13 T residues in the intron just downstream of the forward primer. For all further experiments the Pfu Turbo high-fidelity DNA polymerase was used.
Figure 2.
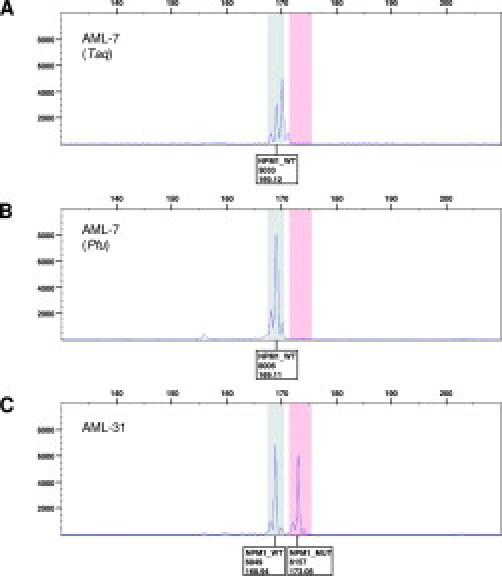
Capillary electrophoresis of a representative wild-type (A, B) and mutant sample (C). The wild-type sample in A has been amplified with TaqDNA polymerase. The samples in B and C have been amplified with Pfu Turbo DNA polymerase. Boxes under the peaks indicate allele type, peak height, and calculated fragment length.
Detection of NPM1 Mutations in AML Samples
Thirty-three whole blood or bone marrow aspirate samples from patients with AML were tested for the presence of NPM1 exon 12 mutations by the capillary electrophoresis assay. A total of nine mutant AML samples were identified, and we also confirmed the mutation previously reported to be present in the cell line OCI-AML3.12 All five CD34-negative samples were mutant, which is in agreement with previous studies that show an association between the presence of NPM1 exon 12 mutations and CD34 negativity.5 All mutant samples and 24 AML samples found to be wild type by the capillary electrophoresis assay were confirmed by direct DNA sequence analysis of the PCR product. Eight of the nine mutant samples were found to carry a type A mutation, and one had the rare type Nm mutation.6 Representative capillary electrophoresis results of a wild-type (AML-7) and mutant (AML-31) AML sample are shown in Figure 2, B and C. The calculated fragment lengths are close to the expected values. DNA sequence analysis of the wild-type (AML-7) sample and the cloned mutant allele of the type A mutant sample (AML-31) are shown in Figure 3. In three cases paired formalin-fixed paraffin-embedded clot sections were also available for analysis and gave congruent results (two wild types and one type Nm mutant).
Figure 3.
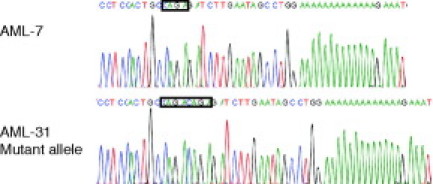
DNA sequence analysis of a wild-type (AML-7) and mutant (AML-31) sample. For AML-31 the mutant allele was first cloned and then sequenced. The reverse PCR primer was used as a sequencing primer yielding the inverse complement of the sequence shown in Figure 1. The unduplicated wild-type and duplicated mutant tetranucleotide sequence is boxed.
Intronic Length Polymorphisms
In five of the 33 clinical samples, both wild type and mutant, we observed a length polymorphism of +1 or −1 in the wild-type fragments, resulting in fragment lengths ranging from 168 to 170 bp (Figure 4). In the AML-40 wild-type sample two wild-type fragments of 168 and 169 bp are present, whereas in the wild-type sample AML-37 two wild-type fragments of 169 and 170 bp are present (Figure 4A). An additional wild-type sample (AML-5, data not shown) had a single peak at 168 bp. It is not clear if this sample is homozygous for a −1 length polymorphism or if one allele has been deleted. The mutant sample AML-60 and the cell line OCI-AML3 harbor single wild-type fragments of 168 and 170 bp, respectively, differing from the expected value of 169 bp (Figure 4B). Cloning followed by DNA sequence analysis of samples with aberrant wild-type peaks revealed length polymorphisms in the poly-T track in the intronic portion of the amplified fragment. Fragments of 168 bp had 12 T residues and fragments of 170 bp had 14 T residues (Figure 5). Based on the range of fragment sizes observed due to the polymorphisms in the intron, we expect wild-type fragments to occur in a range of 168 to 170 bp. By analogy, we expect mutant fragments (+4 or very rarely +5 net insertions4) to occur in a range of 172 to 175 bp, although so far we only observed mutant fragments of 173 bp. From four tests run on different days the average calculated fragment lengths ±2 SD were as follows: 168.1 ± 0.232 bp, 169.17 ± 0.111 bp, and 170.19 ± 0.159 bp, respectively, for the various wild-type fragments, and 173.31 ± 0.173 bp for the mutant fragment. For allele identification, the GeneMapper software was set up with a wild-type bin of 167.5 to 170.5 bp and a mutant bin of 171.5 to 175.5 bp.
Figure 4.
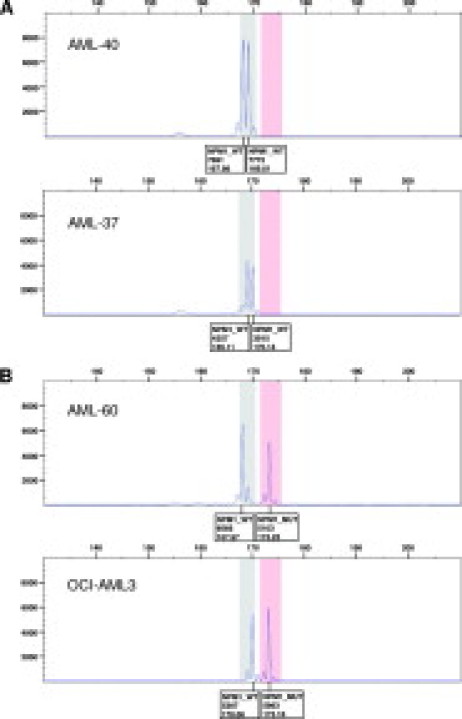
Capillary electrophoresis of samples with aberrant wild-type peaks. A: Wild-type samples AML-40 and AML-37. B: Mutant sample AML-60 and mutant cell line OCI-AML3. Boxes under the peaks indicate allele type, peak height, and calculated fragment length.
Figure 5.
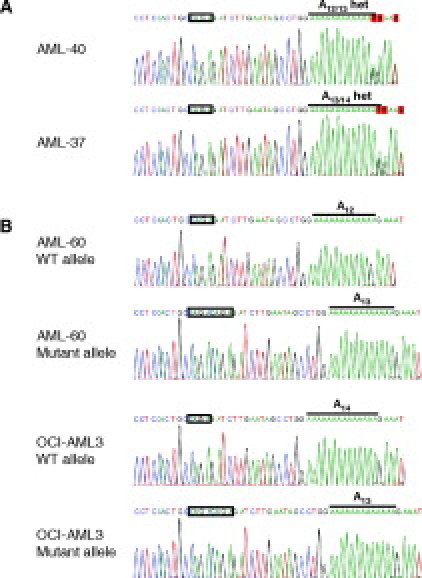
DNA sequence analysis of samples with aberrant wild-type peaks. Sequencing was performed with the reverse PCR primer, yielding the inverse complement of the sequence shown in Figure 1. The unduplicated wild-type and duplicated mutant tetranucleotide sequences are boxed. The poly-T track is indicated with a bold line. A: Wild-type samples AML-40 and AML-37. Total PCR products were sequenced. A heterozygous poly-T track length is observed due to heterozygosity of the samples. B: Mutant sample AML-60 and mutant cell line OCI-AML3. The mutant and wild-type alleles were cloned (see Materials and Methods) and sequenced separately.
Analytical Sensitivity
To determine the detection limit of this test we serially twofold diluted NPM1 mutated OCI-AML3 cells into NPM1 wild-type cells (SUDHL-4) and prepared genomic DNA from the cell mixtures. A NPM1 fragment was amplified as above, and the PCR products were analyzed by capillary electrophoresis. Figure 6 shows the results from four dilutions, 8-, 16-, 32-, and 64-fold, of the positive cell line OCI-AML3. The constant 169-bp wild-type peak in Figure 6 is derived from the SUDHL-4 diluent cell line. We reproducibly detected a mutant peak at 173 bp when the positive cell line was diluted up to 32-fold. A no-template or wild-type control did not harbor any peaks in the mutant fragment range (data not shown).
Figure 6.
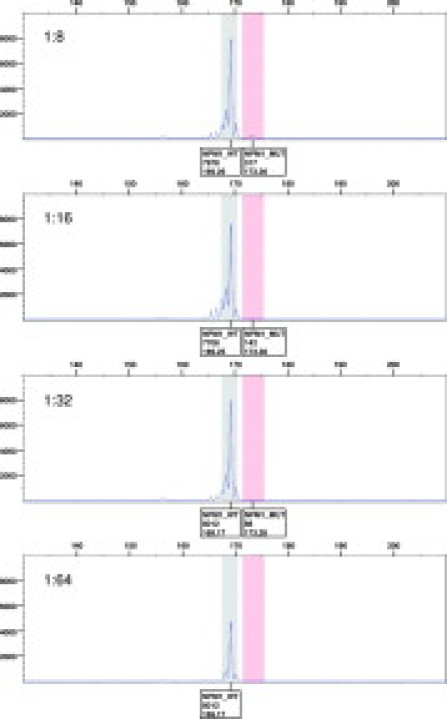
Capillary electrophoresis of serially diluted mutant cell line samples. OCI-AML3 cells were serially twofold diluted into NPM1 wild-type SUDHL-4 cells. Results from the 8-, 16-, 32-, and 64-fold dilutions are shown. Boxes under the peaks indicate allele type, peak height, and calculated fragment length.
Discussion
We have developed a simple and sensitive test for the detection NPM1 exon12 mutations in AML specimens that involves PCR amplification from genomic DNA and product analysis by capillary electrophoresis. NPM1 mutations are identified by the appearance of an additional fragment of increased length relative to the wild-type fragment. The wild-type fragment was used as an internal quality control for test performance, since all reported NPM1 mutations are heterozygous, consistent with homozygous NPM1 mutations being deleterious or embryonic lethal.13 Our test will detect all known exon 12 mutations and is sensitive enough to detect one mutated cell in 20 normal cells. Moreover, the short size of the PCR fragment also allows for successful detection of NPM1 mutations using DNA prepared from formalin-fixed paraffin-embedded cells. Since all AML-associated NPM1 mutations have the same effect on protein function,6 it is not necessary to identify the specific type of mutation present by sequence analysis.
A key design feature of our test was placing the forward PCR primer in intron 11 to eliminate any potential interference from several related pseudogene-like reverse-transcribed elements in the genome. Although this approach added a stretch of repetitive T sequences to the amplicons initially causing varying amounts of +1 and −1 bp fragment size variation, the subsequent use of a high-fidelity polymerase eliminated most of this effect. In addition, the minimal remaining one nucleotide noise did not interfere with the identification of even low amounts of mutant DNA, since the mutant and wild-type peaks are usually separated by 4 to 5 bp. It is interesting that in approximately 10% to 15% of cases we identified +1 and −1 bp length polymorphisms of this poly-T track in wild-type alleles from both mutated and nonmutated specimens. We have not yet detected 1 bp length polymorphisms in the poly-T tracks of any mutant fragments. However, since only small numbers of mutated cases were analyzed, additional studies would be required to determine whether these 1-bp polymorphisms could potentially affect the acquisition of exon 12 mutations.
NPM1 exon 12 mutations can also be detected by immunohistochemical staining based on the resulting mislocalization of the mutant protein to the cytoplasm.5 However, this method may be difficult to interpret due to close packing of blasts and/or the blasts having little cytoplasm.14 In addition, a nucleic acid-based test adds the advantage of being able to clearly identity small numbers of mutated cells admixed with high numbers of nonmutated cells. Noguera et al15 have also recently reported a PCR capillary electrophoresis-based test for detection of NPM1 exon 12 mutations starting with RNA isolated from AML samples. However, the DNA-based test we have described has the advantage of not requiring a reverse transcription step and can be performed on paraffin-embedded tissue specimens. This later feature affords the opportunity to retrospectively test for NPM1 mutations from archived diagnostic bone marrow specimens.
In conclusion, the test we have described for detecting NPM1 mutations in AML specimens is straightforward to perform, has good sensitivity, and works for most types of clinical specimens, including fixed tissue. The availability of such a test, when coupled with FLT-3 mutation analysis, should have considerable clinical utility in choosing treatment options for patients with AML and can also help with proper interpretation of ambiguous immunohistochemical staining results to assess cytoplasmic NPM1 localization.
Footnotes
Supported by the ARUP Institute for Clinical and Experimental Pathology, LLC.
References
- 1.Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–2333. [PubMed] [Google Scholar]
- 2.Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, Haferlach T, Hiddemann W, Falini B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–3739. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 3.Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, Ehninger G. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML) Blood. 2006;107:4011–4020. doi: 10.1182/blood-2005-08-3167. [DOI] [PubMed] [Google Scholar]
- 4.Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, Bullinger L, Frohling S, Dohner H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 5.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 6.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109:874–885. doi: 10.1182/blood-2006-07-012252. [DOI] [PubMed] [Google Scholar]
- 7.Palmisano M, Grafone T, Ottaviani E, Testoni N, Baccarani M, Martinelli G. NPM1 mutations are more stable than FLT3 mutations during the course of disease in patients with acute myeloid leukemia. Haematologica. 2007;92:1268–1269. doi: 10.3324/haematol.11202. [DOI] [PubMed] [Google Scholar]
- 8.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 9.Kondo T, Minamino N, Nagamura-Inoue T, Matsumoto M, Taniguchi T, Tanaka N. Identification and characterization of nucleophosmin/B23/numatrin which binds the anti-oncogenic transcription factor IRF-1 and manifests oncogenic activity. Oncogene. 1997;15:1275–1281. doi: 10.1038/sj.onc.1201286. [DOI] [PubMed] [Google Scholar]
- 10.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004;24:985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 12.Quentmeier H, Martelli MP, Dirks WG, Bolli N, Liso A, Macleod RA, Nicoletti I, Mannucci R, Pucciarini A, Bigerna B, Martelli MF, Mecucci C, Drexler HG, Falini B. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19:1760–1767. doi: 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- 13.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Rassidakis GZ, Medeiros LJ. Nucleophosmin gene mutations in acute myeloid leukemia. Arch Pathol Lab Med. 2006;130:1687–1692. doi: 10.5858/2006-130-1687-NGMIAM. [DOI] [PubMed] [Google Scholar]
- 15.Noguera NI, Ammatuna E, Zangrilli D, Lavorgna S, Divona M, Buccisano F, Amadori S, Mecucci C, Falini B, Lo-Coco F. Simultaneous detection of NPM1 and FLT3-ITD mutations by capillary electrophoresis in acute myeloid leukemia. Leukemia. 2005;19:1479–1482. doi: 10.1038/sj.leu.2403846. [DOI] [PubMed] [Google Scholar]