

Abstract
Lung adenocarcinomas responsive to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors possess EGFR mutations and often increased EGFR copy number. We prospectively studied 334 clinical cases using polymerase chain reaction-based assays to detect deletions within exon 19 and the L858R mutation in exon 21, which together account for approximately 90% of EGFR mutations. Seventy-eight (23%) of these tumors had an EGFR mutation, with 55 (71%) exon 19 deletions and 23 (29%) exon 21 L858R mutations. We were able to compare mutant and normal EGFR alleles and found a preferential amplification of the mutant allele. The association of mutations with EGFR amplification (determined by chromogenic in situ hybridization) and EGFR expression (determined by immunohistochemistry) was further examined in a subset of 60 tumors. EGFR amplification (≥5 EGFR signals per nucleus) was seen in 15 of 29 (52%) EGFR-mutated tumors but in only five of 31 (6%) non-mutated tumors (P = 0.006). EGFR overexpression was strongly associated with amplification but was statistically independent of EGFR mutation. Most patients with EGFR mutations (17 of 29, 59%) never smoked compared with 13% (four of 31) of patients lacking such mutations (P = 0.0003). The association of amplification with smoking status was marginal and was nonexistent with EGFR expression. Thus, these results indicate that EGFR amplification, preferentially of the mutant allele, often accompanies EGFR mutation, whereas EGFR immunohistochemical staining associates with amplification but cannot predict EGFR mutation status.
Recent studies have shown that somatic mutations in the EGFR TK domain in patients with lung adenocarcinoma are associated with sensitivity to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib.1,2,3 The epidermal growth factor receptor (EGFR, HER-1/ErbB1) is a receptor tyrosine kinase (TK) of the ErbB family, which consists of four closely related receptors: HER-1/ErbB1, HER-2/neu/ErbB2, HER-3/ErbB3, and HER-4/ErbB4. The two most common EGFR mutations are short in-frame deletions of exon 19 and a point mutation (CTG to CGG) in exon 21 at nucleotide 2573, which results in substitution of leucine by arginine at codon 858 (L858R). Together, these two mutations account for ∼90% of all EGFR mutations in non-small cell lung cancer (NSCLC). Screening for these mutations in patients with NSCLC can be used to predict which patients will respond to TKIs. So far, most large series have used direct sequencing to detect these mutations. We developed polymerase chain reaction (PCR)-based clinical diagnostic tests for the two EGFR hotspot mutations4 and report our initial experience with 334 prospective clinical cases. We also report on the associations of EGFR mutation, amplification, and protein expression in NSCLC.
Materials and Methods
Tumor Samples
Three hundred thirty-four lung cancer samples, mainly adenocarcinoma, were received in the Memorial Sloan-Kettering Cancer Center Laboratory of Diagnostic Molecular Pathology over a consecutive 12-month period. Tumor DNA was extracted from formalin-fixed paraffin-embedded tissue or frozen fine needle biopsies or frozen surgical resections using standard methods.
EGFR Mutational Analysis
EGFR Exon 19 Deletion Assay
The assay is based on length analysis of fluorescently labeled PCR products.4 By its design, it should also detect the much rarer EGFR exon 19 insertions.5 Briefly, a 207-bp genomic fragment including all of exon 19 is amplified, using the following primers: EGFR-Ex19-FWD1, 5′-GCACCATCTCACAATTGCCAGTTA-3′, and EGFR-Ex19-REV1, 5′-Fam-AAAAGGTGGGCCTGAGG-TTCA-3′.4 PCR products were subjected to capillary electrophoresis on an ABI 3730xl Genetic Analyzer (Applied Biosystems, Foster City, CA). For all cases with exon 19 deletions, the proportion of mutant and normal EGFR alleles was determined by comparison of peak heights on the electrophorogram. The ratio between mutant and normal EGFR alleles was calculated. As clinical samples always contain admixed non-neoplastic elements, sometimes abundant, any ratio >1.5 was interpreted as evidence of EGFR mutant allele amplification in this assay. We have found this ratio to be highly reproducible for a given tumor DNA sample (results not shown).
EGFR Exon 21 L858R Mutation Assay
This mutation is detected by a PCR-restriction fragment length polymorphism assay, based on a Sau96I restriction site created by the mutation (2573T→G).4 Briefly, a 222-bp genomic fragment including all of exon 21 was amplified using primers EGFR-Ex21-FWD1, 5′-CCTCACAGCAGGG-TCTTCTCTGT-3′, and EGFR-Ex21REV1, 5′-Fam-TCAGGAAAAT-GCTGGCTGACCTA-3′. If the 2573T→G mutation is present, after digestion, a 173-bp wild-type product and 87-bp mutant PCR product were produced. The digested fluorescently labeled PCR products are analyzed by capillary electrophoresis. These two assays are more sensitive than direct sequencing and can detect mutations in the presence of up to 90% non-neoplastic cells.4
Construction of Tissue Microarray
A subset of 60 tumors (29 EGFR mutated, 31 EGFR non-mutated) was selected for tissue microarray construction based on paraffin block availability for tumors with EGFR mutations. The 31 cases without EGFR mutation were matched for age, sex, and histology to the 29 EGFR mutated cases. The tissue microarray was constructed using triplicate 0.6-mm tissue cores. Three cores from different areas were selected for each tumor.
EGFR Chromogenic in Situ Hybridization (CISH)
CISH for EGFR was performed according to the manufacturer's instructions (Zymed Laboratories Inc., South San Francisco, CA). Briefly, 4- to 5-μm sections were incubated at 55°C overnight. After deparaffinization in xylene and graded ethanols, heat pretreatment was carried out in the pretreatment buffer (Zymed Laboratories Inc.) at 98–100°C for 15 minutes. The tissue was digested with pepsin for 10 minutes at room temperature. After application of Zymed SpotLight digoxigenin-labeled EGFR probe (Zymed Laboratories Inc.), the slide was coverslipped and edges sealed with rubber cement. The slide was heated at 95°C for 5 minutes followed by overnight incubation at 37°C using a moisturized chamber. A posthybridization wash was performed the next day and followed by immunodetection using the CISH polymer detection kit (Zymed laboratories Inc.) and the signal enumerated on a standard light microscope using a 40X objective. Gene copy numbers in 30 tumor cell nuclei were counted for each tissue core, and the average gene copies per nucleus were used as CISH result for that tissue core. The highest CISH score among all cores was used as the final result for that tumor. The results of CISH were interpreted as follows: <5 gene copies per nucleus, no amplification; 5–10 gene copies per nucleus, low-level amplification; and >10 gene copies per nucleus, high-level amplification.6
EGFR Immunohistochemistry
Immunohistochemistry (IHC) staining for EGFR was performed using monoclonal EGFR antibody 31G7 (Zymed Laboratories Inc.) according to the manufacturer's instructions. EGFR results were scored as follows: 0, no membrane staining; 1+, faint, partial membrane staining; 2+, weak, complete membrane staining in >10% of tumor cells; 3+, intense complete membrane staining in >10% of tumor cells. Tumors with a score 2+ or 3+ were interpreted as positive for overexpression.6,7 The highest score obtained among different cores of the same tumor was used as the final EGFR IHC result of that tumor.
Statistical Analysis
Statistical analyses were performed using Fisher's exact test or χ2 test. Statistical significance was determined by a two-tailed P < 0.01.
Results
Clinical Testing Experience in 334 Consecutive Cases
EGFR exon 19 deletions or exon 21 L858R were identified in 23% (78 of 334) of our consecutive lung cancer samples, over 90% of which were pure adenocarcinomas (Figure 1). Among the cases with mutations, 55 (71%) had an exon 19 in-frame deletion [deletion sizes: 9 bp (n = 3), 15 bp (n = 41), 18 bp (n = 9), 24 bp (n = 2)] and 23 (29%) had the exon 21 L858R point mutation. One patient had two synchronous primaries with different exon 19 alterations, a 15-bp deletion and an 18-bp insertion. The majority (64%) of the patients with mutations were women. Histologically, 42 (54%) of 78 cases were invasive adenocarcinoma, and 31 (40%) cases were either bronchioloalveolar carcinoma (BAC) or adenocarcinoma, mixed subtype with BAC (Figure 1). Age, sex, and histology did not differ significantly between patients whose tumors had exon 19 deletions compared to exon 21 L858R mutations.
Figure 1.
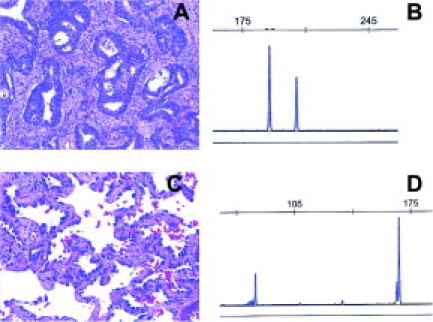
A and B: Adenocarcinoma with 15-bp EGFR exon 19 deletion. The mutant peak is to the left of the 207-bp normal peak. C and D: BAC with EGFR exon 21 L858R mutation. The 87-bp product of this PCR-RFLP assay represents the mutant allele (see Materials and Methods for details).
Operationally, all assays were performed in duplicate (ie, total four PCRs per sample) and all results were concordant in the duplicate reactions. The assays can be completed in 2 days on a rush basis but the turnaround time was generally 7 days because of weekly batching. Most of the specimens were submitted as paraffin-embedded material from surgical resections or, less commonly, from Tru-cut biopsies. Paraffin blocks containing more than 50% tumor were selected by a pathologist for DNA extraction. Ten 5-μ-thick sections were collected as curls in 1.5-ml tubes in each case. If the proportion of the tumor was less than 50%, then the tumor was manually microdissected using a sterile scalpel from 15 unstained sections using a stained section as a guide. In some cases, cell blocks from fine needle aspiration cytology specimens were used (after confirmation of adequacy of tumor content by a cytopathologist). In one case, only air-dried Giemsa-stained fine needle aspiration cytology smears were available. In this case, DNA was extracted from the smear using the PUREGENE kit (Gentra, Minneapolis, MN). Overall, only two samples received during this 12-month period gave no result due to insufficient DNA for PCR amplification. Both were fine needle aspiration cell blocks with inadequate cellularity. All cases received as frozen samples were successfully studied.
Association between EGFR Mutation and EGFR Amplification
In the 55 cases with exon 19 deletions, the relative levels of mutant and wild-type allele were determined by comparing peak heights in the electrophoretogram tracings of the exon 19 deletion assay. These ratios were highly reproducible (not shown) and were greater than, for instance, those typically associated with preferential PCR amplification of the shorter allele at a given polymorphic short tandem repeat locus (ratios generally <1.5). Therefore ratios >1.5 were interpreted as evidence of increased copies of the mutant allele in the tumor cells, given that the mutations are almost always heterozygous and that admixed non-neoplastic cells contain two normal alleles. In 15 of 55 cases (27%) the peak height of the mutant allele was greater than that of the normal EGFR allele (Figure 2). The ratios of peak heights ranged from 1.6 to 7.0 (mean, 2.5; median, 2.1). However, this may be an underestimation of the prevalence of EGFR mutant allele copy number gains because of varying degrees of dilution with DNA from admixed non-neoplastic cells.
Figure 2.
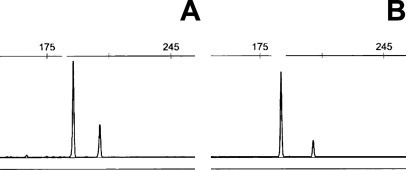
A and B: Cases with EGFR exon 19 deletions (of different sizes) showing three- and fivefold copy number gains of the mutant allele relative to the 207-bp normal allele.
To further assess the prevalence of EGFR amplification, we performed EGFR CISH analysis on a tissue microarray containing 29 EGFR mutated and 31 EGFR non-mutated cases. The clinical characteristics of the NSCLC cases represented on the tissue microarray are listed in Table 1. These cases were matched for age, sex, and histology. Twenty cases were scored as amplified (≥5 signals per nucleus) but only four of 20 showed 7 or more signals (7, 7.2, 8, and 11 signals). Fifty-two percent of tumors with EGFR mutations had EGFR amplification (15 of 29, 52%) as compared to 16% (five of 31) of non-mutated cases (P = 0.006) (Figure 3). There was no significant difference in the frequency of EGFR amplification between EGFR exon 19 deletion cases and L858R cases: among the 10 cases EGFR L858R, seven (70%) were amplified, compared to eight of 19 (42%) cases with EGFR exon 19 deletion (P = 0.25). The proportion of exon 19 deletion cases amplified by CISH was not significantly higher (eight of 19 versus 15 of 55; P = 0.26) than that determined above by the comparison of peak heights in the PCR-based assay. This suggests that most or all of the extra EGFR gene copies seen by CISH in cases with exon 19 deletions are gains of the mutant allele.
Table 1.
Characteristics of 60 Patients Included on Tissue Microarray
| Non-mutated EGFR (n = 31, 52%) | Mutated EGFR (n = 29, 48%) | P value | |
|---|---|---|---|
| Age (years) | |||
| Median | 70 | 69 | |
| Mean | 68 | 66 | |
| Range | 42–85 | 38–88 | |
| Sex | |||
| Male | 7 (23%) | 7 (24%) | |
| Female | 24 (77%) | 22 (76%) | 1.00 |
| Smoking history | |||
| Former and current | 27 (87%) | 12 (41%) | |
| Never | 4 (13%) | 17 (59%) | 0.0003 |
| Stage at presentation | |||
| I | 13 (42%) | 9 (31%) | |
| II | 5 (16%) | 4 (14%) | |
| III | 9 (29%) | 13 (45%) | |
| IV | 4 (13%) | 3 (10%) | 0.43* |
| Site of tumor | |||
| Primary | 31 (100%) | 27 (93%) | |
| Metastasis | 0 (0%) | 2 (7%) | 0.23 |
| Histology | |||
| Adenocarcinoma | 12 (39%) | 12 (41%) | |
| BAC or adenocarcinoma with BAC feature | 18 (58%) | 16 (55%) | |
| Other | 1 (3%) | 1 (4%) | 1.00† |
| EGFR gene amplification | |||
| Amplified | 5 (16%) | 15 (52%) | |
| Not amplified | 26 (84%) | 14 (48%) | 0.006 |
Fisher's exact test result for comparison of stage I versus II, III, and IV.
Fisher's exact test result for comparison of adenocarcinoma, BAC, or adenocarcinoma with BAC feature versus other.
Figure 3.
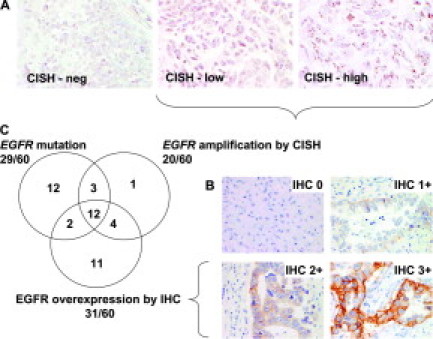
A: Chromogenic in situ hybridization showing no amplification, low-level amplification, and high-level amplification of the EGFR gene in three different lung adenocarcinomas. B: Lung adenocarcinomas with different levels of EGFR IHC staining: 0; 1+; 2+; 3+. Examples of both EGFR mutant and EGFR non-mutant cases are shown. C: Venn diagram showing relationships between EGFR mutations, EGFR amplification, and EGFR overexpression in a set of 60 lung tumors.
Association between EGFR Amplification and EGFR Protein Expression
Eighty percent (16 of 20) of EGFR-amplified tumors were IHC-positive as compared with 13 of 40 (33%) EGFR nonamplified tumors (P = 0.0008) (Table 2 and Figure 3). The proportion of EGFR-amplified cases increased from 11% to 67% as EGFR IHC staining increased from 0 to 3+ (Table 2). All four EGFR-amplified cases showing 7 or more signals per nucleus were also IHC-positive (one 2+, three 3+). Notably, the EGFR IHC results showed no significant association with EGFR mutation status (Table 3). These interrelationships are depicted in Figure 3.
Table 2.
Association of EGFR Gene Amplification and Protein Expression
| IHC | Gene amplification | No gene amplification | Total | P value |
|---|---|---|---|---|
| 0 | 1 (11%) | 8 | 9 | |
| 1+ | 3 (14%) | 19 | 22 | |
| 2+ | 6 (43%) | 8 | 14 | |
| 3+ | 10 (67%) | 5 | 15 | 0.0008* |
| Total | 20 (33%) | 40 | 60 |
EGFR IHC: 0–1+, negative; 2–3+, positive. Fisher's exact test result for comparison of EGFR IHC score versus amplification status.
Table 3.
Association of EGFR Mutations and Protein Expression
| IHC | Mutation | No mutation | Total | P value |
|---|---|---|---|---|
| 0 | 4 (44%) | 5 | 9 | |
| 1+ | 11 (50%) | 11 | 22 | |
| 2+ | 5 (36%) | 9 | 14 | |
| 3+ | 9 (60%) | 6 | 15 | 1.0* |
| Total | 29 (48%) | 31 | 60 |
EGFR IHC: 0–1+, negative; 2–3+, positive. Fisher's exact test result for comparison of EGFR IHC score versus mutation status.
Association between EGFR Mutation and Smoking History
Smoking history data were available for all 60 patients represented on the tissue microarray. Most patients (17 of 29, 59%) whose tumors bore EGFR mutations were never smokers, but this was the case for only four of 31 (13%) without EGFR mutations (P = 0.0003). In contrast, never-smoker status was not significantly associated with EGFR amplification status (P = 0.10) or EGFR IHC results (P = 0.79).
Discussion
Two EGFR mutations, exon 19 deletion and exon 21 L858R mutation, account for about 90% of all EGFR mutations reported in lung adenocarcinoma and are known to be predictive of response to the EGFR TKIs gefitinib and erlotinib.8,9 Screening for these mutations in patients with lung adenocarcinoma can be used to predict which patients will respond to the EGFR TKIs, and numerous testing methods have been proposed.10 With PCR-based tests for these mutations, our data showed a relatively high frequency of EGFR mutations (23%) for a North American patient population. This may reflect some referral bias for patients more likely to have EGFR mutations (using histology, smoking history, or ethnicity). A recent report from another high-volume North American clinical laboratory also described a 23% EGFR mutation rate in routine testing.11 Thus, judicious ordering of these tests can lead to more efficient use of clinical laboratory resources than originally estimated on the basis of a projected mutation rate of 10% in unselected populations of patients with non-small cell lung cancer. The prevalence of EGFR mutations in our clinical experience may also reflect the somewhat higher sensitivity of PCR-based tests compared to direct sequencing. These PCR-based tests can detect a lower proportion of mutant alleles than direct sequencing.4 Others have noted that a small but significant proportion of mutated cases are missed by direct sequencing of clinical tumor samples but can be detected by more sensitive techniques.12,13
The interrelationships and clinical significance of EGFR mutation, amplification, and protein expression are complex and remain controversial.7,14,15,16,17 In these studies, increased EGFR gene copy number/amplification has been reported in seven to 44% of cases. This range may be due to variations in techniques, criteria for determining amplification, and interobserver variability. In our study, CISH-detected EGFR amplification was found in 32% of lung adenocarcinomas, in keeping with previous studies based on fluorescence in situ hybridization. A recent systematic comparison of EGFR fluorescence in situ hybridization and CISH has validated the use of the latter in this setting.18 Using CISH, we found that amplification of EGFR in tumors with EGFR mutation is common (52%) and is more frequent (P = 0.006) than in cases lacking EGFR mutations. We propose that the reported predictive value of EGFR amplification for EGFR TKI response is at least in part due to its strong association with EGFR mutation. Moreover, because EGFR mutation assays are susceptible to false-negative results due to admixed non-neoplastic cells but EGFR fluorescence in situ hybridization assays are not, we speculate that a subset of EGFR-amplified cases that lack mutations by direct sequencing may contain mutations that would be detectable by more sensitive methods.
EGFR overexpression in NSCLC has been reported in 16 to 62% of cases.7,15,19,20 This range in values likely reflects the use of a variety of antibodies, protocols, and interpretation criteria, as well as subjectivity in scoring. EGFR overexpression was seen in 48% of tumors in our analysis, which correlated well with EGFR gene amplification (P = 0.0008). This correlation is consistent with previous studies.7,15,19 In contrast, we found no significant association between EGFR protein overexpression and EGFR mutation. Finally, it should also be noted that the 31 EGFR non-mutant cases were tested by direct sequencing for KRAS mutations, a known strong negative predictor of EGFR TKI response.21,22 This revealed KRAS mutations in eight of 31 samples (results not shown), of which five were positive by EGFR IHC, further underscoring the drawbacks of EGFR IHC as a way of casting a “wider net” for patients potentially responsive to these agents.
EGFR mutation was significantly associated with a history of never smoking (P = 0.0003), which was similar to previous reports.3,23 In contrast, never-smoker status was only marginally associated with EGFR amplification status (P = 0.10) and not at all with EGFR IHC results (P = 0.79).
Our clinical testing experience demonstrates that molecular testing of lung tumors for drug-sensitive EGFR mutations is a feasible, reliable, and relatively efficient process. Moreover, a routine turnaround time of about a week means that the results can be used to help guide treatment decisions regarding the use of gefitinib or erlotinib without clinically significant delays. To further ensure that this information is readily available in the chart at time of disease recurrence, we have recently implemented reflex EGFR mutation testing of all resected lung adenocarcinomas. While a negative test result does not currently eliminate the possibility of benefit from these drugs, a positive test can aid oncologists in several ways. First, as documented by several prospective clinical trials examining mutations and response rates, the presence of an EGFR mutation is associated with an aggregate 75% response rate.8,9,24 Thus, clinicians can feel more confident in choosing EGFR TKIs even as first-line therapy. Second, although never-smoker patients are reported to have a higher incidence of EGFR mutations, only 50% of never-smokers have such mutations and correspondingly, only about half of never-smokers respond to EGFR TKIs. Thus, documenting the lack of an EGFR mutation provides better justification for a never-smoker to switch therapy earlier rather than later in the disease course. As patients with mutations who respond to therapy often experience rapid disease progression after discontinuation of drug, a positive test provides a rationale for clinicians to continue TKI treatment even if there is gradual disease progression; in this setting, rather than discontinuing TKI, additional agents are added but mutant EGFR suppression is maintained. Finally, combining EGFR mutation testing with testing for KRAS mutations, which are mutually exclusive with the former and with response to EGFR TKIs,21,22 can help to further enhance response prediction and inform clinical decision-making.
In conclusion, lung adenocarcinoma with EGFR mutations is a distinct biological subset as evidenced by its strong association with never-smoker status and known high response rate to EGFR TKIs. Amplification of EGFR in NSCLC with EGFR mutation is common and, at least in exon 19 deletion cases, usually affects the mutant allele. Preferential amplification of the mutant allele has also been previously observed by other methods.25 We propose that the predictive value of EGFR amplification for EGFR TKI response is more likely a result of its association with EGFR mutation. EGFR overexpression by IHC is associated with EGFR amplification but is of no utility in predicting the presence of EGFR mutations.
Acknowledgements
We thank Yuanyuan Ma and Tao Zheng for expert technical assistance.
Footnotes
Supported by Joan's Legacy Foundation (to W.P.), the Doris Duke Charitable Foundation Clinical Scientist Development Award (to W.P.), and NIH grant PO1 CA129243 (to M.L. and M.G.K.).
A.R.L. and D.C. contributed equally to this work.
References
- 1.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski MF, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and correlate with sensitivity of tumors to gefitinib (Iressa) and erlotinib (Tarceva) Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan Q, Pao W, Ladanyi M. Rapid PCR-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn. 2005;7:396–403. doi: 10.1016/S1525-1578(10)60569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 6.Bhargava R, Gerald WL, Li AR, Pan Q, Lal P, Ladanyi M, Chen B. EGFR gene amplification in breast cancer: correlation with EGFR mRNA and protein expression and HER2 status and absence of EGFR activating mutations. Mod Pathol. 2005;18:1027–1033. doi: 10.1038/modpathol.3800438. [DOI] [PubMed] [Google Scholar]
- 7.Dacic S, Flanagan M, Cieply K, Ramalingam S, Luketich J, Belani C, Yousem SA. Significance of EGFR protein expression and gene amplification in non-small cell lung carcinoma. Am J Clin Pathol. 2006;125:860–865. doi: 10.1309/H5UW-6CPC-WWC9-2241. [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25:587–595. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 9.Ladanyi M, Pao W: Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol (in press) [DOI] [PubMed]
- 10.Pao W, Ladanyi M. Epidermal growth factor receptor mutation testing in lung cancer: searching for the ideal method. Clin Cancer Res. 2007;13:4954–4955. doi: 10.1158/1078-0432.CCR-07-1387. [DOI] [PubMed] [Google Scholar]
- 11.Sequist LV, Joshi VA, Janne PA, Bell DW, Fidias P, Lindeman NI, Louis DN, Lee JC, Mark EJ, Longtine J, Verlander P, Kucherlapati R, Meyerson M, Haber DA, Johnson BE, Lynch TJ. Epidermal growth factor receptor mutation testing in the care of lung cancer patients. Clin Cancer Res. 2006;12:4403s–4408s. doi: 10.1158/1078-0432.CCR-06-0099. [DOI] [PubMed] [Google Scholar]
- 12.Janne PA, Borras AM, Kuang Y, Rogers AM, Joshi VA, Liyanage H, Lindeman N, Lee JC, Halmos B, Maher EA, Distel RJ, Meyerson M, Johnson BE. A rapid and sensitive enzymatic method for epidermal growth factor receptor mutation screening. Clin Cancer Res. 2006;12:751–758. doi: 10.1158/1078-0432.CCR-05-2047. [DOI] [PubMed] [Google Scholar]
- 13.Thomas RK, Nickerson E, Simons JF, Janne PA, Tengs T, Yuza Y, Garraway LA, LaFramboise T, Lee JC, Shah K, O'Neill K, Sasaki H, Lindeman N, Wong KK, Borras AM, Gutmann EJ, Dragnev KH, DeBiasi R, Chen TH, Glatt KA, Greulich H, Desany B, Lubeski CK, Brockman W, Alvarez P, Hutchison SK, Leamon JH, Ronan MT, Turenchalk GS, Egholm M, Sellers WR, Rothberg JM, Meyerson M. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med. 2006;12:852–855. doi: 10.1038/nm1437. [DOI] [PubMed] [Google Scholar]
- 14.Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Shibata T, Sakiyama T, Yoshida T, Tamura T. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:6829–6837. doi: 10.1200/JCO.2005.01.0793. [DOI] [PubMed] [Google Scholar]
- 15.Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA, Jr, Varella-Garcia M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–655. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 16.Hirsch FR, Varella-Garcia M, McCoy J, West H, Xavier AC, Gumerlock P, Bunn PA, Jr, Franklin WA, Crowley J, Gandara DR. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin Oncol. 2005;23:6838–68345. doi: 10.1200/JCO.2005.01.2823. [DOI] [PubMed] [Google Scholar]
- 17.Bell DW, Lynch TJ, Haserlat SM, Harris PL, Okimoto RA, Brannigan BW, Sgroi DC, Muir B, Riemenschneider MJ, Iacona RB, Krebs AD, Johnson DH, Giaccone G, Herbst RS, Manegold C, Fukuoka M, Kris MG, Baselga J, Ochs JS, Haber DA. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005;23:8081–8092. doi: 10.1200/JCO.2005.02.7078. [DOI] [PubMed] [Google Scholar]
- 18.Sholl LM, John IA, Chou YP, Wu MT, Goan YG, Su L, Huang YT, Christiani DC, Chirieac LR. Validation of chromogenic in situ hybridization for detection of EGFR copy number amplification in nonsmall cell lung carcinoma. Mod Pathol. 2007;20:1028–1035. doi: 10.1038/modpathol.3800946. [DOI] [PubMed] [Google Scholar]
- 19.Hirsch FR, Varella-Garcia M, Bunn PA, Jr, Di Maria MV, Veve R, Bremmes RM, Baron AE, Zeng C, Franklin WA. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21:3798–3807. doi: 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 20.Pastorino U, Andreola S, Tagliabue E, Pezzella F, Incarbone M, Sozzi G, Buyse M, Menard S, Pierotti M, Rilke F. Immunocytochemical markers in stage I lung cancer: relevance to prognosis. J Clin Oncol. 1997;15:2858–2865. doi: 10.1200/JCO.1997.15.8.2858. [DOI] [PubMed] [Google Scholar]
- 21.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13:2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 23.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 24.Miller VA, Riely GJ, Zakowski MF, Li AR, Patel JD, Heelan RT, Kris MG, Sandler AB, Carbone DP, Tsao A, Herbst RS, Heller G, Ladanyi M, Pao W, Johnson DH. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26:1472–1478. doi: 10.1200/JCO.2007.13.0062. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama T, Kondo M, Goto Y, Fukui T, Yoshioka H, Yokoi K, Osada H, Imaizumi K, Hasegawa Y, Shimokata K, Sekido Y. EGFR point mutation in non-small cell lung cancer is occasionally accompanied by a second mutation or amplification. Cancer Sci. 2006;97:753–759. doi: 10.1111/j.1349-7006.2006.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]